
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalysed decarbonylative C(sp2)–H alkylation of indolines with alkyl carboxylic acids and carboxylic anhydrides under redox-neutral conditions†
Hirotsugu
Suzuki
 ,
Yuya
Kawai
,
Yosuke
Takemura
and
Takanori
Matsuda
*
,
Yuya
Kawai
,
Yosuke
Takemura
and
Takanori
Matsuda
*
Department of Applied Chemistry, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: mtd@rs.tus.ac.jp
First published on 16th March 2022
Abstract
We developed a rhodium-catalysed decarbonylative C(sp2)–H alkylation method for indolines. This reaction facilitates the use of alkyl carboxylic acids and their anhydrides as a cheap, abundant and non-toxic alkyl source under redox-neutral conditions, featuring the introduction of a primary alkyl chain, which cannot be addressed by previous radical-mediated decarboxylative reaction. Through a mechanistic investigation, we revealed that an initially formed C-7 acylated indoline was transformed into the corresponding alkylated indoline via a decarbonylation process.
Indoles and their derivatives are ubiquitous structural motifs found in various natural products and pharmaceuticals.1 As such, efficient synthetic methods to access densely functionalised indoles have been extensively developed.2 Among them, site-selective C–H functionalisation of indoles allows the forging of new chemical bonds directly from a simple starting material for the rapid assembly of molecular complexity.3 However, selective activation of the C(7)–H bond is still challenging because the reactivity of the benzenoid moiety is lower than that of the pyrrole one.4 Consequently, ligand-directed C-7 functionalisation of indolines has emerged as a promising alternative to the corresponding functionalisation of indoles.5,6
The C-7 alkylation has also attracted considerable attention within the field of C–H functionalisation of indolines, utilising a series of alkylating reagents: activated alkenes,7 diazo compounds bearing an electron-withdrawing group,8 cyclopropanols9 and aziridines.10,11 However, these alkylating reagents cannot yield indolines bearing a simple alkyl chain without using any specific functional groups. Recently, Punji et al. addressed this problem by using simple primary and secondary alkyl chlorides in their iron-catalysed alkylation, but its highly basic conditions might narrow the substrate scope (Scheme 1a).12
 | ||
| Scheme 1 Transition metal-catalysed C-7 alkylation of indolines. | ||
Alkyl carboxylic acids have recently garnered significant attention as an alkylating reagent owing to their advantages, such as being inexpensive, abundant in nature, stable under benchtop conditions and easy to handle.13,14 Despite these advantages, only one precedent of C-7 alkylation of indolines has been reported to date: the palladium-catalysed oxidative decarboxylative alkylation of indolines with alkyl carboxylic acids (Scheme 1b).15 In this method, primary alkyl carboxylic acids could not be coupled with indolines because of the low stability of the primary alkyl radical species. Moreover, the oxidative conditions sometimes reduce the generality of the reaction and complicate the isolation procedure. Considering these limitations, we developed the redox-neutral decarbonylative alkylation of indolines by employing (in situ-formed) carboxylic anhydrides (Scheme 1c).16
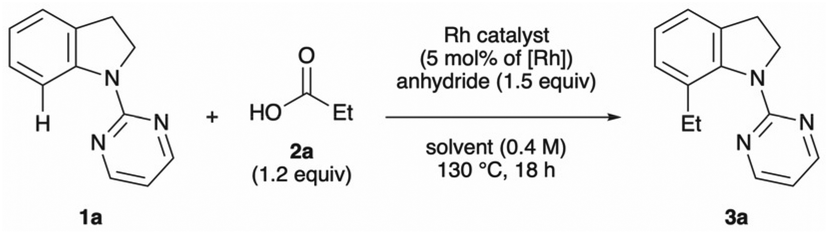
Inspired by previous work on decarbonylative alkylation,13 we began our investigation by employing 1-(pyrimidin-2-yl)indoline (1a), propionic acid (2a) and pivalic anhydride with various rhodium salts as a catalyst (Table 1). An initial experiment was performed in 1,4-dioxane at 130 °C for 18 h in the presence of [RhCl(CO)2]2 (2.5 mol%), producing the desired 7-ethyl-1-(pyrimidin-2-yl)indoline (3a) in good yield (entry 1). Other rhodium catalysts, namely [RhCl(cod)]2, [Rh(OAc)(cod)]2, [Rh(cod)2]BF4, [Rh(cod)2]OTf and RhCl(PPh3)3, yielded little of the desired C–7 alkylated product (entries 2–6). Among the solvents tested, 1,2-dichloroethane (DCE) was proved to be the best choice (entries 7–10). Other anhydrides, acetic anhydride, di-tert-butyl dicarbonate (Boc2O) and the combination of Boc2O and pivalic acid, decreased the yield (entries 11–13). Lower reaction temperature (110 °C) was inadequate for the reaction to promote (entry 14). Increasing the amount of 2a and pivalic anhydride resulted in an improved yield of the coupling product (entry 15). A control experiment showed that both the rhodium catalyst and pivalic anhydride are essential for the reaction to proceed (entries 16 and 17).
