
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of phenoxathiins using an iron-catalysed C–H thioarylation†
Amy C.
Dodds
and
Andrew
Sutherland
 *
*
School of Chemistry, The Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Andrew.Sutherland@glasgow.ac.uk
First published on 8th February 2022
Abstract
Phenoxathiins are an important class of sulfur-containing heterocycle, found as the core component in numerous pharmaceutically active agents and materials. Despite this importance, there are relatively few methods for the synthesis of these heterocycles that avoid complex starting materials, harsh conditions or precious transition metals. We report a two-step synthesis of phenoxathiins from phenols using iron and copper-mediated reactions. The first step involves the accelerated ortho-thioarylation of phenols using N-(2-bromophenylthio)succinimide, catalysed by the Lewis acid, iron(III) triflimide and the Lewis base, bis(4-methoxyphenyl)sulfane. In the second step, the thioarylated products were converted to a series of phenoxathiins using a copper-mediated, Ullmann-type, C–O bond forming cyclisation reaction. The synthetic utility of this two-step approach for the preparation of biologically relevant phenoxathiins was demonstrated using natural product-based phenols.
Introduction
Sulfur-containing heterocycles are an important compound class,1 found as key structural components in a wide variety of pharmaceuticals and materials.2 In particular, tricyclic systems such as phenoxathiins have attracted attention due to possessing optical properties and biological activities (Scheme 1a). For example, boronic ester analogues have been developed as room-temperature phosphorescent materials,3,4 while 2-substituted phenoxathiins have been shown to act as inhibitors of thrombin5 and monoamine oxidase A,6 and as antifungal agents.7 The S-oxides of phenoxathiins have also been used for the C–H functionalisation of arenes.8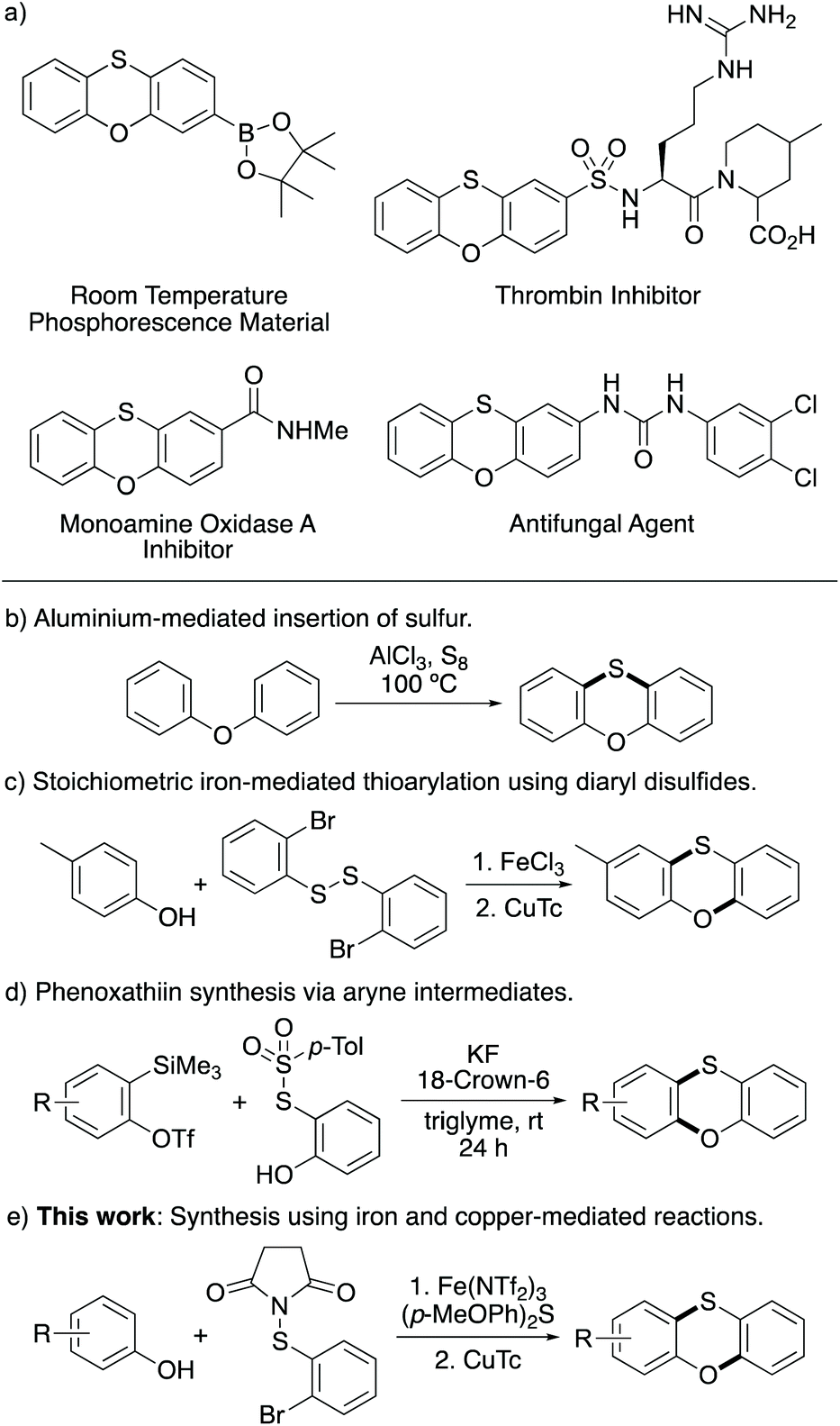 | ||
| Scheme 1 a) Structures of phosphorescent and biologically active phenoxathiins. (b–e) Methods for the synthesis of phenoxathiins. | ||
