
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of spirocyclic dihydropyrazoles from tosylhydrazones and electron-deficient alkenes†
Timothy L.
Wootton
 and
Daniel M.
Allwood
*
and
Daniel M.
Allwood
*
Biomolecular Sciences Research Centre, Sheffield Hallam University, Howard Street, Sheffield, S1 1WB, UK. E-mail: D.Allwood@shu.ac.uk
First published on 4th March 2022
Abstract
Spirocycles represent a diverse class of molecules which have received significant attention in the pharmaceutical industry due to their broad biological activities and inherent molecular three–dimensionality. Herein, we demonstrate a procedurally simple method for the preparation of a range of spirocyclic dihydropyrazoles. The protocol utilises bench stable cyclic tosylhydrazones, which are trivial to prepare from the parent cyclic ketone without need for purification, and commercially available electron deficient alkenes. The synthetic utility of the core scaffold is also demonstrated to highlight potential for applications in medicinal chemistry and drug development programmes.
Introduction
In the past decade, there has been an increased focus on molecular three-dimensionality in the drug discovery process, due to the observation that the mean fraction of sp3-hybridised atoms in drug candidates tends to increase through the clinical trials process, as a result of the attrition of less selective “flat” compounds.1,2 As a response, there has been a drive in the pharmaceutical industry toward increasing the molecular three-dimensionality of screening libraries and discovery molecules. However, bond formation to sp3-hybridised atoms (particularly carbon) represents more of a synthetic challenge than planar or linear counterparts where transition metal-catalysed cross coupling methodologies are frequently utilised. Therefore, novel synthetic methodologies for the preparation of highly three-dimensional core scaffolds are of significant interest to both academic and industrial medicinal chemists.1–8One strategy for increasing molecular three-dimensionality is the incorporation of spirocyclic ring junctions into the core scaffold of the drug, since the rings joined by the spirocyclic atom are mutually orthogonal and thereby effectively engage all three dimensions. Spirocycles therefore represent an inviting synthetic target for medicinal chemists due to the inherent three-dimensionality present but also the broad range of biological activities demonstrated by this compound class.9–14
Many compounds containing spirocyclic scaffolds have been approved as drugs for the treatment of a broad range of disease states and some representative examples are shown in Fig. 1.
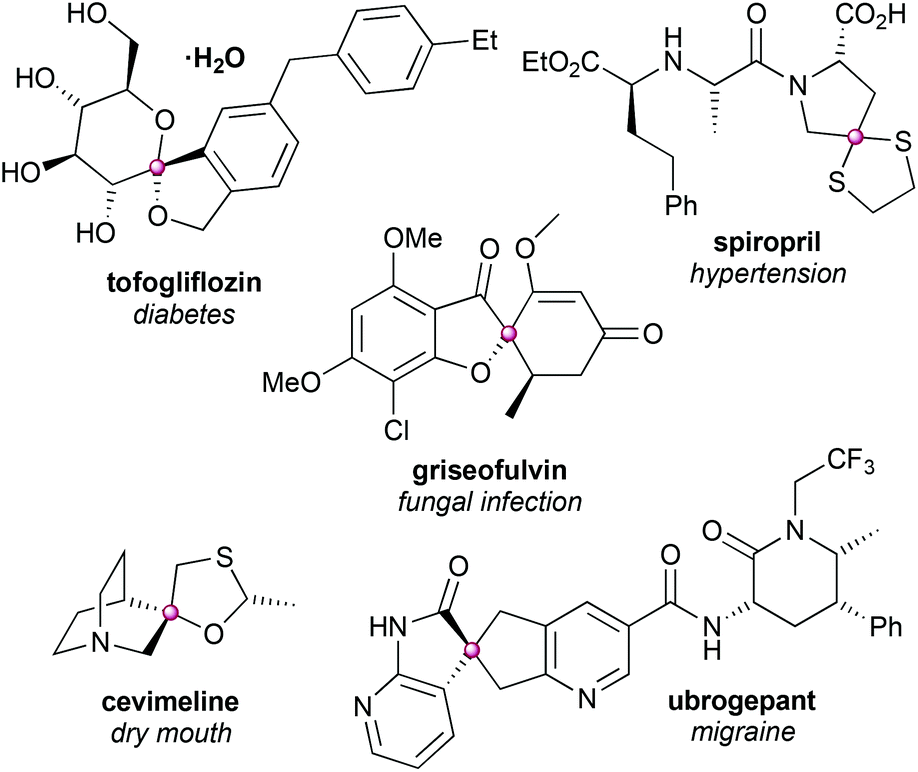 | ||
| Fig. 1 Examples of approved drug molecules featuring a spirocyclic ring junction.9–14 | ||
Spirocycles such as cyclic acetals may be easily prepared by traditional acid-catalysed routes15–17 or using gold-mediated spirocyclisations,18–20 however acetals are frequently hydrolytically labile, which can cause problems in molecules designed for use in medicine. As such, more stable spirocyclic moieties are typically utilised in drug candidates and there is significant interest in the preparation of this type of robust core scaffold.
