
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphine addition to dehydroalanine for peptide modification†
Minglong
Liu
 a,
Miha
Sovrovic
b,
Hiroaki
Suga
c and
Seino A. K.
Jongkees
*abc
a,
Miha
Sovrovic
b,
Hiroaki
Suga
c and
Seino A. K.
Jongkees
*abc
aDepartment of Chemistry and Pharmaceutical Sciences, Amsterdam Institute of Molecular and Life Sciences, VU Amsterdam, De Boelelaan 1108, 1081HZ Amsterdam, The Netherlands. E-mail: s.a.k.jongkees@VU.nl
bDepartment of Chemical Biology & Drug Discovery, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
cDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, 113-0033 Tokyo, Japan
First published on 24th March 2022
Abstract
Thiols are a functional group commonly used for selective reactions in a biochemical setting because of their high nucleophilicity. Phosphorus nucleophiles can undergo some similar reactions to thiols, but remain underexploited in this setting. In this work we show that phosphine nucleophiles react cleanly and quickly with a dehydroalanine electrophile, itself generated from cysteine, to give a stable adduct in a peptide context. NMR reveals the product to be a phosphonium ion and indicates some backbone conformational constraint, possibly arising from transient carbonyl coordination. The reaction proceeded quickly, with a pseudo-first order rate constant of 0.126 min−1 at 1 mM peptide (80% conversion in 10 min), and with no detectable side products on the peptide. A broad peptide sequence scope and water-soluble phosphines with alkyl as well as aromatic groups were all shown to react efficiently. Phosphine addition proved to be efficient on nisin as a model Dha-containing biologically-derived peptide and on an mRNA-displayed peptide, as well as on TCEP-modified agarose for peptide capture from solution. This reaction thus presents a promising approach for modification of peptides for cargo attachment or altered physical properties in peptide discovery.
Cysteines provide a convenient amino acid for selective modification of peptides and proteins, with the high nucleophilicity of its thiol commonly exploited in nucleophilic substitution of halides and in conjugate additions.1 This reactivity can also be reversed, with bis-alkylation of cysteine providing convenient access to a uniquely electrophilic amino acid in dehydroalanine that can react with exogenous nucleophiles (including other thiols).2
While investigating the use of such a thiol conjugate addition to dehydroalanine,3 it was observed that a side product of mass +250 Da occasionally formed in preference over the thiol adduct, suggesting a fast side-reaction. This product was hypothesized to be a stable phosphonium adduct of tris-carboxyethylphosphine (TCEP) to the dehydroalanine, from residual TCEP added to keep the thiol reduced. Given that this formed from residual TCEP with exclusion of detectable thiol adduct, we sought to more rigorously investigate the use of this reaction for peptide modification. Conjugate additions of phosphorus nucleophiles to α,β-unsaturated compounds are not a new reaction, but possible side-reactions of the phosphine reducing agent TCEP are often overlooked when compared to thiol agents such as β-mercaptoethanol or dithiothreitol. Our observations are also consistent with other reports of TCEP (and derivatives) adding to a Lysine acrylamide derivative incorporated in proteins by genetic code reprogramming4 or a side-reaction occuring with TCEP and the alkylating drug Busulfan5 (Scheme 1), although these products were only characterized by mass spectrometry.