|
|
||||
|---|---|---|---|---|
| Entry | Rh catalyst | Solvent | Anhydride | Yield (%)b |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.24 mmol), Rh catalyst (5 mol% of [Rh]) and Piv2O (0.3 mmol) were reacted in solvent (0.5 mL) at 130 °C for 18 h, unless otherwise noted. b Yield was determined by 1H NMR analysis using 1,2,4,5-tetramethylbenzene as an internal standard. The value in parentheses indicates isolated yield. c At 110 °C. d 2a (0.4 mmol) and Piv2O (0.5 mmol) were used. | ||||
| 1 | [RhCl(CO)2]2 | 1,4-Dioxane | Piv2O | 69 |
| 2 | [RhCl(cod)]2 | 1,4-Dioxane | Piv2O | 0 |
| 3 | [Rh(OAc)(cod)]2 | 1,4-Dioxane | Piv2O | 8 |
| 4 | [Rh(cod)2]BF4 | 1,4-Dioxane | Piv2O | Trace |
| 5 | [Rh(cod)2]OTf | 1,4-Dioxane | Piv2O | Trace |
| 6 | RhCl(PPh3)3 | 1,4-Dioxane | Piv2O | Trace |
| 7 | [RhCl(CO)2]2 | Toluene | Piv2O | 59 |
| 8 | [RhCl(CO)2]2 | DCE | Piv2O | 83 |
| 9 | [RhCl(CO)2]2 | MeCN | Piv2O | 24 |
| 10 | [RhCl(CO)2]2 | DMF | Piv2O | Trace |
| 11 | [RhCl(CO)2]2 | 1,4-Dioxane | Ac2O | 50 |
| 12 | [RhCl(CO)2]2 | 1,4-Dioxane | Boc2O | 31 |
| 13 | [RhCl(CO)2]2 | 1,4-Dioxane | Boc2O + PivOH | 16 |
| 14c | [RhCl(CO)2]2 | 1,4-Dioxane | Piv2O | 41 |
| 15d | [RhCl(CO)2]2 | DCE | Piv2O | 91(92) |
| 16 | – | DCE | Piv2O | 0 |
| 17 | [RhCl(CO)2]2 | DCE | Trace | |
Having determined the optimised reaction conditions, we explored the decarbonylative alkylation of various alkyl carboxylic acids 2 with 1a (Table 2).17 Acetic acid, n-octanoic acid, isovaleric acid and 3-phenylpropionic acid delivered the corresponding C-7 alkylated indolines 3b–e in 73–85% yields. Moreover, the reaction with 3-phenylpropionic acid proceeded smoothly on 1 mmol scale, showing the feasibility for a large scale synthesis (3e). Alkyl substituents bearing phenoxide, phthalimide and methyl ester moieties were tolerated, providing the corresponding alkylated indolines in moderate to good yields (3f–h). 2-(4-Methoxyphenyl)acetic acid afforded an acceptable yield of 3i. Subsequently, the substrate scope of the indoline derivatives was examined. The reaction of 2- and 3-substituted indolines produced the desired alkylated indolines 3j–l in 81–96% yields. 4-Methylindoline rendered the desired product 3m in high yield. Indolines bearing an electron-donating and -withdrawing group at the 5-position, including methoxy, methyl, fluoro and chloro groups, underwent the alkylation with high yields (3n–q). 6-Fluoroindoline was compatible with the reaction conditions (3r), while 6-methylindoline failed to react with 3-phenylpropionic acid. Notably, the reaction was not restricted to indolines; a 2-substituted indole and a carbazole reacted with an alkyl carboxylic acid in a similar fashion to yield 3t and 3u in 73% and 52%, respectively. Further, C-2 alkylation proceeded with 1-(pyrimidin-2-yl)indole, furnishing 73% of 3v. The decarbonylative arylation of 1a proceeded smoothly by subjecting benzoic acids to the optimised reaction conditions (3w and 3x).

