
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in asymmetric synergistic catalysis – the judicious combination of selected chiral aminocatalysts with achiral metal catalysts
Christian D.-T.
Nielsen†‡
 ,
Joshua D.
Linfoot‡
,
Alexander F.
Williams
and
Alan C.
Spivey
*
,
Joshua D.
Linfoot‡
,
Alexander F.
Williams
and
Alan C.
Spivey
*
Imperial College London, White City Campus, Molecular Sciences Research Hub (MSRH), 82 Wood Lane, London W12 0BZ, UK. E-mail: a.c.spivey@imperial.ac.uk
First published on 10th March 2022
Abstract
In this review we survey recent synergistic applications of a chiral organocatalyst with an achiral metal to perform stereoselective transformations of synthetic utility (since 2016). The transformations are classified by the modes of reactivity deployed, focussing on organocatalytic activation of carbonyl substrates as chiral nucleophiles via the α-position (e.g., as enamines) and as chiral electrophiles via the β-position (e.g., as iminium ions) combined with complementary activation of their reaction partners by an achiral metal co-catalyst (e.g., Pd or Cu-based). Corresponding radical reactions are also presented in which photocatalysis mediated by achiral metal complexes replaces the metal co-catalyst. Certain privileged structures are revealed and opportunities to develop this exciting field are highlighted.
 Christian D.-T. Nielsen | Christian obtained his MSci from Imperial College London in 2015 with a year abroad at NTU in Singapore. He stayed at Imperial for his PhD under the supervision of Alan Spivey and Jordi Bures, spending time with Miquel Pericas (ICIQ) and Ben List (MPI), investigating organocatalytic mechanisms. Currently, he is carrying out postdoctoral research with Franziska Schoenebeck (RWTH Aachen) in transition metal catalysis with computational analysis. |
 Joshua D. Linfoot | Joshua was awarded an MSci in Chemistry with Industrial Experience from the University of Birmingham in 2018, and spent his industrial year working in drug discovery for Evotec in Abingdon, Oxfordshire. He is currently in the final year of his iCASE-funded PhD at Imperial College London, and is investigating metabolic insecticide resistance using chemical proteomics under the supervision of Alan Spivey and Edward Tate, with co-supervision from Christoph Zimmer and Peter Kilby from his sponsor company, Syngenta. |
 Alexander F. Williams | Alexander obtained his MSci from the University of Bristol in 2016, spending a year in industry working as a medicinal chemist at GSK in Stevenage. He moved to Imperial College for his PhD under the supervision of Chris Cordier and Alan Spivey, investigating palladium catalysed C–H functionalisations. Currently, Alexander is working as a Process Chemist at Pharmaron UK in Hoddesdon. |
 Alan C. Spivey | Alan Spivey (http://www.imperial.ac.uk/people/a.c.spivey) has been professor of synthetic chemistry at Imperial College London since 2008. Previously, he was a lecturer then reader in the Department of Chemistry, University of Sheffield. His research encompasses the development of new reactions, synthesis of complex molecules of biomedical interest, asymmetric organocatalysis, chemical biology and medicinal chemistry. For further details of his groups research activities see: http://www.imperial.ac.uk/spiveygroup. |
1. Introduction
Synergistic catalysis (also known as cooperative catalysis) may be defined as the simultaneous activation of both components in an ionic (or radical) reaction by separate and distinct catalysts.1,2 While the application of two catalysts as opposed to one may at first appear non-ideal, this tactic has proven fruitful in terms of controlling the reactivity of both the electrophilic and nucleophilic components, simultaneously but separately, during the course of a reaction. Methods involving the combination of metal catalysts and organocatalysts3–6 as well as dual metal systems7,8 have been developed (Scheme 1a). | ||
| Scheme 1 (a) The overall tactic of enantioselective synergistic catalysis. (b) Enantioselective synergistic catalysis types discussed within this review. | ||
Asymmetric transformations mediated by metal/organocatalyst combinations may be achieved in three ways. Firstly, a chiral metal may be employed with an achiral organocatalyst. Secondly, an achiral metal may be used in conjunction with a chiral organocatalyst (which is the focus of this review). Finally, both catalysts may be chiral. In the case of both catalysts bearing chiral information, stereodivergent catalysis is possible in which all stereoisomers of the product may be accessed by selection of the appropriate enantiomer of each catalyst.9
By modulation of both the HOMO and LUMO (or SOMO) of the nucleophile and electrophile respectively, new reactivity under mild reaction conditions may be enabled. General synergistic organocatalysis with a variety of activation modes has been reviewed by Carlone.10 However, since the comprehensive reviews by Córdova,11–13 in which the use of amino- and metal-catalysis was specifically inspected, there has been continued interest in the unison of these components. Recently, Snaddon has reviewed the general combination of chiral tertiary amine Lewis base catalysis with chiral transition metal catalysis.14 This review will inspect developments since 2016 in the deployment of a chiral aminocatalyst with an achiral metal catalyst. Specifically, the reactions we will analyse here are examples of ‘Type I’ systems in which each catalyst acts independently and fully cooperatively.15 We will not inspect relay catalysis in which one substrate goes through an initial reaction, in the presence of other components, before undergoing a subsequent second transformation. Our survey is divided into four sections, addressing respective activation modes of the substrates (Scheme 1b). In section 1 we detail ionic reactions in which a chiral aminocatalyst raises the HOMO and an achiral metal lowers the LUMO of respective reactants. In section 2 we detail cases where the opposite applies, i.e., a chiral aminocatalyst lowers the LUMO and an achiral metal raises the HOMO of respective reactants. In sections 3 and 4 we consider the analogous reactivity under a radical paradigm: i.e., reactions of aminocatalytically activated nucleophiles reacting with electrophilic radicals and then aminocatalytically activated electrophiles reacting with nucleophilic radicals.
2. Ionic reactivity
2.1. Nucleophile activation involving organocatalytic enol/enamine formation
Saturated and α,β-unsaturated carbonyls can be activated to act as chiral nucleophiles at their α and β-positions, respectively, following reaction with chiral Lewis base catalysts (i.e., HOMO-raising catalysis). These nucleophilic complexes can then react efficiently with an achiral metal complexed π-(allyl) or allenylidene species (i.e., LUMO-lowering catalysis) to give a chiral product. Success in this area has been reported using chiral amine, N-heterocyclic carbene (NHC) and isothiourea catalysts activating enals and esters, in concert with Pd and Cu activated allylic, propargylic and allenic electrophiles. This approach can be viewed as building upon the seminal work of Scheidt16 and others17 who employed NHCs with Lewis acid activated carbonyls as well as reactions of enamines with Pd π-(allyl) systems, as pioneered by Córdova.18In 2016, Glorius used a chiral triazolium catalyst and Pd(PPh3)4 to catalyse the formation of chiral benzazepines from enals and allyl carbamates (Scheme 2).19
 | ||
| Scheme 2 Synthesis of benzazepines using a chiral triazolium Lewis base catalyst and Pd(PPh3)4, by Glorius.19 | ||
The stereochemistry-determining step involved the chiral NHC-homoenolate 2 formed by 1,2-addition into the enal, reacting at the β-position with the electrophilic π-(allyl)Pd species 1 formed by oxidative addition/decarboxylation to the allyl carbamate. The cyclisation step from zwitterion 3, in which the N-tosyl anion closes onto the acyltriazolium moiety to form the 7-membered ring, is a kinetically favoured 7-exo-trig process that releases the NHC back into the catalytic cycle.20 Mechanistic work indicated that the NHC was also an important ligand for the Pd, with detection of the ligated Pd-species by high-resolution mass spectrometry (HR-MS).21 Furthermore, the NHC catalyst loading was shown to affect the overall reaction outcome. Only trace amounts of product were observed at low NHC loadings indicating the importance for reaction productivity. A positive nonlinear effect was observed between the enantiopurity of the catalyst and the ee of the product showing that this complex was involved in the stereodetermining step. Indeed, the well-known ability of NHCs to ligate late transition metals is one of the challenges in implementing this chemistry.22,23 Glorius managed to circumvent this problem by applying strongly-binding and electron-rich BINAP as the ligand on Pd.24 However, a strong match-mismatch phenomenon was observed between the now chiral Pd and chiral NHC (65% difference in yield and 44% decrease in ee between enantiomers of BINAP). This highlights the challenge of applying NHCs in synergistic catalysis.