Prior work from our group demonstrated the synthesis of diverse spirocyclic pyrazoles from tosylhydrazones and alkynes (Scheme 1). These molecules represent a highly three–dimensional heterocyclic core scaffold, however it was observed that some spirocyclic pyrazoles (particularly those composed of more strained ring systems, or which were electronically activated by adjacent π systems) underwent spontaneous sigmatropic rearrangement to the corresponding fused bicyclic scaffolds.4
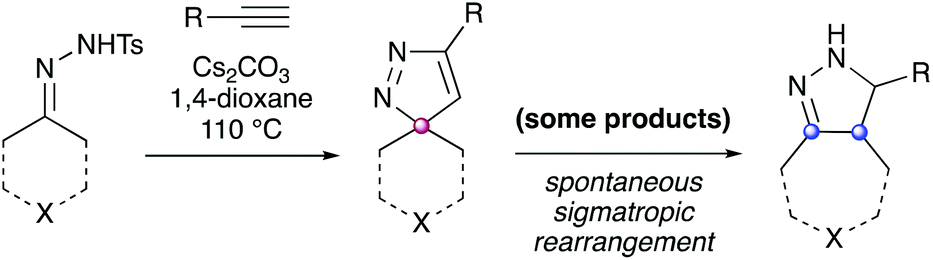 | ||
| Scheme 1 Prior work on the synthesis of spirocyclic (purple) pyrazole compounds from alkynes. Some examples were observed to spontaneously rearrange to the corresponding fused bicyclics (blue).4 | ||
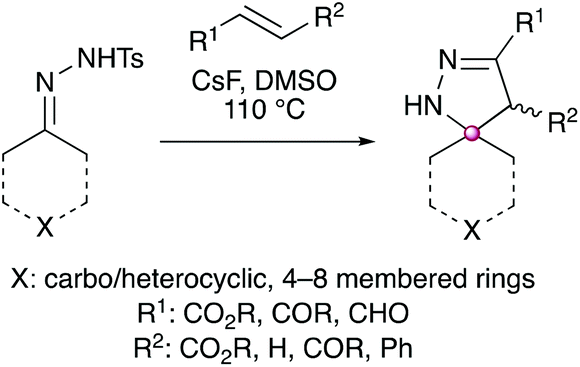
We therefore envisaged that removing a π bond from the pyrazole moiety would negate the ability of the spirocyclic scaffold to undergo sigmatropic rearrangement and therefore lead to a more robust three-dimensional core scaffold. The synthesis of these compounds was proposed to be achieved using an analogous method by removing a π bond from the starting material alkyne, and instead using an alkene (Scheme 2).
 | ||
| Scheme 2 Proposed synthesis of a more robust dihydropyrazole core scaffold, which is unable to undergo sigmatropic rearrangement to a fused bicyclic system. | ||
Synthetic routes to spirocyclic dihydropyrazoles are sparsely represented in the current literature21–24 aside from a prominent example from Wu et al. (Scheme 3).21 This reaction employs tosylhydrazones and α,β-unsaturated ketones to provide dihydropyrazoles in good-to-excellent yields. However, the substrate scope of this reaction is limited to phenyl and methyl ketones, thereby limiting the synthetic versatility of the new dihydropyrazole core scaffold.
 | ||
| Scheme 3 Synthesis of dihydropyrazoles by Wu et al.21 | ||
Herein, we present a complementary, procedurally simple, method for the synthesis of spirocyclic dihydropyrazole scaffolds bearing more synthetically tractable functional groups to allow a broader range of further synthetic modifications for medicinal chemistry applications (Scheme 4).