Alkyl carboxylic anhydrides underwent this decarbonylative alkylation reaction in a similar manner (Table 3). The alkylation of propionic and acetic anhydrides with 1a proceeded smoothly to provide the corresponding products in moderate to high yields (3a and 3b, respectively). To our delight, branched carboxylic anhydrides participated in the reaction to furnish the products 3y–A in 71–85% yields. Application of the reaction conditions to benzoic anhydride derivative successfully led to the formation of the C-7 arylated indoline 3w.
| a Reaction conditions: 1a (0.3 mmol), 4 (0.6 mmol) and [RhCl(CO)2]2 (2.5 mol%) were reacted in DCE (0.75 mL) at 150 °C for 18 h. |
|---|

|
A series of control experiments were performed to gain insight into the reaction mechanism (Scheme 2). Initially, a set of H/D exchange experiments was conducted in the presence/absence of 2a and pivalic anhydride (Scheme 2a). The addition of D2O to the standard reaction conditions resulted in the incorporation of deuterium into the recovered starting material. Moreover, H/D exchange of 1a was observed by heating 1a, [RhCl(CO)2]2 and D2O (5.0 equiv.) in DCE at 130 °C. These results indicate that the C(sp2)–H bond activation step is reversible. When the alkylation reaction was halted after 1 h, a significant amount of 7-acylated indoline 5 was detected (Scheme 2b). As ketone 5 appears to be a putative intermediate in the formation of 3a, 5 was subjected to the standard reaction conditions without an alkyl carboxylic acid and pivalic anhydride (Scheme 2c). The clean conversion of 5 into 3a was confirmed, indicating that 3a arose from the catalytic decarbonylation of 5.18 In the radical trapping experiment with BHT, no significant decrease of the yield was observed in both alkyl carboxylic acid and anhydride cases,19 suggesting the involvement of an alkyl radical species is unlikely (Scheme 2d). Moreover, the kinetic isotope effect experiment revealed that the C–H bond cleavage step is likely to be the rate-determining step (Scheme 2e).
 | ||
| Scheme 2 Control experiments. | ||
Based on the aforementioned experimental results and previous reports,13,14,16 we propose the reaction mechanism depicted in Fig. 1. The reaction starts with the formation of mixed anhydride 2′ followed by oxidative addition of the C–O bond to rhodium(I) A to furnish acylrhodium(III) carboxylate B. Coordination of the indoline 1 to B promotes a C-7 selective C–H activation, which might proceed via an electrophilic mechanism,13c yielding six-membered rhodacycle C. Reductive elimination from C affords the acylated product 5. Besides, acylrhodium(III) C undergoes deinsertion of CO and subsequent reductive elimination, leading to the formation of the C-7 alkylated indole 3 along with the regeneration of active Rh(I) catalyst A.