Snaddon has utilised isothioureas as aminocatalysts in combination with transition metals in related chemistry. Thus, the enantioselective α-allylation of aryl acetic esters by allyl mesylates and carbonates, mediated by the chiral benzotetramisole catalyst BTM and XantphosPd G3 was first disclosed in 2016 (Scheme 3).25
 | ||
| Scheme 3 Allylation of aryl acetic esters using a chiral isothiourea Lewis base catalyst and XantphosPd G3, by Snaddon.25 | ||
The BTM catalyst was originally introduced by Birman who first suggested that the acyl ammonium salt intermediate in asymmetric acylation reactions catalysed by this isothiourea benefitted from a rigidifying nO → σ*C–S anomeric effect,26,27 a feature that was subsequently corroborated computationally by Romo28 and by Smith.29 Snaddon suggests a similar interaction between the anionic oxygen of the C1 ammonium enolate and the BTM during this synergistic process.30 Allylation then occurs on the Re face of the enolate, opposite to the phenyl group.25,31 Allyl mesylate was the preferred reagent for simple allylation, but the introduction of substituted allyl units necessitated a switch to allyl tert-butylcarbonates. In all cases, the use of pentafluorophenol (Pfp) esters was important for product generation by enabling ‘phenolate rebound’ in which the aryloxide is a suitable nucleofuge to form the chiral enolate and can subsequently turnover the Lewis base catalyst post-allylation.32 Conveniently, the Pfp ester can also undergo a variety of subsequent transformations including reduction, transesterification and hydrolysis with retention of stereochemical information. Snaddon showcased the synthetic power of this synthetic method with the synthesis of indomethacin Pfp ester 4, formed from a substrate in which the aryl acetic acid precursor was an indole derivative.
Having identified a suitable catalytic duality between the BTM isothiourea to form a C1 ammonium enolate and Pd to form an allyl electrophile, Snaddon was able to further extend the scope of this reaction class. Specifically, proelectrophiles incorporating Si,33 B,34 benzylic phosphonates35 as well as aryl pyrroles36 as pronucleophiles all proved compatible with this chemistry.
Despite the success of the described systems, allyl electrophiles bearing substitution at the 2 position generally proved unsuited to this reaction manifold. This failure was attributed to unfavourable steric congestion preventing formation of the active π-(allyl)Pd electrophile with the large bidentate Xantphos ligating Pd.30 Due to the modularity of the developed synergistic system, it was hypothesised that a judicious change of the ligand on Pd might allow this electrophile class to participate successfully. Gratifyingly, by applying the small monodentate P(2-thienyl)3 ligand, 2-subsituted allyl pro-electrophiles were able to engage with the Pfp ester derived enolates (Scheme 4).30
 | ||
| Scheme 4 Allylation of esters with 2-substituted allyl pro-electrophiles using a chiral isothiourea Lewis base catalyst and Pd2(dba)3/(2-thienyl)3P, by Snaddon.30 | ||
Transamidation of the Pfp ester, in situ was shown to be possible (e.g., → 5) and alkenyl esters possessing enolisable δ-hydrogens could be formed smoothly (e.g., → 6). DFT calculations supported the proposal of independent reaction components coming together in the C–C bond forming step. In the case of an inner sphere attack, in which the enolate is within a seven-membered palladacycle with a Pd–O bond, the energy of this transition state was predicted to be 17.6 kcal mol−1 higher in energy than the unbound outer sphere attack (Scheme 4, inset).
As has been described by Trost and Tsuji, the attack of a nucleophile on a π-(allyl)Pd electrophile is dominated by the electronic and steric nature of the π-(allyl)Pd(II) species,37,38 but generally occurs to give the linear products.39–41 In order to modulate the regioselectivity of nucleophilic attack and so the linear or branched nature of the product, bespoke ligand–catalyst systems are often applied. The requirement for tailored systems may be exemplified by the varying outcome using Snaddon's original conditions (BTM and XantphosPd G3) in the allylation of esters with a variety of electron deficient alkenes (Scheme 5).42
 | ||
| Scheme 5 Identification of a unified catalyst system for allylation of Pfp esters, by Snaddon.42 | ||
However, with the validation that the two catalytic cycles operated independently (vide supra), Snaddon reasoned that a unified Pd catalyst system could be identified to deliver products in high yield and ee with the E-double bond configuration. In this regard, the syn/anti reactivity and ratio of the π-(allyl)Pd(II) component, as previously noted by Bosnich, appeared to be crucial.43,44 Pd[(P-2Th3)3] was identified as a privileged catalyst system enabling the allylation of esters with a variety of electron deficient allyl groups and with short reaction times. By comparing a variety of Xantphos derivatives, this system is proposed to be successful by combining both low steric demand and strong π-accepting character.
An emerging pressure within synergistic catalysis (and synthesis broadly) is “pot economy”.45 A common rearrangement of amides (which have been shown to be accessible in one-pot from Pfp esters, i.e., → 5, Scheme 4) is the Hofmann rearrangement. Snaddon has successfully applied this rearrangement to his ester allylation method. By addition of ammonia to the Pfp ester products post allylation, the resultant primary amides rearrange to give N-substituted linear homoallylic carbamates/amines upon treatment, in situ with phenyliodine(III) bis(trifluoroacetate) (PIFA) as oxidant (Scheme 6).46
 | ||
| Scheme 6 Sequential one-pot synergistic allylation of aryl acetic esters [using a chiral isothiourea Lewis base catalyst and Pd(Xantphos)] then Hofmann rearrangement to furnish homoallylic linear and branched amines, by Snaddon.47 | ||
A variety of alcohols were shown to successfully trap the isocyanate intermediate furnishing a variety of common carbamate protected amines such as N-CBz protected product 7. Additionally, water could be employed to deliver the free amine 8 directly. Alkenyl acetic acid esters were also competent pronucleophiles, undergoing highly regioselective allylation to give e.g., diene 9. The incorporation of the α,β-unsaturated ester in 10 is noteworthy as using a traditional disconnection in which a metal allyl nucleophile would add into the imine could prove challenging due to requirement of chemoselective addition (i.e., imine vs. α,β-unsaturated ester).
Taking inspiration from Hartwig's work,48 Snaddon also carried out this type of transformation using a cyclometallated phosphoramidite (R)-Ir catalyst 11, and demonstrated that the branched products still underwent Hofmann rearrangement. Accordingly, all four stereoisomers of the branched homoallylic amines were accessible individually by selection of the appropriate enantiomer combination of the two synergistic chiral catalysts. Potential developments of this work include trapping the Hofmann intermediate isocyanate with an amine (which may originate from the aforementioned prior allylation/Curtius rearrangement process) to generate a chiral urea, which have been shown to be potent hydrogen bond donors.49
The enabling potential of Snaddon's synergistic approach is underscored by its application in the enantioselective total syntheses of alkaloids. Specifically, C2/3 indole acetic acids were used to access Strychos alkaloids while Chelidonium alkaloids were accessed by the aforementioned branched selective allylation and Hofmann rearrangement.50
Evidently, the use of an isothiourea-Pd catalyst system is a powerful solution to the parasitic coordination of NHCs to Pd. However, an alternative solution is the selection of an alternative metal catalyst.
Building on Shi's report,51 Gong has reported the enantioselective Cu-catalysed α-amidation of Pfp-esters to hydantoins, in which the amine source is di-tert-butyldiaziridinone.52 The reaction was shown to involve a Cu-amide radical (detected by EPR) and features an unusual C–N bond formation between an isothiourea-derived C1-ammonium enolate and a Cu-amide, which confirms the compatibility of Cu-catalysis and Lewis base catalysis. In this regard, Gong has subsequently formed a Cu allenylidene intermediate which undergoes nucleophilic attack from an isatin derived enal (Scheme 7).53
 | ||
| Scheme 7 Decarboxylation and [3 + 3]/[3 + 4] cyclisation to form spirooxindole lactones and spirobenzazepinones using a chiral triazolium Lewis base catalyst and Cu(MeCN)4PF6, by Gong.53 | ||
Subtle changes in catalyst and conditions facilitated highly stereoselective [3 + 3] cycloadditions with ethynylethylene carbonates and [3 + 4] cycloadditions to form spirobenzazepinones. However, MS suggested the presence of NHC binding with the Cu catalyst. Indeed, a non-linear effect (NLE) was observed implicating multiple NHCs participating in stereoinduction.