 | ||
| Scheme 4 Synthesis of spirocyclic dihydropyrazoles bearing pendant functional groups for further synthetic modification. | ||
Results and discussion
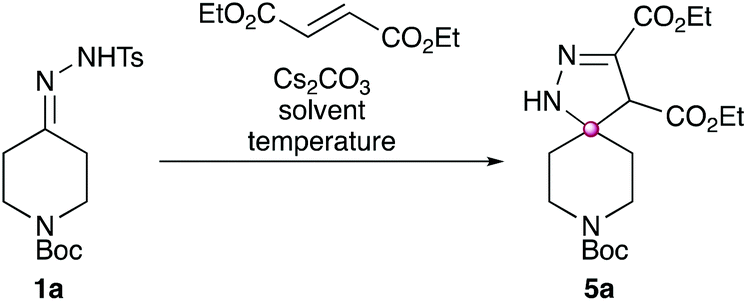
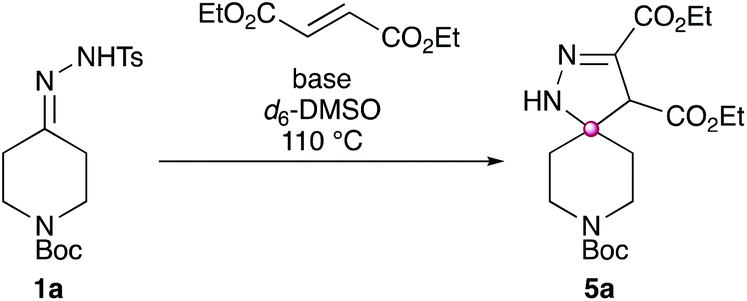
Investigations began by treating the model substrate 1a with a range of electronically diverse alkenes (Scheme 5) using conditions previously developed by our group for the analogous synthesis of spirocyclic pyrazoles.4 | ||
| Scheme 5 Initial investigations into alkene electronics. Four separate reactions were run with alkenes: 3,4-dihydro-2H-pyran (2, 0%), 2,6-dimethyl-4H-pyran-4-one (3, 0%), styrene (4, 0%) and diethyl fumarate (5a, 57%). Conditions: 1a (0.1 mmol), alkene (0.15 mmol), Cs2CO3 (0.15 mmol), 1,4-dioxane (0.5 mL), N2 atmosphere, sealed tube, 110 °C, 18 h. | ||
No spirocyclic product was observed in any of the reactions involving the highly electron rich alkene (3,4-dihydro-2H-pyran, product 2), a donor–acceptor “push–pull” alkene (2,6-dimethyl-4H-pyran-4-one, product 3) or a moderately electron deficient alkene (styrene, product 4). However, when a strongly electron deficient alkene was used (diethyl fumarate), a 57% isolated yield of the desired spirocyclic dihydropyrazole (5a) was obtained.
Optimisation of the reaction conditions began with a solvent screen (Table 1, entries 2–13) where polar aprotic solvents generally performed the best, with DMSO providing the highest yield at 74%. Since the yields of the optimisation reactions were being calculated using 1H NMR, it was decided that using d6-DMSO as the reaction solvent would expedite the analysis process for the remaining optimisation reactions.
|
|
|||
|---|---|---|---|
| Entry | Solvent | Temp (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.25 mmol), Cs2CO3 (1.5 equiv.), diethyl fumarate (1.5 equiv.), solvent (1 mL), sealed tube, N2 atmosphere, 22 h. b Yield determined by 1H NMR using 1,4-dinitrobenzene as internal standard. c Isolated yield. d Complete conversion, no product but complex mixture observed. e Reaction run for 48 h. f Solvent degassed, dried over 4 Å molecular sieves and stored under N2. | |||
| 1 | 1,4-Dioxane | 110 | 57c |
| 2 | Toluene | 110 | 29 |
| 3 | DMF | 110 | 52 |
| 4 | Acetone | 100 | 22 |
| 5 | MeCN | 100 | 41 |
| 6 | H2O | 110 | 0d |
| 7 | 1,2-DCE | 100 | 27 |
| 8 | DMSO | 110 | 74 |
| 9 | EtOH | 100 | 41 |
| 10 | THF | 80 | 0d,e |
| 11 | MTBE | 75 | 47 |
| 12 | Et2O | 50 | 0d |
| 13 | PhCF3 | 110 | 33 |
| 14 | d 6-DMSO | 110 | 84 |
| 15 | DMSOf | 110 | 85 |
However, upon switching from h6-DMSO to d6-DMSO, an increase in yield to 84% was observed (Table 1, entry 14). This was postulated to be due to the reduced water content in the sealed bottle of d6-DMSO compared with the open bottle of h6-DMSO lacking a septum. This was confirmed by running the reaction in h6-DMSO which had been degassed, dried over 4 Å molecular sieves and stored under a N2 atmosphere prior to use, which provided an equivalent increased yield (Table 1, entry 15).