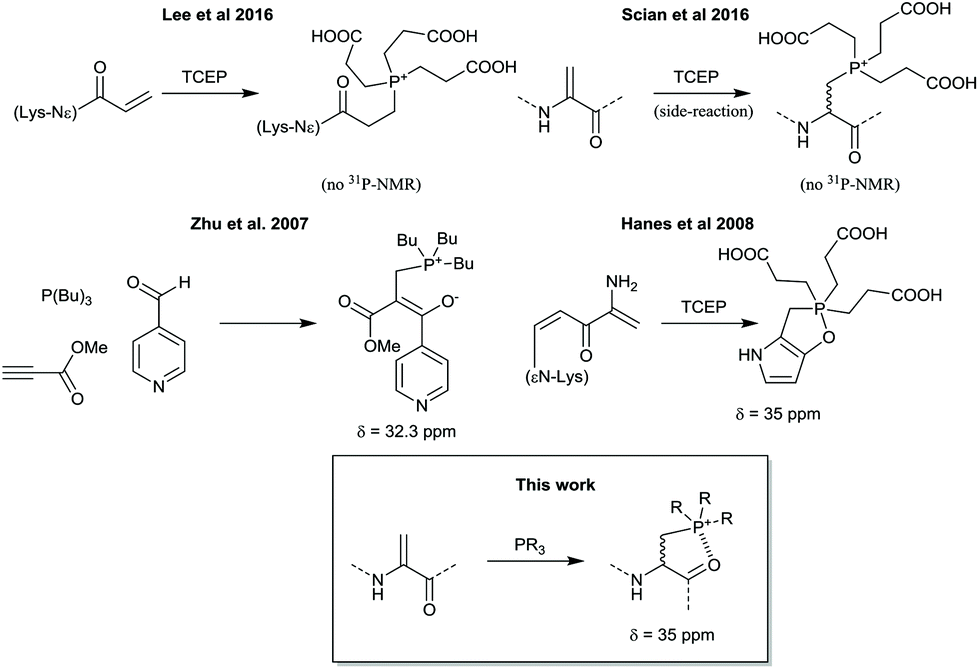 | ||
| Scheme 1 Previous reports of phosphine adducts to α,β-unsaturated carbonyls in proteins (upper), and products of such addition reactions with additional conformational constraints (lower), as compared to the current work. | ||
We note that several similar products have been reported in the literature as either a stable phosphonium ion enolate pair6 or as a cyclic phosphorane7 (Scheme 1), and so we were particularly interested to see if the phosphine addition would provide conformational constraints to the peptide backbone and also whether the backbone carbonyl might retain any enolate character. In order to fully characterise the product, a short peptide P1 was designed and synthesised that would be water soluble (prefering 2,3-diaminopropionic acid over lysine to minimize NMR peak overlaps), be traceable by absorbance at 280 nm (A280), and would give relatively simple peaks in 1H NMR to facilitate an unequivocal structural assignment while still in a relevant peptide setting (Fig. 1). The test peptide was converted to its dehydrolalanine form P2 using dibromovalerate (DBV) before addition of excess TCEP and overnight reaction to ensure full conversion (validated by 31P NMR of the reaction mixture) with subsequent purification by preparative HPLC. The resulting product P3 was characterised by 1H, and 13C NMR as well as relevant 2D experiments (COSY, HSQC, HMBC). These clearly showed a phosphonium ion as the product and no enolate present, most notably characterised by a proton on the alpha carbon at 4.9 ppm as well as the absence of any 13C peaks between 55 and 115 ppm and the presence of diastereomers arising from proton addition to the alpha carbon (Fig. 1 and ESI†). It is, however, noteworthy that in the dehydroalanine intermediate the methyl peak arising from the alanine located immediately C-terminally of the dehydroalanine showed splitting into two doublets (relative integrals 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) that we take to be indicative of known conformational effects of this residue8 (slow interconversion of amide bond isomers, also represented in the peak from the olefinic proton). These same peaks in the phosphine adduct show a similar pattern and ratio (further split by the two diastereomers resulting from the phosphonium adduct), suggesting that the phosphine adduct is perturbing the usual amide bond conformational dynamics. We cautiously interpret this as suggesting some transient interaction of a carbonyl oxygen with the phosphonium ion, but we also cannot say if this is from the N- or C-terminal residue (a pseudo 6- or 5-membered ring, respectively).
:3) that we take to be indicative of known conformational effects of this residue8 (slow interconversion of amide bond isomers, also represented in the peak from the olefinic proton). These same peaks in the phosphine adduct show a similar pattern and ratio (further split by the two diastereomers resulting from the phosphonium adduct), suggesting that the phosphine adduct is perturbing the usual amide bond conformational dynamics. We cautiously interpret this as suggesting some transient interaction of a carbonyl oxygen with the phosphonium ion, but we also cannot say if this is from the N- or C-terminal residue (a pseudo 6- or 5-membered ring, respectively).