2.2. Electrophilic activation involving organocatalytic iminium ion formation
Chiral aminocatalysts may also perform as LUMO-lowering agents, in which a carbonyl containing reactant is rendered more electrophilic by conversion to an iminium species.54 Such catalysts are often proline-based, with diphenylprolinol silyl ethers such as the Jørgensen–Hayashi catalyst being particularly popular.54 This mode of activation can be synergistically combined with activation of a pro-nucleophile using a Pd, Cu or Lewis acid co-catalyst. This section explores the versatility of this tactic in the context of iminium ions derived from enals. Much of the early work in this area was carried out by Córdova, who for example reported enantioselective conjugate addition of Me2PhSi-B(pin) to enals catalysed by Jørgensen–Hayashi catalyst/copper(I) chloride in 2011.55 The concept can be extended to cascade processes in which the enamine intermediate formed from initial attack on the iminium ion is captured by a pendant electrophile. A pioneering non-enantioselective example of this was Dixon's formation of cyclopentenes from enones and propargylated carbon acids catalysed by pyrrolidine/copper(II) triflate in 2008.56The potential of this reaction manifold has been further extended recently. Following their benchmark cooperative catalysis disclosure in 2014,57 in which prolinol iminium ions derived from cinnamaldehydes condensed stereoselectively with nucleophilic 2-alkyl-nitrobenzoxazoles, Rios et al. have now extended this approach to the reaction of 2-chloroalkyl nitrobenzoxazoles. This modification of the nucleophilic component enables a cascade process to ensue, in which 2 new C–C bonds are generated in a cyclopropanation reaction.58 In this triple catalytic cycle, coordination of Pd(II) to the benzoxazole nitrogen and resulting activation of this system facilitates base-mediated isomerization to the corresponding enamine species to form the benzylic nucleophile. Separately, condensation of the enal with a Jørgensen–Hayashi type prolinol organocatalyst (12) to give an iminium ion, activates the electrophilic component. A second coordination of the palladium to the distal iminium π-system, proposed based on observations by Córdova on a related system, arranges the two substrates such that they are poised for a 3-exo-tet cyclisation to afford a cyclopropane, which upon hydrolysis provides the aldehyde product (Scheme 8).
 | ||
| Scheme 8 Diastereoselective addition of 2-chloroalkyl benzoxazoles to enals to give cyclopropanes using prolinol-type aminocatalyst and Pd(OAc)2, by Rios.58 | ||
This reaction was found to be reasonably diastereoselective, with 1H NMR analysis confirming a bias towards the cinnamyl aryl ring and benzoxazole adopting a syn relationship, which is the opposite to what occurs in MacMillan cyclopropanation and thus offers complimentary synthetic utility.59 Yields and diastereoselectivities were consistently high for substituted cinnamaldehydes containing ortho- or para-electron withdrawing groups, and enantioselectivities of up to >99% ee were achieved. The major diastereomer in all cases had the aldehyde anti relative to the other ring substituents, and this diastereomer could be isolated from the others by column chromatography. Use of an alkyl enals resulted in complex mixtures being obtained, however presence of an ester at the β-carbon of the enal afforded the (1R,2R,3S)-diastereomer selectively, albeit with diminished enantioselectivity. This was speculatively attributed to in situ epimerisation of the product (i.e., fumarate mono-aldehyde → 13, Scheme 8). A notable limitation of these reactions is that only strong electron-withdrawing substituents on the benzoxazole allow the reaction to proceed; unsubstituted benzoxazole is unreactive. Later computational interrogation of this mechanism using pyrrolidine as a model organocatalyst suggested that after the Michael addition step, which is expected to occur on the iminium ion's less hindered Re face for the real system, gives a pair of diastereomers at C2. The final stereochemical outcome arises from the stereospecific inversion of this centre occurring from the Si face of the trans-enamine for the 2R diastereomer but from the Re face for the 2S diastereoisomer.60 This model did not incorporate the secondary coordination of the palladium to the iminium ion as previously postulated.
Cyclopropanes can themselves be used as substrates in synergistic catalysis, as demonstrated by the works of Vitale,61 Rios,62,63 Jørgensen64 and Veselý65 in the formal [3 + 2]-cycloaddition of vinylcyclopropanes with enals to afford cyclopentane products with up to four contiguous stereogenic centres (Scheme 9).
 | ||
| Scheme 9 Formal [3 + 2]-cycloadditions of vinylcyclopropanes across enals to afford decorated cyclopentane products with 2 new C–C bonds and up to 4 stereogenic centres using prolinol-type aminocatalyst and Pd2(dba)3, by Vitale (a), Rios (b and d), Jørgensen (c) and Veselý (e).61–65 *Pd2(dba)3·CHCl3 with 10 mol% dppe, and p-nitrobenzoic acid. | ||
These domino reactions are proposed to proceed by oxidative addition of Pd into the cyclopropane to generate a zwitterionic π-allyl–Pd intermediate with the negative charge stabilised by two adjacent electron-withdrawing substituents, whilst in parallel the enal is once again activated by a chiral prolinol organocatalyst 12 to form a transient iminium species. The electron density from the anionic centre in the dipole can then add into the iminium ion in a Michael process to afford an enamine, which reacts intramolecularly with the π-allyl–Pd species in a 5-exo-trig cyclisation. Protonation and subsequent reductive elimination furnishes the cyclopentane product decorated with an allyl and aldehyde group on adjacent carbons in a syn relative configuration. This stereochemistry in the cyclisation step may be partly influenced by steric repulsion between the π-allyl–Pd species and the bulky enamine, but is also preferred on electronic grounds (i.e., favourable distal attack of the enamine on the Pd centre).
Vitale and colleagues explored this reactivity in 2016 using activated enals and vinylcyclopropanes bearing electron-withdrawing gem-dicyano units (Scheme 9a).61 After selecting chloroform-coordinated Pd2(dba)3 as the transition metal precatalyst, aminocatalyst screening was performed using the Jørgensen–Hayashi catalyst and a selection of MacMillan's imidazolidinones. However, in all cases, no desired product was formed despite complete consumption of the vinylcyclopropane. This was attributed to the tendency of this moiety to undergo unproductive polymerisation under the influence of Pd(0) in the absence of a reactive enough acceptor molecule. Fortunately, bolstering iminium ion formation by addition of a substoichiometric Brønsted acid rectified this. Dppe gave the best yields and stereoselectivities compared to other mono- or bis-phosphines. After optimisation, high yields, good diastereocontrol and excellent enantioselectivity were obtained in a relatively short reaction time for a range of cinnamaldehydes bearing electron-rich and electron-poor aryl groups (→ 14–15). Aliphatic dienals were not tolerated, however. Importantly, a plethora of alternative electron-withdrawing groups in the vinylcyclopropane were found to be compatible with this process, including 1,3-indanone (→ 16), N,N′-dimethylbarbituric acid, Meldrum's acid (→ 17) and dimethylmalonate containing scaffolds.
Rios et al. published their work on this reaction class later that year, focusing on 1,3-indanone as the electron-withdrawing component (Scheme 9b).62 Concordant with Vitale's observations, phosphine ligands were found to be detrimental to the reactions, which may suggest that the Pd has a potential role as a Lewis acid. EtOAc was the optimal solvent for stereoinduction with the Jørgensen–Hayashi catalyst and under these conditions a range of electron-poor cinnamaldehydes and aliphatic enals gave the products with high levels of stereoselectivity (→ 18–20). 2,4-Hexadienal reacted through its distal alkene and with reduced enantioselectivity, presumably caused by diminished facial selectivity in the stereodetermining transition state (→ 21).
Jørgensen also noted that phosphine ligands led to enal decomposition in this type of reaction, probably via Pd-assisted phosphine SN2′-like ring opening.64 His work demonstrated that geminal dinitriles and nitrile esters can be used as the electron-withdrawing moieties, but showed that in contrast to Vitale's work, the use of a geminal diester starting material in this system apparently provided too much pseudoaxial steric hindrance for the subsequent cyclisation reaction to proceed (→ 22–25, Scheme 9b). Inclusion of substoichiometric benzoic acid permitted full conversion, once again by facilitating iminium ion condensation as a Brønsted acid. For the geminal dinitrile cyclopropane, reaction with a range of cinnamaldehydes and other (hetero)aryl enals gave rise to cyclopentanes with high stereoselectivity. Exchanging one of the nitriles in the starting material with an ester gave an additional quaternary stereocentre in the product, and both methyl and benzyl esters led to similarly good selectivities for reactions with cinnamaldehydes.
In research on the same substrate class by Rios in 2017,63 it was found that by using higher catalyst loadings, a broad scope of cinnamaldehyde components were tolerated with exquisite stereoselectivities, including those bearing electron-donating or withdrawing substituents (→ 26–29, Scheme 9c). It was found that the use of a methyl ester in the starting material provided better enantio- and diastereoselectivities than the ethyl variant as well as a higher yield.
More recently, Veselý made further advancements by extending this reaction paradigm to azlactone cyclopropanes (→ 30–33, Scheme 9d).65 Whilst operationally equivalent, the product of this reaction possesses additional complexity which allows for further elaboration into interesting products such as unnatural quaternary amino acids. However, this approach suffered from long reaction times. A series of cinnamaldehydes containing an electron-withdrawing group at the meta- or para-position, and alkyl enals, engendered moderately diastereoselective and highly enantioselective transformations with a phenyl Erlenmeyer–Plöchl azlactone, although in general electron-poor cinnamaldehydes had less stereocontrol. Remarkably, the ortho-halo substituents caused the reaction to favour the opposite diastereomeric result (32). One limitation of this protocol seems to be the incompatibility of heteroaryl enals, such as thiophene derivatives.
Synergistic catalysis also provides a route towards other interesting spirocycles, as evidenced by Rios’ and Veselý's overall ring contraction of a [5,7]-fused pyrazole-oxacyclic system with concomitant addition to an enal to afford pyrazolone [5,5]-spirocycles.66 For this reaction class, ESI-MS and IRPD analysis suggested that the fused bicycle is activated by Pd insertion with concomitant C–O bond cleavage to give enolate-coordinated π-allyl complex 34, which computational modelling suggested could be formed via an unusual syn-transition state. This zwitterionic species can then engage in stepwise annulation with the iminium ion, which is generated in a parallel catalytic cycle, and the subsequent enamine (Scheme 10).