Despite the interest in phenoxathiin applications, there are relatively few methods for the synthesis of this heterocycle. Traditional methods utilised an aluminium-mediated sulfur insertion reaction of diaryl ethers under harsh conditions (Scheme 1b).9 More recently, several groups reported a two-step approach involving transition metal-mediated thioarylation reactions of phenols as the key step.10 Takaki and co-workers used stoichiometric amounts of iron trichloride and diaryl disulfides as the thioarylating agent (Scheme 1c).10a Palladium catalysed thioarylation using an N-(arylthio)succinimide and trifluoroacetic acid as the solvent has also been utilised.10b Other strategies to access phenoxathiins include a base-mediated coupling and cyclisation of 2-sulfanylphenol with 1,2-dihaloarenes,11 while an approach using N-benzyl dithiocarbamate salts as a sulfur source for copper-catalysed reaction with cyclic diaryliodoniums has also been reported.12 Other single-step processes include the reaction of aryne intermediates with 2-hydroxyaryl 4-toluenethiosulfonates, which allowed the preparation of a wide range of phenoxathiins under mild conditions (Scheme 1d).13
Some of these methods allow the general synthesis of phenoxathiins bearing various substituents and functional groups. However, these methods can require harsh conditions,9,10b highly functionalised starting materials,11–13 stoichiometric amounts of metal reagent10a or the use of precious transition metal catalysts.10b,c In recent years, we have reported the synthesis of various benzannulated heterocycles using earth-abundant, non-precious transition metal catalysts. This approach utilised the super Lewis acid, iron(III) triflimide for the activation of N-halosuccinimides and the regioselective halogenation of arenes, followed by copper-catalysed intramolecular cyclisation to form the heterocycles.14 More recently, we showed that iron(III) triflimide could also be used to activate N-(arylthio)succinimides for the regioselective thioarylation of arenes.15 Based on this work, we proposed that iron(III)-catalysed ortho-thioarylation of para-substituted phenols with N-(2-bromophenylthio)succinimide would generate an intermediate, that following a copper-mediated Ullmann-type cyclisation would allow the rapid synthesis of phenoxathiins. Herein, we now report this two-stage approach for the synthesis of phenoxathiins from phenols. As well as demonstrating the acceleration of the iron-catalysed thioarylation step using the Lewis base catalyst, bis(4-methoxyphenyl)sulfane (Scheme 1e), we also describe the straightforward application of this two-stage approach for the incorporation of the phenoxathiin motif into biologically active compounds.