 | ||
| Fig. 1 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we developed a rhodium-catalysed decarbonylative alkylation approach for indolines under redox-neutral conditions. Alkyl carboxylic acids, which are cheap, abundant and stable, function as ideal alkylating reagents with the assistance of pivalic anhydride to provide the C-7 alkylated indolines in good to high yields. Additionally, a related decarbonylative alkylation reaction with symmetrical carboxylic anhydrides is achieved under acid/base- and redox-neutral conditions. Our reaction facilitates the use of primary carboxylic acids, which are difficult to use in the previous radical-mediated decarboxylative process, as alkylating reagents. Further investigation of decarbonylative alkylation is currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP21K14633 and JP21K05061.Notes and references
- (a) W. Xu, D. J. Gavia and Y. Tang, Nat. Prod. Rep., 2014, 31, 1474 RSC; (b) M.-Z. Zhang, Q. Chen and G.-F. Yang, Eur. J. Med. Chem., 2015, 89, 421 CrossRef CAS PubMed; (c) V. M. Norwood and R. W. Huigens, ChemBioChem, 2019, 20, 2273 CrossRef CAS PubMed.
- (a) K. Krueger, A. Tillack and M. Beller, Adv. Synth. Catal., 2008, 350, 2153 CrossRef CAS; (b) D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195 CrossRef CAS PubMed; (c) S. Kotha and C. Chakkapalli, Chem. Rec., 2017, 17, 1039 CrossRef CAS PubMed; (d) S. W. Youn and T. Y. Ko, Asian J. Org. Chem., 2018, 7, 1467 CrossRef CAS; (e) A. K. Clarke, H. E. Ho, J. A. Rossi-Ashton, R. J. K. Taylor and W. P. Unsworth, Chem. – Asian J., 2019, 14, 1900 CrossRef CAS PubMed.
- (a) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618 CrossRef CAS; (b) D. V. Vorobyeva and S. N. Osipov, Synthesis, 2018, 50, 227 CrossRef CAS; (c) T. A. Shah, D. Bhusan, S. Pradhan and T. Punniyamurthy, Chem. Commun., 2019, 55, 572 RSC; (d) S. Pradhan, P. B. De, T. A. Shah and T. Punniyamurthy, Chem. – Asian J., 2020, 15, 4184 CrossRef CAS PubMed; (e) J. Wen and Z. Shi, Acc. Chem. Res., 2021, 54, 1723 CrossRef CAS PubMed; (f) P. Bhattacharjee and U. Bora, Org. Chem. Front., 2021, 8, 2343 RSC.
- For selected examples on C(7)–H selective functionalisation of indoles, see: (a) A. J. Borah and Z. Z. Shi, J. Am. Chem. Soc., 2018, 140, 6062 CrossRef CAS PubMed; (b) X. D. Qiu, P. P. Wang, D. Y. Wang, M. Y. Wang, Y. Yuan and Z. Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 1504 CrossRef CAS PubMed; (c) C. N. Kona, Y. Nishii and M. Miura, Angew. Chem., Int. Ed., 2019, 58, 9856 CrossRef CAS PubMed; (d) X. W. Han, Y. Yuan and Z. Z. Shi, J. Org. Chem., 2019, 84, 12764 CrossRef CAS PubMed; (e) S. J. Fang, G. B. Jiang, M. Li, Z. Y. Liu, H. F. Jiang and W. Q. Wu, Chem. Commun., 2019, 55, 13769 RSC; (f) P. Chen, Y. H. Hao, X. X. Wang, D. Yuan, Y. M. Yao and L. Ackermann, Chem. – Eur. J., 2019, 25, 7292 CrossRef CAS PubMed; (g) I. Choi, A. M. Messinis and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 12534 CrossRef CAS PubMed.
- For recent reviews on C(7)–H selective functionalisation of indolines, see: (a) T. A. Shah, P. B. De, S. Pradhan and T. Punniyamurthy, Chem. Commun., 2019, 55, 572 RSC; (b) R. A. Jagtap and B. Punji, Asian J. Org. Chem., 2020, 9, 326 CrossRef CAS; (c) S. Pradhan, P. B. De, T. A. Shah and T. Punniyamurthy, Chem. – Asian J., 2020, 15, 4184 CrossRef CAS PubMed.