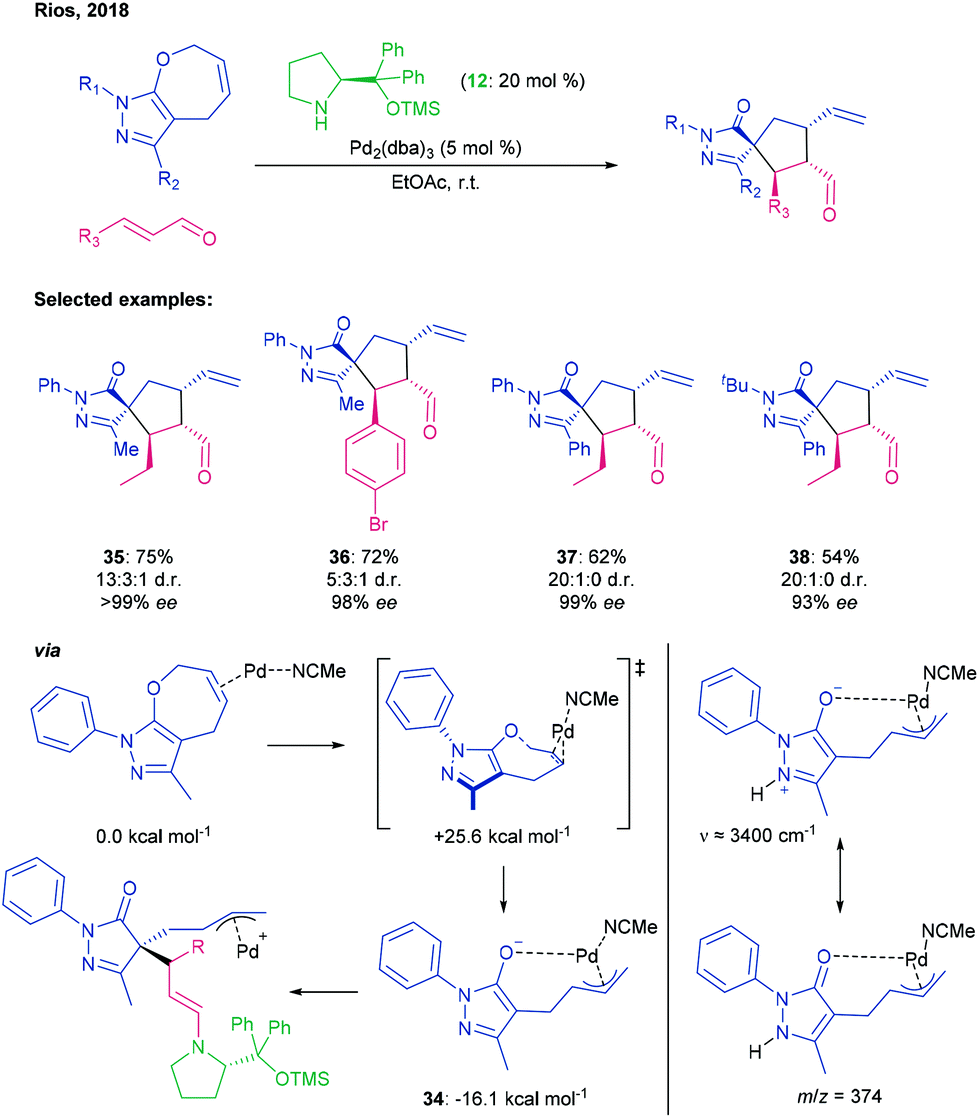 | ||
| Scheme 10 Reaction of [5,7]-fused pyrazole-oxacycles with enals to afford pyrazolone [5,5]-spirocyclic pyrazolones using a prolinol-type aminocatalyst and Pd2(dba)3, with analysis of Pd-containing intermediates and a key transition state by ESI-MS, IRPD and computation, by Rios.66 | ||
Using a 3-methyl-1-phenylpyrazole derivative as a representative substrate, enal scope was explored and found to tolerate a range of aliphatic chain lengths (→ 35) with excellent enantio- and diastereoselectivities. Aryl enals that were meta- or para-substituted with electron-withdrawing or donating groups (→ 36) were also acceptable substrates, furnishing products with comparable enantioselectivities but with poorer diastereocontrol. Introducing a 3-phenyl group in place of the methyl group in the pyrazole led to good yields and high degrees of stereocontrol with aliphatic enals (→ 37), but with lower de with cinnamaldehydes. Exchanging the 1-phenyl group with either a tert-butyl (→ 38) or 2,4-dinitrophenyl group maintained the ee, but diminished yields and a reduction in dr for aryl enals. Pyrazolone products could be further converted via reduction, oxidation, cyclisation, halogenation, metathesis and Suzuki reactions with retention of optical purity.
More recently, Veselý and colleagues demonstrated an analogous reaction of related [5,7]-fused thiazole-oxacyclic ring systems under similar conditions, which furnished structurally related spirothiazolones with comparable yields and stereoselectivities. It was found that switching the solvent from ethyl acetate to acetonitrile enhanced the diastereocontrol, and that the Pd catalyst loading could be reduced to 2% without reduction in reaction efficiency or enantioselectivity.67
Rios and Veselý then went on to demonstrate that [5,5]-spirobicyclic pyrazolones could also be synthesised in an intermolecular Michael/Conia-ene cascade using synergistic Pd and iminium ion catalysis, with an alkyne-containing monocyclic pyrazolone starting material acting as a ring-opened analogue of the [5,7]-bicycle in their original approach.68 This transformation only required low loadings of both catalysts to proceed whilst still giving the spiropyrazolone scaffolds 39–42 in impressive ee and with acceptable diastereocontrol. A range of aliphatic enals and substituted cinnamaldehydes were found to be compatible with this reaction, as were heteroaryl enals (40), a glyoxylate derivative (41) and skipped dienals. Heteroaromatics were found to result in higher diastereoselectivities, whereas the reaction with an ortho-substituted cinnamaldehyde, while still highly diastereoselective, suffered from reduced yield possibly due to unfavourable steric interactions. Changing the C-alkyl substituent from methyl to phenyl had a dramatic effect on reaction rate, with full conversion unable to be achieved even after 7 days; an ethyl group gave comparable results. Introduction of electron-withdrawing or donating para-substituents on the N-aryl group (→ 42) was tolerated with little perturbation of the observed stereoselectivities. Further transformations on the aldehyde product included Pinnick oxidation to the corresponding carboxylic acid which enabled the absolute configuration to be determined by a single crystal anomalous dispersion X-ray crystallography experiment. NMR kinetics studies led the authors to conclude that the Michael reaction was reversible and rate-determining and that the Conia-ene was irreversible and stereo-determining, thus setting up Curtin–Hammett kinetic regime (Scheme 11). The authors of this work illustrated the Pd as being coordinated to the alkyne externally during the carbocylisation, however calculations for related processes suggest that a more elaborate Pd(0)/Pd(II) cycle might occur.69
 | ||
| Scheme 11 A Michael/Conia-ene cascade process to convert pyrazolone alkynes and enals into spiropyrazolones using a prolinol-type aminocatalyst and Pd2(dba)3, and the postulated synergistic dual catalytic cycles, by Veselý.68 | ||
Numerous examples exist of multicomponent reactions catalysed in a synergistic fashion. Jørgensen has reported the reaction of alkyl quinolines with two equivalents of cinnamaldehydes in a sequential double Michael addition and subsequent aldol dehydration, furnishing cyclohexyl enals with three contiguous stereocentres with high levels of stereoselectivity (Scheme 12).70
 | ||
| Scheme 12 Double Michael addition of alkylquinolines into cinnamaldehydes, followed by aldol dehydration to afford cyclohexene derivatives using a prolinol-type aminocatalyst and InCl3, by Jørgensen.70 | ||
In these transformations, the Lewis acidic InCl3 coordinates to the quinoline nitrogen, increasing the acidity of the alkyl α-protons and allowing enamine formation. Simultaneously, the enal is rendered more electrophilic by iminium ion formation. Double Michael addition of the enamine to two iminium ion-activated enals affords an enamine that undergoes dehydrative annulation via a 6-exo-trig aldol reaction to furnish the cyclohexene product. In terms of enal scope, this process was compatible with a range of substituted (hetero)aryl substrates to give their corresponding cyclohexenes (43–45) with high dr, and with the major diastereomer heavily enantioenriched. Substitution at the 6-poisition of the quinoline with EDGs or EWGs (→ 44) was also tolerated, as was the introduction of halogens at the 4- or 7-positions. Excitingly, further deviations from the model system such as using 2-ethylquinoline with a dienal, or using isoquinolines or quinoxalines (→ 45), were also successful. Monoaddition to enals can be favoured by performing the reaction on a 4-alkylquinoline or a 2-alkyl-8-chloroquinoline (→ 46), or by control of stoichiometry and exclusion of the benzoic acid.