Results and discussion
Initial development of an iron(III) triflimide-catalysed thioarylation with N-(2-bromophenylthio)succinimide (2) focused on the use of p-cresol (1a) as a suitable starting material (Table 1).16 A reaction was attempted using 10 mol% of iron(III) triflimide, which was generated in situ from iron(III) chloride and the readily available ionic liquid, [BMIM]NTf2 (entry 1). The reaction showed no further conversion after 24 h and gave thioarylated product 4a, in 38% yield. This result demonstrated that further acceleration of this reaction was required to overcome the steric constraints of ortho-thioarylation with N-(2-bromophenylthio)succinimide (2). During the development of our previous iron(III)-catalysed thioarylation reaction,15 kinetic studies revealed that the transformation was autocatalytic, in which electron-rich biaryl sulfane products acted as Lewis base catalysts, resulting in accelerated reactions. Using this information, the iron(III) triflimide-catalysed thioarylation of p-cresol (1a) with N-(2-bromophenylthio)succinimide (2) was repeated using commercially available bis(4-methoxyphenyl)sulfane (3) as a Lewis base catalyst (10 mol%).17 After 2 h, the reaction was found to be complete and gave 4a in 84% yield (entry 2). To verify that the accelerated reaction was due to the combination of both iron(III) triflimide and biaryl sulfane 3, and not just 3, the reaction was repeated using only the Lewis base (entry 3). After 24 h, no reaction was observed, thereby confirming the dual role of both the Lewis acid and Lewis base catalysts in accelerating the reaction. Further optimisation studies investigated the actual reaction end point. Inspection of the reaction mixture using 1H NMR spectroscopy at various time points, revealed that the reaction was complete after only 0.5 h, resulting in the isolation of 4a in 81% yield.
The scope of the combined Lewis acid and Lewis base-catalysed ortho-thioarylation with a range of phenols was then explored (Scheme 2). For most substrates, the reactions were found to be fast and efficient, and gave biaryl sulfanes in good to high yields. As expected, highly activated compounds (1b and 1d) were thioarylated at lower temperatures, while higher temperatures and longer reaction times (1g) were required for phenols bearing electron-withdrawing substituents. The use of this transformation for bi-directional thioarylation was investigated using hydroquinone (1j) as a starting material. This gave bis-thioarylated product 4j in 79% yield, after a 1 h reaction time. A larger scale reaction was also investigated using 2-naphthol (1f). On a 5 mmol scale, the reaction proceeded as normal, providing gram quantities (1.6 g) of 4f in similar yields to the small-scale reaction (95%).
 | ||
Scheme 2 Reaction scope of phenols 1. Isolated yields. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction was done at 40 °C. bReaction was done at rt. cReaction was done at 85 °C. d2.2 equivalents of 2 were used. Reaction was done at 40 °C. bReaction was done at rt. cReaction was done at 85 °C. d2.2 equivalents of 2 were used. | ||
Following the effective synthesis of a range of 2-bromobenzene phenol sulfanes 4, these were converted to the corresponding phenoxathiins 5 using a stoichiometric amount of copper(I) thiophene-2-carboxylate under standard conditions for Ullmann-type cyclisation (Scheme 3).10a,b Irrespective of the electronics or steric bulk of the substituents, all cyclisations proceeded efficiently, allowing the synthesis of various phenoxathiins 5 in high yields.18 This reaction also allowed the clean, double-cyclisation of 4j and access to 5,12-dioxa-7,14-dithiapentacene (5j) in 82% yield. A one-pot process that combined the thioarylation and cyclisation steps was also investigated. However, despite testing a range of solvents, the two processes were found to be incompatible due to solubility issues of the starting materials and reagents.