A base screen using the optimised solvent revealed poorer yields with small alkali metal carbonates (Table 2, entries 2 and 3), with the yield increasing with ionic radius (Table 2, entries 4, 5 and 1). This is consistent with our previous findings that large ionic radius carbonate bases provide the best yield in reactions of unstabilised tosylhydrazones.25,26 We propose that this is due to the lack of a tight ion pair between the hydrazonyl anion and the larger metal cation after deprotonation, making the former more reactive. However, in this case, the trend in yield is less pronounced than in our previous studies, presumably due to the cation-coordinating nature of the solvent in this reaction having a sequestering effect, even on the smaller cations.
|
|
||
|---|---|---|
| Entry | Base | Yieldb (%) |
| a Reaction conditions: 1a (0.25 mmol), base (1.5 equiv.), diethyl fumarate (1.5 equiv.), d6-DMSO (1 mL), sealed tube, N2 atmosphere, 22 h. b Yield determined by 1H NMR using 1,4-dinitrobenzene as internal standard. c Reaction run for 48 h. d Complete conversion, no product but complex mixture observed. | ||
| 1 | Cs2CO3 | 84 |
| 2 | Li2CO3 | 43 |
| 3 | Na2CO3 | 20 |
| 4 | K2CO3 | 58 |
| 5 | Rb2CO3 | 85 |
| 6 | LiOH·H2O | 79 |
| 7 | NaOH | 71 |
| 8 | KOH | 75 |
| 9 | CsOH·H2O | 85 |
| 10 | MgCO3 | 58 |
| 11 | SrCO3 | 10 |
| 12 | BaCO3 | 0c |
| 13 | DIPA | 82 |
| 14 | NEt3 | 13 |
| 15 | KOtBu | 0d |
| 16 | TBAF | 55 |
| 17 | CsF | 93 |
Stronger hydroxide bases provided similar high yields (Table 2, entries 6–9), but Cs2CO3 was preferred to avoid cross-reactivity with base sensitive functional groups when exploring the scope of the reaction.
Further exploration of the base with alkali earth metal carbonates revealed consistently poorer yields (Table 2, entries 10–12). The secondary amine base N,N-diisopropylamine (DIPA) provided an equivalent yield (Table 2, entry 13) but interestingly, the tertiary amine triethylamine performed very poorly (Table 2, entry 14).
Using a stronger base (KOtBu) provided complete conversion of the starting material but did not provide the dihydropyrazole product (Table 2, entry 15), presumably due to Bamford–Stevens type processes, so further strong bases were discounted.
TBAF provided a moderate yield (Table 2, entry 16) before CsF provided a further improved yield of 93% (Table 2, entry 17). At this stage, CsF was selected as the optimal base and reaction optimisation moved on to assessing temperature and stoichiometry.