Rios developed a similar process the following year, in which 2-acyl azines were employed to generate their corresponding enolates as nucleophiles, also with excellent enantioselectivity and diastereocontrol (Scheme 13).71
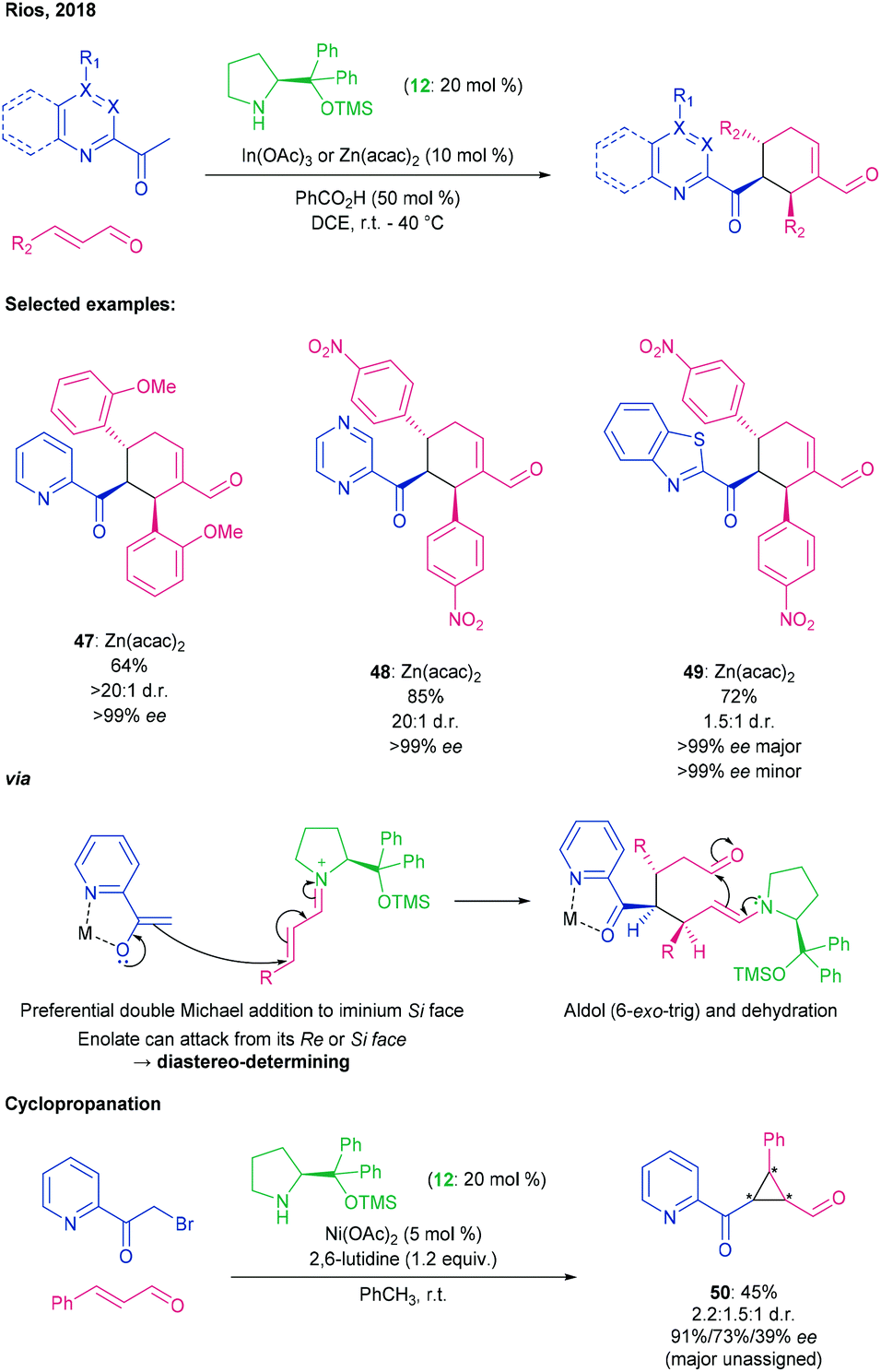 | ||
| Scheme 13 Double Michael addition and subsequent aldol dehydration between acyl azine enolates and cinnamaldehydes to give cyclohexenes using a prolinol-type aminocatalyst and either In(OAc)3 or Zn(acac)2, by Rios.71 | ||
Examination of a series of Lewis acidic metal reagents for 2-acyl azine enolate formation, revealed that oxophilic Lewis acids promoted 1,2-addition whereas Zn(acac)2 and In(OAc)3 promoted the desired reaction. In the proposed mechanism, the enal is activated by conversion to the iminium ion and the 2-acyl azine is activated towards enolate formation by chelation of the metal between the carbonyl oxygen and the azine nitrogen. Double Michael addition, aldol ring-closure, then dehydration gave rise to the cyclohexyl enal moiety as for the quinoline derivatives.
A series of cinnamaldehydes containing EWGs or EDGs were tested with 2-acetylpyridine (→ 47), and all of them gave good dr and ee. Addition of a methyl to the para-position of the pyridine was tolerated, and other azines such as pyrazines (→ 48) and pyrimidines were successful. Acetylbenzothiazole could even be used as an azine surrogate (→ 49), albeit with compromised diastereocontrol. Extending the acyl chain length to 2-propionylpyridine led to essentially racemic product from single Michael addition being isolated, which was expected due to increased steric hindrance. Performing the reaction on 2-(bromoacetyl)pyridine, this time with a Ni Lewis acid, allowed a cyclopropane to be formed after the first Michael addition (→ 50), but the necessity to include a base (2,6-lutidine) was detrimental to the stereochemical outcome. The importance of the bidentate chelation of the Lewis acid was exemplified in reactions with 3-acetyl and 4-acetyl pyridines, neither of which could form this chelate and both of which gave no reaction under the standard conditions.
Taking inspiration from Jørgensen's 2-alkylquinoline reactions, Rios later expanded on the concept by incorporating an aldehyde at the 3-position, priming the quinoline to react intermolecularly with a cinnamaldehyde in a Michael/aldol cyclisation cascade, which after dehydration gives dihydroacridines with high stereoselectivities (Scheme 14).72
 | ||
| Scheme 14 Condensation of 2-alkylquinoline aldehydes with cinnamaldehydes to give dihydroacridines using a prolinol-type aminocatalyst and Yb(OTf)3, by Rios.72 | ||
As before, coordination of the quinoline nitrogen to a Lewis acid enhances the α-methylene acidity, allowing formation of an enamine that undergoes Michael addition into the enal, which is activated as an iminium ion. The resulting enamine from this addition can then condense with the intramolecular quinoline aldehyde, which is rendered more electrophilic by protonation from the orthonitrobenzoic acid additive, to afford the dihydroacridine This reaction can produce up to 2 new stereocentres. The selective iminium ion condensation with the enal rather than the quinoline aldehyde is key to the stereochemical outcome. A broad scope of substituted cinnamaldehydes was tolerated, as were other (hetero)aryl enals (→ 51), and all gave enantioenriched samples of their respective products in good yields. Enantiodiscrimination when using a butyl enal was poor, however. Introduction of substituents at the 5-, 6-, 7- and 8-positions of the quinoline (→ 52) did not cause any change in stereoselectivity but generally gave lower yields. Extension of the alkyl chain at the 2-position gave rise to a second stereocentre (e.g., → 53). This stereocentre epimerised during column chromatography, leading to the thermodynamically preferable anti isomer, albeit with good ee. Various functional group interconversions could be performed on products with maintenance of enantiopurity.
DFT calculations were performed using an indium Lewis acid rather than Yb to avoid relativistic complications, and using pyrrolidine instead of the prolinol catalyst with the proviso that the iminium ion in the real system would be expected to be attacked from the Re face as it is sterically less hindered.54 The study suggested that the lowest energy transition state for the Michael addition involves the indium-coordinated quinoline enamine, associated with benzoic acid via the aldehyde, experiencing a stabilising London dispersion interaction between its aromatic rings and that of the iminium ion. The most favourable transition state for the subsequent cyclisation was calculated to arise from the enamine Si face approaching the aldehyde Re face, affording an intermediate with (S,S,R)-stereochemistry. However, since these two new stereocentres are lost upon dehydration, the only relevant factor in determining enantioselectivity is the steric influence of the organocatalyst in the Michael addition, and the predicted effect of this is reflected in the experimentally observed outcome.