 | ||
| Scheme 3 Synthesis of phenoxathiins 5. Isolated yields. a2 equivalents of CuTc were used. | ||
Having developed an efficient two-step synthesis of phenoxathiins using a commercially available Lewis base and earth abundant transition metals, the synthetic utility of this approach for incorporating this heterocyclic motif within biologically active compounds was investigated. Initially, this strategy was used for the synthesis of novel, phenoxathiin-derived α-amino acids (Scheme 4). Application of the Lewis acid and Lewis base catalysed thioarylation of commercially available tyrosine derivative 6 gave biaryl sulfane 7 in 80% yield. It should be noted that an attempt of this transformation using only iron(III) triflimide showed no reaction after 24 h. Cyclisation using copper(I) thiophene-2-carboxylate gave phenoxathiin 8 in 93% yield. Ester hydrolysis, followed by acid-mediated removal of the Cbz-protecting group gave parent α-amino acid 9 in 58% overall yield from tyrosine 6. Diversification of the heterocyclic motif was achieved by oxidation to sulfone 10. Treatment of 8 with hydrogen peroxide in the presence of glacial acetic acid gave sulfone 10 in 95% yield. A similar deprotection strategy gave phenoxathiin-dioxide 11 in 55% yield over the five steps.
 | ||
| Scheme 4 Synthesis of phenoxathiin-derived amino acids. | ||
A further demonstration of the two-step synthesis of phenoxathiins was applied to the estrogen steroid hormone, estradiol (Scheme 5). While all previous thioarylation reactions gave a single product, application of the optimised method with 17-β-estradiol-17-acetate (12)19 gave two products via reaction at the 2- or 4-ortho positions. Analysis of the crude reaction mixture by 1H NMR spectroscopy showed a 6:1 ratio of compounds. The major product, 2-regioisomer 13 was readily separated by flash column chromatography and isolated in 73% yield. Copper(I)-mediated Ullmann-type cyclisation, followed by removal of the acetate protecting group under basic conditions gave steroid-containing phenoxathiin 15 in 52% yield over the three steps.
 | ||
| Scheme 5 Synthesis of phenoxathiin-derived steroid 15. | ||
Based on the results from this study and the control experiments presented in Table 1, a mechanism involving both catalysts has been proposed (Scheme 6). Following formation of iron(III) triflimide from iron(III) chloride and [BMIM]NTf2, the strongly Lewis acidic iron(III) cation activates N-(2-bromophenylthio)succinimide (2).20,21 While this intermediate can undergo slow thioarylation with the phenol, reaction instead with bis(4-methoxyphenyl)sulfane (3) forms a cationic disulfide intermediate. This charged species reacts significantly more quickly with the phenols, forming the ortho-thioarylated products and regenerating the Lewis base catalyst. Evidence of the cationic disuflide or a similarly reactive intermediate was observed from kinetic studies during our previous work of iron(III)-catalysed thioarylation reactions.15 These studies showed that in the absence of a Lewis base, the transformations were autocatalytic with an induction period, requiring the formation of a more reactive intermediate before rate acceleration. Addition of a Lewis base biaryl sulfide showed no induction period, confirming the role of an activated sulfide intermediate in the rate acceleration. Gustafson and co-workers have also proposed cationic disulfide intermediates during their Lewis base and Brønsted acid catalysed thioarylation processes.17
 | ||
| Scheme 6 Proposed catalytic pathway for Lewis acid and Lewis base catalysed thioarylation. | ||
Conclusions