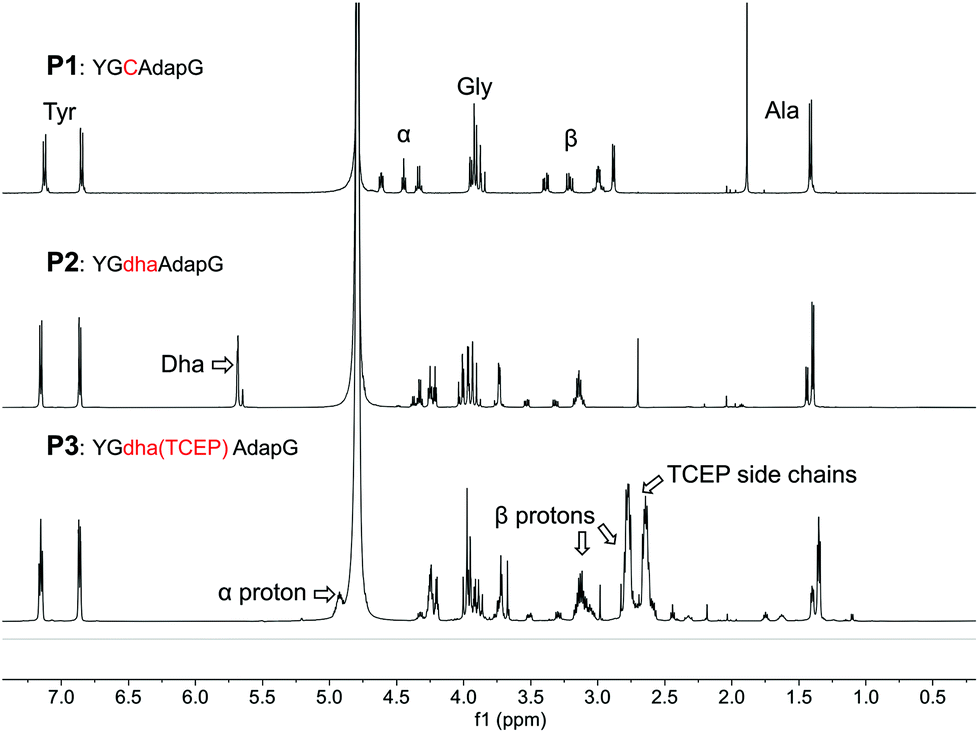 | ||
| Fig. 1 1H NMR spectra for a short model peptide, showing formation of dehydroalanine from cysteine and subsequent conversion to a phosphonium ion with TCEP. Relevant peaks for the cysteine-derived amino acids are indicated in the lower panel. Additional spectra in ESI† (13C, 31P NMR, COSY, HSQC, HMBC). In the peptide sequence, single-letter codes are used for canonical amino acids while non-canonical amino acids are shown as lower case triplets dap for 2,3-diaminopropionic acid and dha for dehydroalanine. | ||
Given that the product is a potentially reactive phosphonium ion, we investigated the stability of the product in a range of conditions (Table 1): high and low pH (∼1, 3.7, 10, and ∼14), high temperature (60 and 90 °C), nucleophiles (azide, dithiothreitol), electrophiles (acrylamide), oxidation (peroxide), and reduction (dithiothreitol). Decomposition of P3 was assessed by HPLC at 5 and 24 hours (H2O2 overlapped with the P3 peak and so intregrals from the extracted ion chromatograms were used and dithiothreitol slightly degraded the tryptophan used as standard for quantification, but available data clearly showed both to be at least largely stable). Degradation was observed in two cases, at 90 °C (−17 Da, deamination) and at pH 14 (isobaric, isomerisation), which appear to be related to changes elsewhere in the peptide and not the TCEP adduct. The phosphonium product thus proved to be remarkably stable. Also notable is that re-elimination of the phosphonium cation does not appear to take place even with extended treatment with high pH.