3. Radical reactivity
3.1 Photoredox activated catalysis involving enamines
Electron rich enamines readily interact with radical reagents generated using photoredox chemistry. This was recognised in 2008 by MacMillan who introduced the concept of cooperative organocatalytic and photoredox cycles following this reaction manifold.73 As photoredox processes have proliferated in recent years, so has their combination with organocatalysts to allow asymmetric processes.74,75In 2017, Tung and co-workers were able to use a tricatalytic system to achieve the asymmetric cross-dehydrogenative coupling of tetrahydroisoquinolines to ketones (→ 54–56, Scheme 15).76
 | ||
| Scheme 15 Alkylation of ketones by reaction of in situ generated enamines with photocatalytically formed iminium ions using a chiral cyclohexane-1,2-diamine catalyst and Ru(bipy)3Cl2/Co(dmgH)2Cl2 as photocatalyst, by Tung et al.76 | ||
The combination of an achiral Ru(byp)3Cl2 photocatalyst, a Co co-catalyst, and 3-nitrobenzoic acid as proton source allowed for oxidation of the amine component to an iminium ion using blue LED light. This species was then trapped in an asymmetric manner using an enamine formed from a chiral cyclohexane-1,2-diamine catalyst. Enantioselectivity was believed to be derived from minimisation of steric and electrostatic interactions between the two cationic species via the transition state shown in Scheme 15. In contrast to previous reports of similar processes, the syn-diastereomers were formed selectively.77 A range of substituted cyclohexanones (54), pyranones (55), cyclopentanones (56) and N-aryl-tetrathydroisoquinolones were formed in high yield, dr and ee. Acyclic ketones were also tolerated, though with a reduction in ee.
In 2018, MacMillan et al. disclosed a method for the enantioselective α-benzylation of aldehydes using heterobenzylic alcohols as alkylating agents products in excellent yields and ees, using cooperative photoredox and organocatalysis (Scheme 16).78
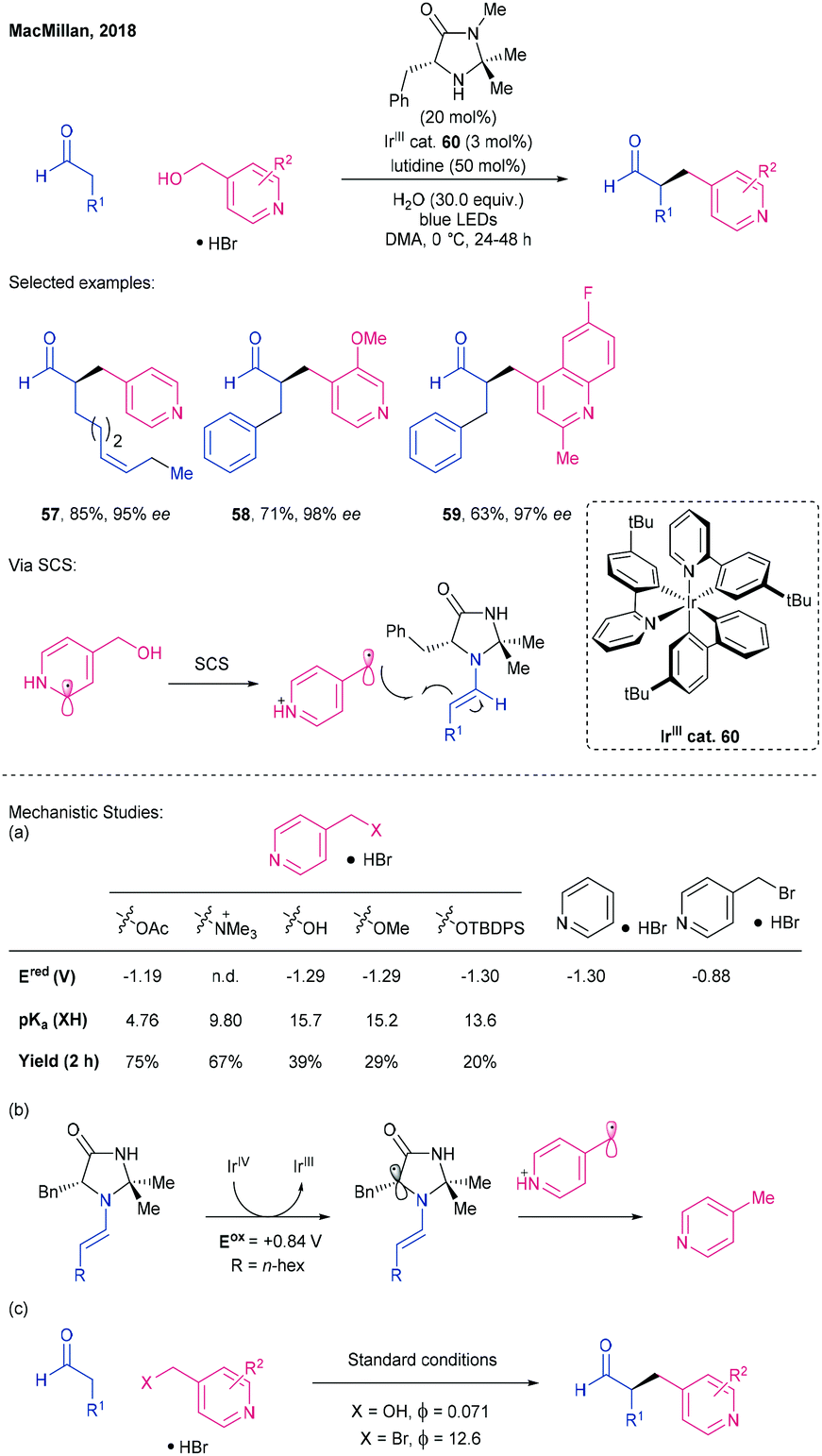 | ||
| Scheme 16 Alkylation of aldehydes with benzyl alcohols as radical precursors, by harnessing a spin-centre shift based mechanistic pathway, using a chiral imidazolidinone catalyst and Ir photocatalyst 60, by MacMillan.78 | ||
In these reactions, the achiral IrIII-based photocatalyst 60 in combination with blue LED light effects single electron reduction of the protonated heterobenzylic alcohol to give the corresponding benzyl radical. This radical couples with the chiral imidazolidinone-derived aldehyde enamine to generate a radical that undergoes SET oxidation by the IrIV species formed during prior alcohol reduction to give an iminium ion that is hydrolysed to the functionalised aldehyde product. MacMillan postulated that an ‘open shell’ radical was initially generated at the carbon α to the protonated heterocycle ring nitrogen which then undergoes a rapid ‘biomimetic’ spin-centre-shift (SCS) to the thermodynamically favoured benzylic position. Several diversely substituted 4-hydroxymethyl pyridine and quinoline alkylating agents were combined successfully with a range of substituted aldehydes (→ 57) and hydrocinnamaldehydes (→ 58, 59), including those containing alkene and alkyne substituents, with ees in excess of 96%. The SCS method was also amenable to the use of α-acetoxyketones as alkylating agents (in combination with octanal).
The authors performed some mechanistic studies to probe (a) the proposed SCS mechanism, (b) the origins of a 4-methylpyridine by-product observed during optimisation and (c) whether a radical chain mechanism was taking place. Evidence for a SCS was obtained by comparing the reduction potentials (Ered) of 4-hydroxymethyl pyridine and various O-derivatives (e.g., –OAc, –OH, –OMe, Scheme 16b) against pyridine and 4-(bromomethyl)pyridine. Similar Ered values for the alcohol, its derivatives and pyridine, and a large difference to 4-(bromomethyl)pyridine, suggested that activation of all the electrophiles proceeds via SET to the aromatic core. The methylpyridine by-product was hypothesised to form after direct oxidation of the enamine. This was supported by a study of photocatalyst structure–activity relationship. Photocatalysts with a higher IrIV/IIIEred1/2 (closer to that required to oxidise the enamine, Ered = +0.84 V) saw increased by-product formation and vice versa. Lastly, the low quantum yield value of ϕ = 0.071, consistent with each photon absorbed by the photocatalyst leading to a single product molecule, suggests radical chain propagation is unlikely.
While the aforementioned examples require the interaction of enamines with a metal photosensitiser, the recent work of Davis and Bergmann suggests that modification to the organocatalysts could eliminate the requirement for the separate photocatalysts.79
3.2 Photoredox activated catalysis involving iminium ions
Seminal work by Melchiorre et al. in 2016 showed that iminium ions formed from enones could trap nucleophilic radicals generated using a white LED promoted photoredox processes.80 The α-iminyl radical cations formed after 1,4-addition are highly unstable however, undergoing rapid β-scission to regenerate the starting materials. To overcome this and achieve radical Michael addition for the formation of asymmetric quaternary centres, Melchiorre designed a chiral primary amine organocatalyst (65), with a tethered carbazole which rapidly intercepted this transient radical. The process was exemplified by the β-alkylation of β-substituted cyclic enones by acetals and amines (Scheme 17). | ||
| Scheme 17 β-Alkylation of enones using a chiral cyclohexane-1,2-diamine catalyst (65) and Ir photocatalyst 66, by Melchiorre.80 | ||
In the prototypical case of β-methyl cyclohexanone, iminium ion formation situates the carbazole moiety close to the electron deficient alkene, directing the asymmetric addition. After the conjugate addition of a nucleophilic radical, the α-iminyl radical cation is quenched by rapid SET transfer with the carbazole. The long lived, oxidised carbazole ‘e− hole’ is subsequently quenched by SET with the reduced photocatalyst, completing the synergistic catalytic cycles after product iminium ion hydrolysis.