In summary, a fast and efficient method for the ortho-thioarylation of phenols with N-(2-bromophenylthio)succinimide (2) involving the combined catalysis of the Lewis acid, iron(III) triflimide and the Lewis base, bis(4-methoxyphenyl)sulfane (3) has been developed. The transformation was found to be compatible with a wide-range of substituents and in combination with a copper(I)-mediated Ullmann-type intramolecular cyclisation allowed access to a variety of phenoxathiins. The application of this two-step approach was extended for the synthesis of more complex phenoxathiins using natural product-based phenols. The use of both tyrosine and estradiol derivatives allowed efficient access to the corresponding phenoxathiins. Overall, this work provides a new approach for the synthesis of important heterocycles using a commercially available Lewis base and earth abundant transition metals. Work is currently underway to find new applications of this general strategy.Experimental
All reagents and starting materials, including N-(benzyloxycarbonyl)-L-tyrosine methyl ester (6) and bis(4-methoxyphenyl)sulfane (3) were obtained from commercial sources and used as received. N-(2-Bromophenylthio)succinimide (2)22 and 17-β-estradiol-17-acetate (12)19 were prepared as previously described in the literature. Reactions were performed open to air unless otherwise mentioned. Brine refers to a saturated aqueous solution of sodium chloride. Flash column chromatography was performed using silica gel 60 (35–70 μm). Aluminium-backed plates pre-coated with silica gel 60F254 were used for thin layer chromatography and were visualised with a UV lamp or by staining with potassium permanganate. 1H NMR spectra were recorded on an NMR spectrometer at either 400 or 500 MHz and data are reported as follows: chemical shift in ppm relative to the undeuterated solvent as internal standard (CHCl3, δ 7.26 ppm; CH3OH, δ 3.31 ppm; DMSO, δ 2.50), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or overlap of non-equivalent resonances, integration). 13C NMR spectra were recorded on an NMR spectrometer at either 101 or 126 MHz and data are reported as follows: chemical shift in ppm relative to tetramethylsilane or the solvent as internal standard (CDCl3, δ 77.16 ppm; CD3OD, δ 49.00 ppm; DMSO-d6, δ 39.52), multiplicity with respect to hydrogen (deduced from DEPT experiments, C, CH, CH2 or CH3). Infrared spectra were recorded on a FTIR spectrometer; wavenumbers are indicated in cm−1. Mass spectra were recorded using electron impact or electrospray techniques. HRMS spectra were recorded using dual-focusing magnetic analyser or quadrupole time of flight (Q-TOF) mass spectrometers. Melting points are uncorrected.General procedure A: preparation of sulfenylated products 4
Iron(III) trichloride (10 mol%) was dissolved in [BMIM]NTf2 (30 mol%) and left to stir for 0.5 h at room temperature before being added to a solution of N-(2-bromophenylthio)succinimide (2) (1.2 equiv.) in chloroform (0.6 M in arene), within a sealed vial. The arene (1.0 equiv.) and bis(4-methoxyphenyl)sulfane (3) (10 mol%) were then added and the reaction mixture was left to stir at the required temperature for 1–48 h. The reaction mixture was concentrated in vacuo and purified using flash column chromatography.(2-Hydroxy-5-methylphenyl)(2′-bromophenyl)sulfane (4a)10a
The reaction was performed as described in general procedure A using p-cresol (1a) (31 mg, 0.29 mmol). The reaction mixture was stirred at 75 °C for 0.5 h. Purification by flash column chromatography (hexane/dichloromethane, 4:1) gave (2-hydroxy-5-methylphenyl)(2′-bromophenyl)sulfane (4a) (69 mg, 81%) as a white solid. Mp 67–69 °C; spectroscopic data was consistent with the literature.10aδH (400 MHz, CDCl3) 2.31 (3H, s, CH3), 6.21 (1H, s, OH), 6.60 (1H, dd, J 8.0, 1.5 Hz, 6′-H), 6.96–7.04 (2H, m, 3-H and 4′-H), 7.09–7.15 (1H, m, 5′-H), 7.23 (1H, dd, J 8.3, 2.0 Hz, 4-H), 7.33 (1H, d, J 2.0 Hz, 6-H), 7.53 (1H, dd, J 7.9, 1.3 Hz, 3′-H); δC (101 MHz, CDCl3) 20.5 (CH3), 114.8 (C), 115.7 (CH), 121.1 (C), 126.8 (CH), 127.0 (CH), 128.2 (CH), 131.2 (C), 133.1 (CH), 133.8 (CH), 137.3 (CH), 137.6 (C), 155.5 (C); m/z (ESI) 319 (MNa+. 100%).