A short screen of reaction temperature revealed 110 °C to be optimal, with yields dropping off sharply above this and below 90 °C (Table 3, entries 1–5). A screen of base stoichiometry revealed 1.5 equivalents to provide the highest yield, with a small loss in yield from additional equivalents and fewer equivalents being severely deleterious to the yield (Table 3, entries 1, 6–8). Finally, the reaction was found to be insensitive to increases in alkene stoichiometry, with further equivalents providing a similar yield (Table 3, entry 9), however the yield dropped sharply when the stoichiometry was reduced to 1.0 equivalent (Table 3, entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Equiv. base | Equiv. alkene | Temp (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.25 mmol), CsF, diethyl fumarate, d6-DMSO (1 mL), sealed tube, N2 atmosphere, 22 h. b Yield determined by 1H NMR using 1,4-dinitrobenzene as internal standard. | ||||
| 1 | 1.5 | 1.5 | 110 | 93 |
| 2 | 1.5 | 1.5 | 130 | 39 |
| 3 | 1.5 | 1.5 | 90 | 70 |
| 4 | 1.5 | 1.5 | 70 | 20 |
| 5 | 1.5 | 1.5 | 50 | Trace |
| 6 | 0.5 | 1.5 | 110 | 20 |
| 7 | 1.0 | 1.5 | 110 | 32 |
| 8 | 2.0 | 1.5 | 110 | 78 |
| 9 | 1.5 | 2.0 | 110 | 90 |
| 10 | 1.5 | 1.0 | 110 | 13 |

Further investigations found that the reaction was insensitive to being performed under open reflux conditions (rather than in a sealed tube) and that a N2 atmosphere was not particularly beneficial. All yields in these examples were found to be within ±3% of the optimised conditions (Table 3, entry 1). However, the optimised conditions were retained for exploration of the substrate scope as a precaution.
With optimised conditions in hand (Table 3, entry 1), focus turned to the substrate scope of the reaction, beginning with assessment of a range of cyclic tosylhydrazones in the process (Scheme 6). Tosylhydrazones bearing 6-membered carbo- or hetero-cyclic rings (1a–1d) provided the corresponding spirocyclic dihydropyrazole products in good-to-excellent isolated yields (5a–5d).
 | ||
Scheme 6 Isolated yields from reaction of a range of tosylhydrazones 1a–k with diethyl fumarate. Reaction conditions: 1a–k (1.0 mmol), diethyl fumarate (1.5 equiv.), CsF (1.5 equiv.), dried DMSO (4 mL), 110 °C, 22 h, sealed vial, N2 atmosphere. Diastereomeric ratios (dr) 5e 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 5f 5:4, 5g 5:4; configurations not elucidated. :2, 5f 5:4, 5g 5:4; configurations not elucidated. | ||
The five-membered cyclic analogues generally performed well, with Boc-pyrrolidine (5e) and cyclopentane (5h) dihydropyrazole products being isolated in excellent yields. The tetrahydrothiophene product (5g) was isolated in a moderate yield of 55% while the tetrahydrofuran (5f) performed more poorly at 25%. In previous studies, we have noted the tendency of some small-ring oxygen and sulfur heterocycles to undergo ring-opening elimination processes under these type of reaction conditions, so this may account for the lower yield for these substrates.25 In the scope of four-membered spirocyclic dihydropyrazoles, only oxetane provided isolable product (5i). Thietane, azetidine and cyclobutane analogues provided an inseparable mixture of products. Finally, larger medium-sized carbocyclic rings provided spirocyclic products in good yield (5j and 5k).
What is particularly pleasing about the scope shown in Scheme 6 is that all of the 5- and 4-membered spirocyclic dihydropyrazoles isolated (5e–5i) were stable, in contrast to our previously published work where analogous 5- and 4-membered spirocyclic pyrazoles were observed to spontaneously undergo sigmatropic rearrangement to their fused bicyclic counterparts.4
Analysis of the reaction scope then turned to the alkene substrate. As preliminary investigations had already been performed on the required electronic properties of the alkene (Scheme 5), the scope focused on electron-deficient alkenes and introducing synthetically useful carbonyl-based functional groups (Scheme 7). The model compound 5a could be synthesised using either isomer of the alkene diester, however a higher yield was observed with the trans-isomer (diethyl fumarate) over the cis-isomer (diethyl maleate). The reaction tolerated the removal of one of the ester groups and ethyl acrylate provided the mono-ester product 5l in moderate yield. This was particularly pleasing as the requirement for two electron withdrawing groups on the alkene was potentially limiting in terms of the synthetic utility of the product.