A variety of benzodioxoles (→ 61, 62, using TBADT as photocatalyst) and α-amino radicals (→ 63, 64, using IrIII photocatalyst 66) were shown to add to β-substituted cyclic enones. The cooperative catalytic process allowed asymmetric access to quaternary all-carbon stereocentres, in high yields and ee using mild conditions. During optimisation of the organocatalyst, the authors noticed that modification of the carbazole moiety with –CF3 substituents unexpectedly led to a more active catalyst, in comparison to electron-donating groups. This is contrary to what would have been expected, given the moiety's purported role as a SET reductant and prompted a mechanistic investigation (Scheme 18).81
 | ||
| Scheme 18 Mechanistic studies on the proposed electron-relay during photoredox promoted radical alkylation of iminium ions. (a) electronic effects on the rate of SET, and (b) the suggested 2-step SET process in which quenching of the carbazoliumyl radical cation is rate-limiting (i.e., SET 2), by Melchiorre.81 | ||
Melchiorre et al. synthesised a series of sterically similar yet electronically varied carbazoles (Scheme 18a) allowing Hammett analysis of the effect on reaction rate. Indeed, as was observed during optimisation, electron-withdrawing substituents gave carbazoles with greater reduction potentials and led to increased initial rates of reaction. The authors proposed that the organocatalyst was in fact involved in two SET events in the catalytic cycle (Scheme 18b), one involving oxidation (i.e., quenching the α-iminyl radical cation, SET1) and one involving reduction to regenerate the TBADT photocatalyst (i.e., SET2). The results indicated that the second SET event, between the TBADT-H photocatalyst and the carbazoliumyl radical cation, was the rate limiting step. This was corroborated by spectroscopic studies which detected the long-lived carbazoliumyl species in the reaction mixture. Consequently, the inclusion of electron-withdrawing CF3 substituents favoured the rate-determining TBADT-H reduction, leading to the observed increase in catalytic activity. The authors proposed that the mechanistic insights gained in this study might help drive the development of a next generation of electron relay catalysts.
Yu et al. were able to achieve the asymmetric β-hydroacylation of enals using acyl radicals generated from α-ketoacids using white LED light, an achiral Ru photocatalyst 70, and a prolinol derived organocatalyst (Scheme 19).82
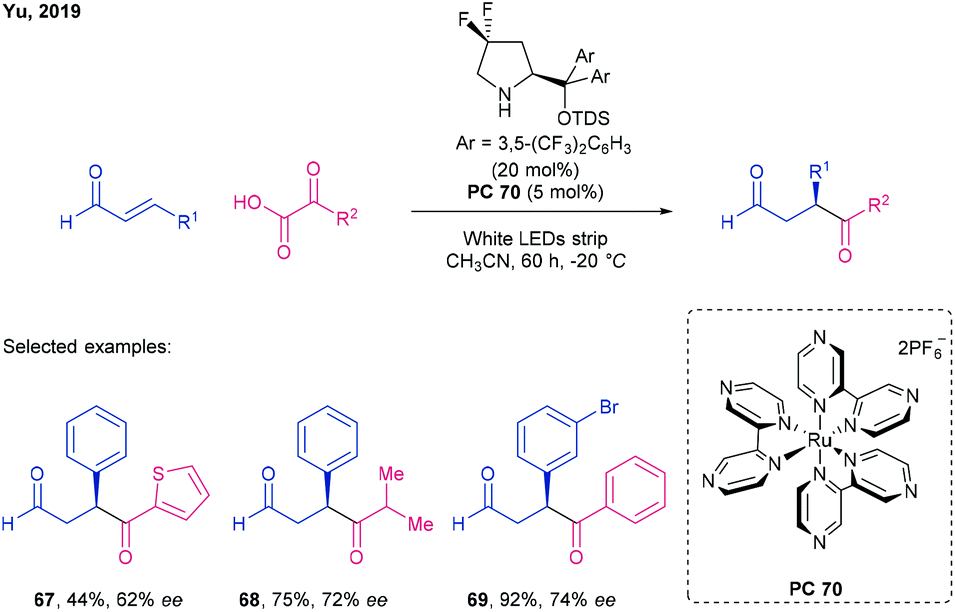 | ||
| Scheme 19 Hydroacylation of enals by α-keto acids using a chiral gem-difluoro-diaryl prolinol organocatalyst and a Ru photocatalyst (66), by Yu.82 | ||
Decarboxylation of the α-ketoacid, catalysed by SET from the Ru photocatalyst, generates an acyl radical species which can subsequently undergo 1,4-addition to an in situ generated iminium ion of the enal. Reduction and hydrolysis of the resulting iminyl radical completes the catalytic cycle. Tuning of the iminium ion electrophilicity by use of a difluorinated Jørgensen catalyst was key to achieving high yields and ee. The reaction scope included (hetero)aromatic (→ 67) and alkyl substituted (→ 68) α-ketoacids, in conjunction with a range of aryl substituted cinnamaldehydes (→ 69). The method is of particular note insofar as it gives access to 1,4 dicarbonyl containing products which require an umpolung retrosynthetic disconnection using classical ionic chemistry (Scheme 20).
 | ||
| Scheme 20 A comparison of polar and radical strategies for accessing 1,4-dicarbonyls and radical precursors used by Yu,82 and other, non-synergistic, methods.83,84 | ||
4. Conclusions
In this article we have briefly surveyed selected synthetic methods which highlight the power of pairing a chiral, typically amino organocatalyst with an achiral metal complex to effect synergistic catalysis. We have focussed on recent developments (since 2016) and examined how under appropriate conditions the organocatalyst can activate a substrate by modulating the energy of either its HOMO or LUMO whilst a metal complex simultaneously and independently performs complementary activation of the reaction partner. These reaction partners can be closed shell ionic species or open shell radicals. So far, successful examples of these types of reaction are relatively rare. This undoubtedly reflects the considerable challenge of engineering conditions under which the respective catalytic cycles can be ‘compartmentalised’ to avoid unwanted cross-reactivity and tuned to operate at compatible rates. It is clear that certain organocatalysts (e.g., Jørgensen–Hayashi-type prolinols), metal complexes (e.g., simple Pd and Cu complexes) and substrates (e.g., enals) can currently be regarded as ‘privileged’ in respect to their successful deployment in the methods reported thus far. We hope that our compilation and analysis will encourage the expansion of this repertoire and lead to the application of synergistic catalysis to new areas of chemical space.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the BBSRC and Syngenta for an iCASE award to JL and Prof. T. N. Snaddon for insightful discussions that helped to refine this manuscript.References
- A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC.
- R. Peters, Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, 2015 Search PubMed.
- Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC.
- Y. Deng, S. Kumar and H. Wang, Chem. Commun., 2014, 50, 4272–4284 RSC.
- S. M. Inamdar, V. S. Shinde and N. T. Patil, Org. Biomol. Chem., 2015, 13, 8116–8162 RSC.
- N. Butt, G. Yang and W. Zhang, Chem. Rec., 2016, 16, 2683–2692 CrossRef CAS PubMed.
- J. Fu, X. Huo, B. Li and W. Zhang, Org. Biomol. Chem., 2017, 15, 9747–9759 RSC.
- U. B. Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S.-g. Lee, Chem. Rev., 2020, 120, 13382–13433 CrossRef CAS PubMed.
- S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef CAS PubMed.
- A. Sinibaldi, V. Nori, A. Baschieri, F. Fini, A. Arcadi and A. Carlone, Catalysts, 2019, 9, 928 CrossRef CAS.
- S. Afewerki and A. Córdova, Chem. Rev., 2016, 116, 13512–13570 CrossRef CAS PubMed.
- S. Afewerki and A. Córdova, Top. Curr. Chem., 2019, 377, 1–27 CrossRef CAS PubMed.
- S. Afewerki and A. Córdova, in Chiral Lewis Acids in Organic Synthesis, ed. J. Mlynarski, Wiley-VCH Verlag, Weinheim, 2017, pp. 345–374 Search PubMed.
- G. J. Knox, L. S. Hutchings-Goetz, C. M. Pearson and T. N. Snaddon, Tertiary Amine Lewis Base Catalysis in Combination with Transition Metal Catalysis, Springer International Publishing, 2020, vol. 378 Search PubMed.
- F. Romiti, J. Del Pozo, P. H. S. Paioti, S. A. Gonsales, X. Li, F. W. W. Hartrampf and A. H. Hoveyda, J. Am. Chem. Soc., 2019, 141, 17952–17961 CrossRef CAS PubMed.