- For recent examples on C(7)–H selective functionalisation of indolines, see: (a) P.-L. Wang, Y. Li, L. Ma, C.-G. Luo, Z.-Y. Wang, Q. Lan and X.-S. Wang, Adv. Synth. Catal., 2016, 358, 1048 CrossRef CAS; (b) Z. Q. Song and A. P. Antonchick, Org. Biomol. Chem., 2016, 14, 4804 RSC; (c) H. Q. Luo, H. D. Liu, Z. P. Zhang, Y. F. Xiao, S. H. Wang, X. Z. Luo and K. J. Wang, RSC Adv., 2016, 6, 39292 RSC; (d) S. Maiti, L. Burgula, G. Chakraborti and J. Dash, Eur. J. Org. Chem., 2017, 2017, 332 CrossRef CAS; (e) H. Jo, J. Park, N. K. Mishra, M. Jeon, S. Sharma, H. Oh, S.-Y. Lee, Y. H. Jung and I. S. Kim, Tetrahedron, 2017, 73, 1725 CrossRef CAS; (f) T. Jeong, S. H. Lee, N. K. Mishra, U. De, J. Park, P. Dey, J. H. Kwak, Y. H. Jung, H. S. Kim and I. S. Kim, Adv. Synth. Catal., 2017, 359, 2329 CrossRef CAS; (g) M. K. Manna, G. Bairy and R. Jana, Org. Biomol. Chem., 2017, 15, 5899 RSC; (h) A. E. Hande and K. R. Prabhu, J. Org. Chem., 2017, 82, 13405 CrossRef CAS PubMed; (i) P. B. De, S. Pradhan, S. Banerjee and T. Punniyamurthy, Chem. Commun., 2018, 54, 2494 RSC; (j) Y. Q. Dong, S. Sun, J. T. Yu and J. Cheng, Tetrahedron Lett., 2019, 60, 1349 CrossRef CAS; (k) R. Du, K. Zhao, J. Liu, F. Han, C. Xia and L. Yang, Org. Lett., 2019, 21, 6418 CrossRef CAS PubMed; (l) H. Q. Luo, Q. Xie, K. Sun, J. B. Deng, L. Xu, K. J. Wang and X. Z. Luo, RSC Adv., 2019, 9, 18191 RSC; (m) S. K. Banjare, P. Biswal and P. C. Ravikumar, J. Org. Chem., 2020, 85, 5330 CrossRef CAS PubMed; (n) Q. K. Yan, H. Huang, H. Zhang, M. H. Li, D. D. Yang, M. P. Song and J. L. Niu, J. Org. Chem., 2020, 85, 11190 CrossRef CAS PubMed; (o) G. Xie, Y. Zhao, C. Cai, G.-J. Deng and H. Gong, Org. Lett., 2021, 23, 410 CrossRef CAS PubMed; (p) S. Maiti, T. Mandal, B. P. Dash and J. Dash, J. Org. Chem., 2021, 86, 1396 CrossRef CAS PubMed.
- (a) S. G. Pan, N. Ryu and T. Shibata, Adv. Synth. Catal., 2014, 356, 929 CrossRef CAS; (b) H. Oh, J. Park, S. H. Han, N. K. Mishra, S. H. Lee, Y. Oh, M. Jeon, G.-J. Seong, K. Y. Chung and I. S. Kim, Tetrahedron, 2017, 73, 4739 CrossRef CAS; (c) C. Pan, Y. Wang, C. Wu and J.-T. Yu, Org. Biomol. Chem., 2018, 16, 693 RSC; (d) S. K. Banjare, R. Chebolu and P. C. Ravikumar, Org. Lett., 2019, 21, 4049 CrossRef CAS PubMed.