| Condition | Recovery 5 hours | Recovery 24 hours |
|---|---|---|
| a Based on ESI-MS due to peak overlap. b Degradation of internal standard prevented accurate quantification. | ||
| pH ∼1 (HCl) | >99% | 95% |
| pH ∼14 (NaOH) | 75% | 43% |
| pH 3.66 (acetate) | >99% | >99% |
| pH 10 (Gly) | 96% | 98% |
| 60 °C | 98% | 95% |
| 90 °C | 65% | 0% |
| NaN3 | 97% | 98% |
| H2O2 | >99%a | >99%a |
| Acrylamide | 98% | >99% |
| Dithiothreitol | >99%b | >99%b |
We next sought to assess the rate of reaction (Fig. 2). Using 1 mM of the same dehydroalanine-containing peptide P2 we measured consumption of peptide by reaction with a small excess of TCEP on HPLC (with quenching of time points by H2O2, which did not overlap with starting material peaks in this case). Around 80% conversion was found after just 10 min, with a pseudo-first order rate constant of 0.126 min−1 (95% C.I. 0.120–0.138; Fig. 2).
 | ||
| Fig. 2 Basic pseudo-first order kinetics for reaction of 1 mM peptide P2 with 3 mM TCEP at pH 8.0 and room temperature. Datapoints represent three independent measurements, and were fit to a single exponential decay. | ||
The influence of the peptide sequence was investigated as an end-point measurement on a small set of potentially challenging peptides (Table 2), investigating product distribution after 2 hours at 42 °C in a two-step one-pot approach together with the bis-amide (dibromo adipic bis-amide/DBAA or dibromovalerate/DBV, for one or two cysteines respectively). Sequences with aromatic amino acids, prolines, and bulky hydrophobic amino acids in the positions flanking the dehydroalanine were chosen for testing, as well as the presence of glutamate and lysine to test for influence of charge repulsion/attraction. Most peptides showed good conversion and yield (80+% by LC-MS), with the worst being P6 (54% yield). This peptide contains an N-terminal valine and a C-terminal proline flanking the dehydroalanine. In some cases two diastereomers could be easily separated on HPLC (e.g.P7 and P11), while in others (e.g.P5 and P6) only a single peak was observed (but could be resolved with a longer HPLC program). Overall the phosphine addition reaction seems to be suited to application in a diverse range of peptides, with no apparent incompatible sequences.
| Peptide | Sequence | Phosphine | Yield |
|---|---|---|---|
| N.R., no reaction. | |||
| P4 | ATGVNQ![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) YG YG |
TCEP | 78% |
| P5 | ATGVPQYG |
TCEP | >99% |
| P6 | ATGVPQYG |
TCEP | 54% |
| P7 | ATGVEQYG |
TCEP | >99% |
| P8 | ATGVQYG |
TCEP | 43% (2 TCEP), 23% (1 TCEP) |
| P9 | ATGVQYG |
TCEP | 68% (2 TCEP) |
| P10 | ATGVIYG |
TCEP | 70% (2 TCEP) |
| P11 | ATGVWYKNG |
TCEP | >99% |
| P12 | ATGVMYQNG |
TCEP | 94% |
| P4 | ATGVNQYG |
TPPMS | 92% |
| P4 | ATGVNQYG |
DPPB | >97% |
| P4 | ATGVNQYG |
TPPTS | 24% |
| P4 | ATGVNQYG |
Trifuryl phosphine | N.R. |
We also investigated reaction with a small set of commercially available water-soluble phosphines using peptide P4 (Table 2). After conversion of the thiol to dehydroalanine, reaction with the aromatic water soluble phosphines diphenylphosphinobenzene-3-sulfonate (TPPMS) and 2-(diphenylphosphino)benzoic acid (DPPB) showed excellent conversion (92% and >97% from the dehydroalanine intermediate, respectively, relative to Tyr internal standard from peak area at 280 nm), while 3,3′,3′′-phosphanetriyltris(benzenesulfonic acid trisodium salt (TPPTS) gave less product (estimated at 24%). Trifuryl phosphine also gave no product, despite appearing sufficiently soluble. The reason for the lower conversions is unclear, but may arise at least in part from unfavourable charge repulsions in TPPTS that the monosulfonated TPPMS and the flexible TCEP can better avoid.