- D. E. A. Raup, B. Cardinal-David, D. Holte and K. A. Scheidt, Nat. Chem., 2010, 2, 766–771 CrossRef CAS PubMed.
- N. T. Patil, Angew. Chem., Int. Ed., 2011, 50, 1759–1761 CrossRef CAS PubMed.
- I. Ibrahem and A. Córdova, Angew. Chem., Int. Ed., 2006, 45, 1952–1956 CrossRef CAS PubMed.
- C. Guo, M. Fleige, D. Janssen-Müller, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2016, 138, 7840–7843 CrossRef CAS PubMed.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC.
- C. Guo, D. Janssen-Müller, M. Fleige, A. Lerchen, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2017, 139, 4443–4451 CrossRef CAS PubMed.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- S. Singha, T. Patra, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2018, 140, 3551–3554 CrossRef CAS PubMed.
- K. J. Schwarz, J. L. Amos, J. C. Klein, D. T. Do and T. N. Snaddon, J. Am. Chem. Soc., 2016, 138, 5214–5217 CrossRef CAS PubMed.
- V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS PubMed.
- P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 CrossRef CAS PubMed.
- M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492–4495 CrossRef CAS PubMed.
- E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H. Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919–6927 RSC.
- K. J. Schwarz, C. M. Pearson, G. A. Cintron-Rosado, P. Liu and T. N. Snaddon, Angew. Chem., Int. Ed., 2018, 57, 7800–7803 CrossRef CAS PubMed.
- L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC.
- W. C. Hartley, T. J. C. O'Riordan and A. D. Smith, Synthesis, 2017, 49, 3303–3310 CrossRef CAS.
- J. W. B. Fyfe, O. M. Kabia, C. M. Pearson and T. N. Snaddon, Tetrahedron, 2018, 74, 5383–5391 CrossRef CAS PubMed.
- W. R. Scaggs and T. N. Snaddon, Chem. – Eur. J., 2018, 24, 14378–14381 CrossRef CAS PubMed.
- K. J. Schwarz, C. Yang, J. W. B. Fyfe and T. N. Snaddon, Angew. Chem., Int. Ed., 2018, 57, 12102–12105 CrossRef CAS PubMed.
- W. R. Scaggs, T. D. Scaggs and T. N. Snaddon, Org. Biomol. Chem., 2019, 17, 1787–1790 RSC.
- B. M. Trost, Tetrahedron, 2015, 71, 5708–5733 CrossRef CAS PubMed.
- J. Tsuji, Tetrahedron, 2015, 71, 6330–6348 CrossRef CAS.
- Q. Cheng, H. F. Tu, C. Zheng, J. P. Qu, G. Helmchen and S. L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed.
- B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed.
- L. Hutchings-Goetz, C. Yang and T. N. Snaddon, ACS Catal., 2018, 8, 10537–10544 CrossRef CAS PubMed.
- P. R. Auburn, P. B. Mackenzie and B. Bosnich, J. Am. Chem. Soc., 1985, 107, 2033–2046 CrossRef CAS.
- P. B. Mackenzie, J. Whelan and B. Bosnich, J. Am. Chem. Soc., 1985, 107, 2046–2054 CrossRef CAS.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- C. M. Pearson, J. W. B. Fyfe and T. N. Snaddon, Angew. Chem., Int. Ed., 2019, 58, 10521–10527 CrossRef CAS PubMed.
- C. Pearson, J. Fyfe and T. N. Snaddon, Angew. Chem., Int. Ed., 2019, 58, 10521–10527 CrossRef CAS PubMed.
- X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87–90 CrossRef CAS PubMed.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed.
- L. S. Hutchings-Goetz, C. Yang, J. W. B. Fyfe and T. N. Snaddon, Angew. Chem., 2020, 132, 17709–17717 CrossRef.
- B. Zhao, H. Du and Y. Shi, J. Am. Chem. Soc., 2008, 130, 7220–7221 CrossRef CAS PubMed.
- J. Song, Z. J. Zhang, S. Sen Chen, T. Fan and L. Z. Gong, J. Am. Chem. Soc., 2018, 140, 3177–3180 CrossRef CAS PubMed.
- Z.-J. Zhang, L. Zhang, R.-L. Geng, J. Song, X.-H. Chen and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 12190–12194 CrossRef CAS PubMed.
- B. S. Donslund, T. K. Johansen, P. H. Poulsen, K. S. Halskov and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 13860–13874 CrossRef CAS PubMed.
- I. Ibrahem, S. Santoro, F. Himo and A. Córdova, Adv. Synth. Catal., 2011, 353, 245–252 CrossRef CAS.
- T. Yang, A. Ferrali, L. Campbell and D. J. Dixon, Chem. Commun., 2008, 2923–2925 RSC.
- M. Meazza, V. Ceban, M. B. Pitak, S. J. Coles and R. Rios, Chem. – Eur. J., 2014, 20, 16853–16857 CrossRef CAS PubMed.
- M. Meazza, M. E. Light, A. Mazzanti and R. Rios, Chem. Sci., 2016, 7, 984–988 RSC.
- R. K. Kunz and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3240–3241 CrossRef CAS PubMed.
- M. Meazza, V. Polo, P. Merino and R. Rios, Org. Chem. Front., 2018, 5, 806–812 RSC.
- M. Laugeois, S. Ponra, V. Ratovelomanana-Vidal, V. Michelet and M. R. Vitale, Chem. Commun., 2016, 52, 5332–5335 RSC.
- M. Meazza and R. Rios, Chem. – Eur. J., 2016, 22, 9923–9928 CrossRef CAS PubMed.
- K. Zhang, M. Meazza, A. Izaga, C. Contamine, M. C. Gimeno, R. P. Herrera and R. Rios, Synthesis, 2017, 49, 167–174 CrossRef CAS.
- K. S. Halskov, L. Næsborg, F. Tur and K. A. Jørgensen, Org. Lett., 2016, 18, 2220–2223 CrossRef CAS PubMed.
- M. Kamlar, M. Franc, I. Císařová, R. Gyepes and J. Veselý, Chem. Commun., 2019, 55, 3829–3832 RSC.
- M. Meazza, M. Kamlar, L. Jašíková, B. Formánek, A. Mazzanti, J. Roithová, J. Veselý and R. Rios, Chem. Sci., 2018, 9, 6368–6373 RSC.
- M. Franc, I. Císařová and J. Veselý, Adv. Synth. Catal., 2021, 363, 4349–4353 CrossRef CAS.
- S. Putatunda, J. V. Alegre-Requena, M. Meazza, M. Franc, D. Rohal'ová, P. Vemuri, I. Císařová, R. P. Herrera, R. Rios and J. Veselý, Chem. Sci., 2019, 10, 4107–4115 RSC.
- S. Santoro, L. Deiana, G. L. Zhao, S. Lin, F. Himo and A. Córdova, ACS Catal., 2014, 4, 4474–4484 CrossRef CAS.
- M. Meazza, F. Tur, N. Hammer and K. A. Jørgensen, Angew. Chem., Int. Ed., 2017, 56, 1634–1638 CrossRef CAS PubMed.
- M. Meazza, G. Sitinova, C. Poderi, M. Mancinelli, K. Zhang, A. Mazzanti and R. R. Torres, Chem. – Eur. J., 2018, 24, 13306–13310 CrossRef CAS PubMed.
- S. Meninno, M. Meazza, J. W. Yang, T. Tejero, P. Merino-Gomez and R. Rios, Chem. – Eur. J., 2019, 25, 7623–7627 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- M. Silvi and P. Melchiorre, Nature, 2018, 554, 41–49 CrossRef CAS PubMed.
- Y.-Q. Zou, F. M. Hörmann and T. Bach, Chem. Soc. Rev., 2018, 47, 278–290 RSC.
- Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694–3698 CrossRef CAS PubMed.
- G. Zhang, Y. Ma, S. Wang, W. Kong and R. Wang, Chem. Sci., 2013, 4, 2645–2651 RSC.
- E. D. Nacsa and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 3322–3330 CrossRef CAS PubMed.
- K. Bergmann and R. L. Davis, Org. Lett., 2021, 23, 7033–7037 CrossRef CAS PubMed.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed.
- A. Bahamonde, J. J. Murphy, M. Savarese, É. Brémond, A. Cavalli and P. Melchiorre, J. Am. Chem. Soc., 2017, 139, 4559–4567 CrossRef CAS PubMed.
- J. J. Zhao, H. H. Zhang, X. Shen and S. Yu, Org. Lett., 2019, 21, 913–916 CrossRef CAS PubMed.
- T. Morack, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 1208–1212 CrossRef CAS PubMed.
- G. Goti, B. Bieszczad, A. Vega-Peñaloza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 1213–1217 CrossRef CAS PubMed.
Footnotes |
| † Current address: RWTH Aachen University, Department of Chemistry, Landoltweg 1a, 52074, Aachen, Germany. |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2022 |