(2-Hydroxy-4,5-dimethylphenyl)(2′-bromophenyl)sulfane (4b)
The reaction was performed as described in general procedure A using 3,4-dimethylphenol (1b) (36 mg, 0.29 mmol). The reaction mixture was stirred at 40 °C for 4 h. Purification by flash column chromatography (hexane/dichloromethane, 17:3) gave (2-hydroxy-4,5-dimethylphenyl)(2′-bromophenyl)sulfane (4b) (72 mg, 80%) as a white solid. Mp 86–88 °C; νmax/cm−1 (neat) 3446 (OH), 3012 (CH), 1479 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1446, 1311, 1204, 1019, 747; δH (400 MHz, CDCl3) 2.21 (3H, s, CH3), 2.29 (3H, s, CH3), 6.13 (1H, s, OH), 6.58 (1H, dd, J 8.0, 1.6 Hz, 6′-H), 6.91 (1H, s, 3-H), 6.96–7.01 (1H, m, 4′-H), 7.07–7.13 (1H, m, 5′-H), 7.26 (1H, s, 6-H), 7.52 (1H, dd, J 7.9, 1.3 Hz, 3′-H); δC (101 MHz, CDCl3) 18.9 (CH3), 20.2 (CH3), 111.4 (C), 116.9 (CH), 120.8 (C), 126.6 (CH), 126.8 (CH), 128.1 (CH), 130.1 (C), 133.0 (CH), 137.5 (CH), 137.9 (C), 142.4 (C), 155.6 (C); m/z (ESI) 330.9763 (MNa+. C14H1379BrNaOS requires 330.9763).
C), 1446, 1311, 1204, 1019, 747; δH (400 MHz, CDCl3) 2.21 (3H, s, CH3), 2.29 (3H, s, CH3), 6.13 (1H, s, OH), 6.58 (1H, dd, J 8.0, 1.6 Hz, 6′-H), 6.91 (1H, s, 3-H), 6.96–7.01 (1H, m, 4′-H), 7.07–7.13 (1H, m, 5′-H), 7.26 (1H, s, 6-H), 7.52 (1H, dd, J 7.9, 1.3 Hz, 3′-H); δC (101 MHz, CDCl3) 18.9 (CH3), 20.2 (CH3), 111.4 (C), 116.9 (CH), 120.8 (C), 126.6 (CH), 126.8 (CH), 128.1 (CH), 130.1 (C), 133.0 (CH), 137.5 (CH), 137.9 (C), 142.4 (C), 155.6 (C); m/z (ESI) 330.9763 (MNa+. C14H1379BrNaOS requires 330.9763).
(2-Hydroxy-5-t-butylphenyl)(2′-bromophenyl)sulfane (4c)
The reaction was performed as described in general procedure A using p-t-butylphenol (1c) (44 mg, 0.29 mmol). The reaction mixture was stirred at 75 °C for 1 h. Purification by flash column chromatography (hexane/dichloromethane, 4:1) gave (2-hydroxy-5-t-butylphenyl)(2′-bromophenyl)sulfane (4c) (61 mg, 62%) as a colourless oil. νmax/cm−1 (neat) 3455 (OH), 2959 (CH), 1486 (CC), 1446, 1181, 1018, 822, 744; δH (400 MHz, CDCl3) 1.32 (9H, s, 3 × CH3), 6.22 (1H, s, OH), 6.57 (1H, dd, J 8.0, 1.5 Hz, 6′-H), 7.01 (1H, td, J 8.0, 1.5 Hz, 4′-H), 7.05 (1H, d, J 8.6 Hz, 3-H), 7.12 (1H, td, J 8.0, 1.5 Hz, 5′-H), 7.47 (1H, dd, J 8.6, 2.5 Hz, 4-H), 7.53 (1H, d, J 2.5 Hz, 6-H), 7.54 (1H, dd, J 8.0, 1.5 Hz, 3′-H); δC (101 MHz, CDCl3) 31.6 (3 × CH3), 34.4 (C), 114.3 (C), 115.5 (CH), 120.9 (C), 126.5 (CH), 126.9 (CH), 128.2 (CH), 130.2 (CH), 133.1 (CH), 134.0 (CH), 137.7 (C), 144.8 (C), 155.4 (C); m/z (ESI) 359.0072 (MNa+. C16H1779BrNaOS requires 359.0076).