Finally, we set out to demonstrate applicability of this reaction. We first showed conversion of dehydroalanine residues in the natural product polycyclic peptide antibiotic Nisin A (Fig. 3). This contains two dehydroalanine residues as well as a dehydrobutyrine, with one dehydroalanine in a linear region and the other in a macrocycle. Chemical modification of natural peptide antibiotics is a promising route to modulate their biophysical properties9 and attach new functionality.10 Incubation with five equivalents of TCEP over five hours shows conversion to modified product, with the doubly modified product dominating (roughly 2:1 relative to singly modified product). Also visible is a small amount of triply-modified product, indicating reaction with dehydrobutyrene is possible but slower.
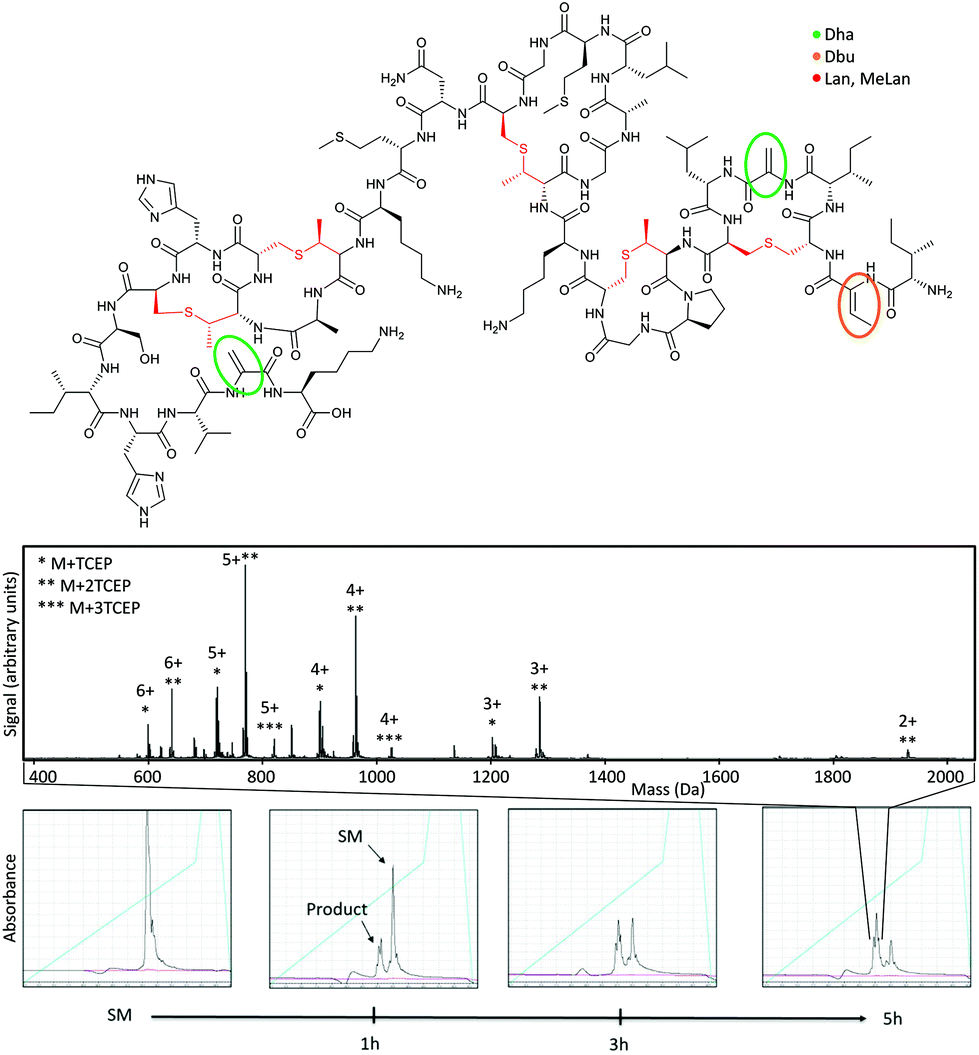 | ||
| Fig. 3 Addition of 5 equivalents TCEP to Nisin A at pH 8.0 and 42 °C. Upper, structure of Nisin A emphasising dehydroalanine (Dha, green), dehydrobutyrine (Dbu, orange) and lanthionine/methyllanthionine bridges (Lan/MeLan, red). Mid, ESI-MS of the product from reaction of TCEP with Nisin A, with peaks labeled by ionization state and number of additions. Lower, HPLC chromatograms showing the progress of TCEP addition to Nisin A at indicated time-points (detection at 215 nm in black and 280 nm in pink, gradient indicated in cyan). | ||
We also tested applicability of this modification reaction in the context of mRNA-displayed peptide libraries. Both TCEP and TPPMS were confirmed by MALDI-TOF-MS to react with in vitro translated peptides and gave clean conversion to the desired products without intermediate purification (Fig. 4). Modification of a peptide directly in display format was further verified by purification of mRNA by gel filtration before modification, followed by cleavage using Tobacco Etch Virus (TEV) protease of the peptide for MALDI-TOF-MS analysis (Fig. 5). This again showed clean conversion, and was also shown be compatible with cyclisation using the bis-NHS reagent disuccinimidyl glutarate (DSG). Finally, we showed that TCEP-modified agarose could efficiently capture Dha-containing peptides from solution (Fig. S24†), which suggests a new covalent-immobilization methodology.