 | ||
| Scheme 7 Isolated yields from reaction of 1a with a range of different alkenes. Reaction conditions: 1a (1.0 mmol), alkene (1.5 equiv.), CsF (1.5 equiv.), dried DMSO (4 mL), 110 °C, 22 h, sealed vial, N2 atmosphere. aDiethyl fumarate used as alkene. bDiethyl maleate used as alkene. | ||
An alkene bearing two ketone functional groups provided product 5m in moderate yield, while cinnamaldehyde selectively formed the regioisomer of 5n shown, with the aromatic ring closest the spirocyclic junction. This is consistent with the previously proposed mechanism where the electron withdrawing group is required to be at that position for the initial conjugate attack of the tosylhydrazonyl anion to occur.21 Interestingly, when a similar cinnamate ester substrate to those used by Wu et al. was assessed, the yield was relatively poor (5o), demonstrating the complementarity of these protocols.21
A further set of studies aimed to assess the downstream synthetic utility of the spirocyclic dihydropyrazole core, particularly 5a, which bears the diester functionality (Scheme 8).
 | ||
| Scheme 8 Exploration of the synthetic utility of model spirocyclic dihydropyrazole 5a. Reaction conditions: (a) NEt3 (2.0 equiv.), DMAP (0.1 equiv.), Boc2O (1.1 equiv.), CH2Cl2, RT, 12 h, 59%. (b) 5% KOH (18.0 equiv.), MeOH, reflux, 72%. (c) BnNH2 (2.0 equiv.), MeOH, 60 °C, 12 h, 75%. (d) NaBH4 (2.0 equiv.), MeOH, RT, 12 h, 30%.27–30 | ||
Initial synthetic studies on 5a found the core to be surprisingly stable and it was resistant to a wide range of reaction conditions including reduction and hydrolysis. However, protection of the amino nitrogen to give 6 rendered the ester groups more reactive, presumably as a result of the reduction in conjugation from the nitrogen lone pair. As a result, both ester groups in compound 6 could be hydrolysed under standard conditions to provide the diacid 7. Treatment of compound 6 with a representative amine provided the monoamide 8 in excellent yield where reaction occurred selectively at the ester group more distant from the spirocyclic junction. Finally, treatment of compound 6 with a complex metal hydride led to the surprising observation that the distal ester group was fully reduced down to the primary alcohol (9), with both the proximal ester and the iminyl π bond remaining intact.
In addition to the analogues presented previously (Scheme 7), these transformations on the model diester 5a demonstrate the potential for further decoration of the spirocyclic dihydropyrazole core and its potential utility in medicinal chemistry projects.
Finally, the reported procedure can be effectively performed as a one-pot process beginning from the parent ketone (Scheme 9). The scalability of the reaction was simultaneously demonstrated by performing this process on a gram scale, resulting in a 65% yield from the starting ketone.
 | ||
| Scheme 9 One-pot transformation of the parent ketone 11 into compound 5a on a gram scale. | ||
Conclusion
In summary, we report a procedurally simple method for formation of highly three-dimensional spirocyclic dihydropyrazole building blocks bearing functionality for further synthetic elaboration. These pharmaceutically-relevant compounds are formed from commercial electron-deficient alkenes and bench-stable tosylhydrazones, which are trivial to synthesise from their respective parent ketones in high yields without purification. A versatile substrate scope is reported, including stable 4- and 5-membered rings at the spirocyclic junction. Finally, the further synthetic utility of this class of spirocycle is demonstrated via orthogonally reactive analogues and example synthetic transformations on the core structure. These building blocks represent a valuable addition to the arsenal of core structures for medicinal chemistry applications.Author contributions
TLW – Investigation, methodology, data curation, formal analysis, validation, visualisation, writing – original draft. DMA – Funding acquisition, conceptualisation, investigation, methodology, data curation, formal analysis, supervision, validation, visualisation, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TLW and DMA thank the SHU BMRC for funding. TLW wishes to thank Jack Slater for advice and support. TLW and DMA also wish to thank the technical team at SHU for their help and assistance throughout this project.Notes and references
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- F. Lovering, MedChemComm, 2013, 4, 515 RSC.
- M. Aldeghi, S. Malhotra, D. L. Selwood and A. W. E. Chan, Chem. Biol. Drug Des., 2014, 83, 450 CrossRef CAS PubMed.
- R. R. Merchant, D. M. Allwood, D. C. Blakemore and S. V. Ley, J. Org. Chem., 2014, 79, 8800–8811 CrossRef CAS PubMed.
- T. J. Richie, S. J. Macdonald, R. J. Young and S. D. Pickett, Drug Discovery Today, 2009, 14, 1011 CrossRef PubMed.