- (a) W. Ai, X. Yang, Y. Wu, X. Wang, Y. Li, Y. Yang and B. Zhou, Chem. – Eur. J., 2014, 20, 17653 CrossRef CAS PubMed; (b) I. E. Iagafarova, D. V. Vorobyeva, D. A. Loginov, A. S. Peregudov and S. N. Osipov, Eur. J. Org. Chem., 2017, 2017, 840 CrossRef CAS.
- X. Zhou, S. Yu, Z. Qi, L. Kong and X. Li, J. Org. Chem., 2016, 81, 4869 CrossRef CAS PubMed.
- P. B. De, S. Atta, S. Pradhan, S. Banerjee, T. A. Shah and T. Punniyamurthy, J. Org. Chem., 2020, 85, 4785 CrossRef CAS PubMed.
- For recent examples on C(sp2)–H alkylation, see: (a) H. Tian, H. Yang, C. Tian, G. An and G. Li, Org. Lett., 2020, 22, 7709 CrossRef CAS PubMed; (b) Y. Cheng, Y. He, J. Zheng, H. Yang, J. Liu, G. An and G. Li, Chin. Chem. Lett., 2021, 32, 1437 CrossRef CAS.
- R. A. Jagtap, P. P. Samal, C. P. Vinod, S. Krishnamurty and B. Punji, ACS Catal., 2020, 10, 7312 CrossRef CAS.
- (a) H. Yang, C. Tian, D. Qiu, H. Tian, G. An and G. Li, Org. Chem. Front., 2019, 6, 2365 RSC; (b) H. Zhao, X. Xu, H. Yu, B. Li, X. Xu, H. Li, L. Xu, Q. Fan and P. J. Walsh, Org. Lett., 2020, 22, 4228 CrossRef CAS PubMed; (c) H. Yu, H. Zhao, X. Xu, X. Zhang, Z. Yu, L. Li, P. Wang, Q. Shi and L. Xu, Asian J. Org. Chem., 2021, 10, 879 CrossRef CAS; (d) Y. He, H. Yang, D. Gao, J. Ma, Y. Shao, G. An and G. Li, Chin. J. Org. Chem., 2021, 41, 4725 CrossRef.
- For selected examples on C(sp2)–H arylations and alkenylations with (in situ formed) carboxylic anhydrides, see: (a) W. Jin, Z. Yu, W. He, W. Ye and W.-J. Xiao, Org. Lett., 2009, 11, 1317 CrossRef CAS PubMed; (b) H. Zhao, X. Xu, Z. Luo, L. Cao, B. Li, H. Li, L. Xu, Q. Fan and P. J. Walsh, Chem. Sci., 2019, 10, 10089 RSC.
- C. Premi, A. Dixit and N. Jain, Org. Lett., 2015, 17, 2598 CrossRef CAS PubMed.
- For our recent works on rhodium-catalysed C–H functionalisation of (hetero)arenes with anhydrides, see: (a) H. Suzuki, Y. Liao, Y. Kawai and T. Matsuda, Eur. J. Org. Chem., 2021, 2021, 4938 CrossRef CAS; (b) H. Suzuki, F. Sasamori and T. Matsuda, Org. Lett., 2022, 24, 1141–1145 CrossRef PubMed.
- The use of α-branched carboxylic acids suffered from the formation of 3d (∼1(0%) as an inseparable byproduct owing to the unselective C–O bond cleavage and the rearrangement of a tert-butyl group.
- For chelation-assisted decarbonylation of unstrained ketones, see: (a) Z.-Q. Lei, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 2690 CrossRef CAS PubMed; (b) T.-T. Zhao, W.-H. Xu, Z.-J. Zheng, P.-F. Xu and H. Wei, J. Am. Chem. Soc., 2018, 140, 586 CrossRef CAS PubMed; (c) T.-Y. Yu, W.-H. Xu, H. Lu and H. Wei, Chem. Sci., 2020, 11, 12336 RSC.
- Acylation of BHT has not been observed in crude 1H NMR analysis.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for new compounds. See https://doi.org/10.1039/d2ob00249c |
| This journal is © The Royal Society of Chemistry 2022 |