 | ||
| Fig. 4 MALDI-TOF-MS showing peptides (fMKYSHCGFLTKENLYFQGSGSGS–OH) derived from in vitro translation (upper), conversion of a single cysteine within that peptide to dehydroalanine (mid), and addition of two different phosphines to this product (lower two panels). Calculated and observed masses are indicated for the relevant peaks along with ionisation state, and phosphine structures are as shown (TCEP and TPPMS). | ||
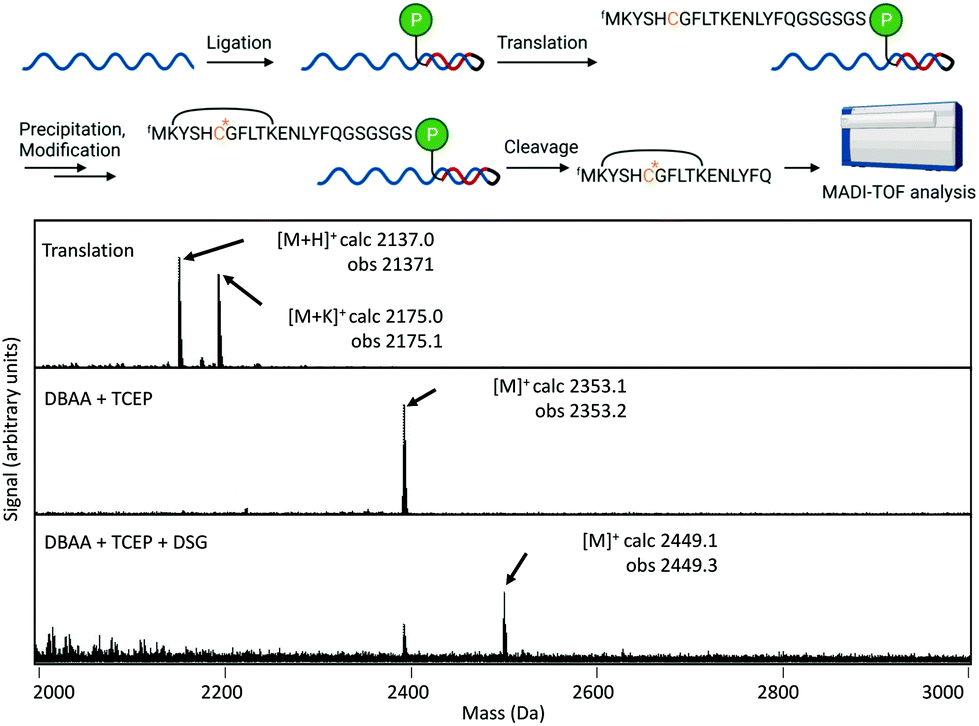 | ||
| Fig. 5 Modification of mRNA-displayed peptide by one-pot conversion of cysteine through dehydroalanine to phosphonium. Upper, cartoon representation of work-flow (blue, mRNA; red, cDNA; P, puromycin). Lower, MALDI-TOF-MS spectra for the peptide alone, after phosphonium formation, and after phosphonium formation followed by cyclization by lysine cross-linking with disuccinimidyl glutarate (DSG). Calculated and observed masses are indicated for the relevant peaks along with ionisation state. | ||
A previous study with the bis-electrophile drug busulfan had shown that an excess of TCEP could intercept dehydroalanine formed from catalytically-active cysteines in a protein,5 with this adduct confirmed indirectly from a deaminated derivative peak in LC-MS/MS. Others have shown addition of TCEP to an acrylamide group as a modification on the side chain of lysine, as an analogue of pyrollysine.4 However, this requires genetic code reprogramming to incorporate the electrophile into the target protein or peptide and so is not broadly accessible. The use of cysteine as a source for the dehydrolalanine electrophile in our work obviates this requirement. The authors of that work postulate that the phosphine addition is uniquely catalysed by the carboxylates in TCEP. Our data here, with a broader range of phosphines, show that this is not the case at least for dehydroalanine. This appears to be a general reaction of any water-soluble phosphine, although we do note some non-reactive species in our work that we cannot yet explain. The same previous report also details a synthetic approach for attaching cargo to the carboxylates of TCEP, suggesting modification of dehydroalanine with TCEP as a convenient strategy for stably attaching multivalent cargos to proteins such as antibodies. Our demonstration of modification with triphenylphosphine derivatives in mRNA-displayed macrocyclic peptides shows that selection of novel peptide sequences