- C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514 RSC.
- S. Lee, J. H. Song, C. M. Park, J.-S. Kim, J.-H. Jeong, W.-Y. Cho and D.-C. Lim, ACS Med. Chem. Lett., 2013, 4, 1054 CrossRef CAS PubMed.
- Y. Dong, T. Furuta, H. Sabit, T. Kitabayashi, S. Jiapaer, M. Kobayashi, Y. Ino, T. Todo, L. Teng, A. Hirao, S. Zhao and M. Nakada, Oncotarget, 2017, 8, 111728–111741 CrossRef PubMed.
- Y. Zheng and C. M. Tice, Expert Opin. Drug Discovery, 2016, 11, 831–834 CrossRef PubMed.
- M. Develoux, Ann. Dermatol. Venereol., 2001, 12, 1317–1325 Search PubMed.
- J. Weber and G. M. Keating, Drugs, 2008, 68, 1691–1698 CrossRef CAS PubMed.
- G. P. Reams, A. Leu, V. Knaus and J. H. Bauer, J. Clin. Pharmacol., 1933, 33, 348–353 CrossRef PubMed.
- D. W. Dodick, R. B. Lipton, J. Ailani, K. Lu, M. Finnegan, J. M. Trugman and A. Szegedi, N. Engl. J. Med., 2019, 23, 2230–2241 CrossRef PubMed.
- L. J. Scott, Drugs, 2020, 80, 323–328 CrossRef PubMed.
- A. S. K. Hashmi, L. Schwarz and J. W. Bats, J. Prakt. Chem., 2000, 342, 40–51 CrossRef CAS.
- (a) S. Yamada, S. Karasawa, Y. Takahashi, M. Aso and H. Suemune, Tetrahedron, 1998, 54, 15555–15566 CrossRef CAS; (b) N. Spiliopoulou, N. F. Nikitas and C. G. Kokotos, Green Chem., 2020, 22, 3539–3545 RSC.
- Y. Zheng, C. Tice and S. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed.
- D. D. Vachhani, M. Galli, J. Jacobs, L. V. Meervelt and E. V. Van der Eycken, Chem. Commun., 2013, 49, 7171–7173 RSC.
- X. Chen, H. Chen, X. Ji, H. Jiang, Z. Yao and H. Liu, Org. Lett., 2013, 8, 1846–1849 CrossRef PubMed.
- Y. Yang, L. Li, Y. He, J. Luo and Y. Liang, Tetrahedron, 2014, 70, 702–707 CrossRef CAS.
- Q. Wu, H. Li, H. Wanga, Z. Zhanga, C. Wanga and Y. Wu, Synlett, 2015, 26, 243–249 CAS.
- R. Huang, H. Tao and C. Wang, Org. Lett., 2017, 19, 1176–1179 CrossRef CAS PubMed.
- A. P. Molchanov, A. V. Stepakov and R. R. Kostikov, Russ. J. Org. Chem., 2004, 40, 1512–1517 CrossRef CAS.
- J. Shao, W. Chen, M. Zhao, K. Shu, H. Liu and P. Tang, Org. Lett., 2018, 20, 3992–3995 CrossRef CAS PubMed.
- D. M. Allwood, D. C. Blakemore, A. D. Brown and S. V. Ley, J. Org. Chem., 2014, 79, 328–338 CrossRef CAS PubMed.
- D. M. Allwood, D. C. Blakemore and S. V. Ley, Org. Lett., 2014, 16, 3064–3067 CrossRef CAS PubMed.
- J. Barluenga, F. Fernandez-Marí, A. L. Viado, E. Aguilar, B. Olano, S. García-Granda and C. Moya-Rubiera, Chem. – Eur. J., 1999, 5, 883–896 CrossRef CAS.
- E. G. Mamedov, Russ. J. Org. Chem., 2007, 43, 192–195 CrossRef.
- P. S. Poon, A. K. Banerjee and M. S. Laya, J. Chem. Res., 2011, 67–73 CrossRef CAS.
- N. P. Grigoryan, S. A. Pogosyan and R. S. Sukasyan, Pharm. Chem. J., 2007, 41, 59–61 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/d2ob00093h |
| This journal is © The Royal Society of Chemistry 2022 |