incorporating this unique functional group would be possible, which is well established as a mitochondrial targeting motif.11 The induced backbone constraints seen in NMR may also lead to unusual peptide conformations and stability. Finally, reaction with commercially available TCEP-modified agarose also appears to be a convenient strategy for peptide or protein immobilization on a solid support.
Overall, we present here an approach for efficient peptide modification by addition of phosphine nucleophiles into a dehydroalanine electrophile, generated from cysteine. This reaction generates a stable product with little to no peptide sequence influence and tolerates both aliphatic and aromatic phosphines. We envisage applications in peptide drug discovery, protein cargo attachment, and protein immobilization on solid support.
Conflicts of interest
There are no conflicts to declare.References
- (a) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed; (b) S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed.
- (a) J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1617–1618 RSC; (b) P. M. Morrison, P. J. Foley, S. L. Warriner and M. E. Webb, Chem. Commun., 2015, 51, 13470–13473 RSC.
- S. A. K. Jongkees, S. Umemoto and H. Suga, Chem. Sci., 2017, 8, 1474–1481 RSC.
- Y.-J. Lee, Y. Kurra and W. Liu, ChemBioChem, 2016, 17, 456–461 CrossRef CAS PubMed.
- M. Scian, M. Guttman, S. D. Bouldin, C. E. Outten and W. M. Atkins, Biochemistry, 2016, 55, 4720–4730 CrossRef CAS PubMed.
- X. F. Zhu, C. E. Henry and O. Kwon, J. Am. Chem. Soc., 2007, 129, 6722–6723 CrossRef CAS PubMed.
- J. W. Hanes, I. Keresztes and T. P. Begley, Angew. Chem., Int. Ed., 2008, 47, 2102–2105 CrossRef CAS PubMed.
- D. E. Palmer, C. Pattaroni, K. Nunami, R. K. Chadha, M. Goodman, T. Wakamiya, K. Fukase, S. Horimoto and M. Kitazawa, J. Am. Chem. Soc., 1992, 114, 5634–5642 CrossRef CAS.
- T. Koopmans, T. M. Wood, P. T. Hart, L. H. J. Kleijn, A. P. A. Hendrickx, R. J. L. Willems, E. Breukink and N. I. Martin, J. Am. Chem. Soc., 2015, 137, 9382–9389 CrossRef CAS PubMed.
- R. H. Vries, J. H. Viel, R. Oudshoorn, O. P. Kuipers and G. Roelfes, Chem. – Eur. J., 2019, 25, 12698–12702 CrossRef PubMed.
- M.-C. Frantz and P. Wipf, Environ. Mol. Mutagen., 2010, 51, 462–475 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterisation, HPLC traces. See DOI: https://doi.org/10.1039/d2ob00410k |
| This journal is © The Royal Society of Chemistry 2022 |