[2-Hydroxy-4,5-(methylenedioxy)phenyl](2′-bromophenyl)sulfane (4d)
The reaction was performed as described in general procedure A using sesamol (1d) (40 mg, 0.29 mmol). The reaction mixture was stirred at room temperature for 3 h. Purification by flash column chromatography (hexane/dichloromethane, 7:3) gave [2-hydroxy-4,5-(methylenedioxy)phenyl](2′-bromophenyl)sulfane (4d) (72 mg, 76%) as a white solid. Mp 95–97 °C; νmax/cm−1 (neat) 3397 (OH), 2899 (CH), 1617 (CC), 1468, 1272, 1181, 1113, 1034, 1016, 935, 856, 760; δH (400 MHz, CDCl3) 5.99 (2H, s, CH2), 6.27 (1H, s, OH), 6.60–6.68 (2H, m, 3-H and 6′-H), 6.92 (1H, s, 6-H), 7.01 (1H, td, J 7.8, 1.6 Hz, 4′-H), 7.11–7.16 (1H, m, 5′-H), 7.52 (1H, dd, J 7.8, 1.3 Hz, 3′-H); δC (101 MHz, CDCl3) 97.9 (CH2), 101.9 (CH), 104.5 (C), 114.7 (CH), 120.8 (C), 126.5 (CH), 127.1 (CH), 128.2 (CH), 133.1 (CH), 137.9 (C), 142.2 (C), 151.6 (C), 154.1 (C); m/z (ESI) 346.9348 (MNa+. C13H979BrNaO3S requires 346.9348).
(2-Hydroxy-5-biphenyl)(2′-bromophenyl)sulfane (4e)
The reaction was performed as described in general procedure A using 4-phenylphenol (1e) (50 mg, 0.29 mmol). The reaction mixture was stirred at 75 °C for 1 h. Purification by flash column chromatography (hexane/ethyl acetate, 49:1) gave (2-hydroxy-5-biphenyl)(2′-bromophenyl)sulfane (4e) (79 mg, 76%) as a colourless oil. νmax/cm−1 (neat) 3440 (OH), 3028 (CH), 1504 (CC), 1472 (CC), 1285, 1174, 1018, 830, 742; δH (400 MHz, CDCl3) 6.40 (1H, s, OH), 6.68 (1H, dd, J 8.0, 1.6 Hz, 6′′-H), 6.99–7.05 (1H, m, 4′′-H), 7.10–7.16 (1H, m, 5′′-H), 7.19 (1H, d, J 8.5 Hz, 3-H), 7.31–7.36 (1H, m, 4′-H), 7.40–7.46 (2H, m, 3′-H and 5′-H), 7.53–7.60 (3H, m, 2′-H, 6′-H and 3′′-H), 7.68 (1H, dd, J 8.5, 2.3 Hz, 4-H), 7.79 (1H, d, J 2.3 Hz, 6-H); δC (101 MHz, CDCl3) 115.9 (C), 116.4 (CH), 121.3 (C), 126.8 (2 × CH), 127.0 (CH), 127.2 (CH), 127.3 (CH), 128.3 (CH), 129.0 (2 × CH), 131.7 (CH), 133.2 (CH), 135.1 (C), 135.7 (CH), 137.2 (C), 139.7 (C), 157.1 (C); m/z (ESI) 378.9761 (MNa+. C18H1379BrNaOS requires 378.9763).
(2-Hydroxynaphthalen-1-yl)(2′-bromophenyl)sulfane (4f)10b
The reaction was performed as described in general procedure A using 2-naphthol (1f) (0.72 g, 5.0 mmol). The reaction mixture was stirred at 75 °C for 1 h. Purification by flash column chromatography (hexane/dichloromethane, 4:1) gave (2-hydroxynaphthalen-1-yl)(2′-bromophenyl)sulfane (4f) (1.6 g, 95%) as a colourless oil. Spectroscopic data was consistent with the literature.10bδH (400 MHz, CDCl3) 6.32–6.37 (1H, m, 6′-H), 6.92–7.00 (2H, m, 4′-H and 5′-H), 7.02 (1H, s, OH), 7.36 (1H, d, J 8.9 Hz, 3-H), 7.40 (1H, ddd, J 8.1, 6.9, 1.2 Hz, 6-H), 7.51 (1H, ddd, J 8.4, 6.9, 1.3 Hz, 7-H), 7.54–7.59 (1H, m, 3′-H), 7.84 (1H, dd, J 8.1, 1.3 Hz, 5-H), 7.96 (1H, d, J 8.9 Hz, 4-H), 8.15 (1H, dd, J 8.4, 1.2 Hz, 8-H); δC (101 MHz, CDCl3) 107.3 (C), 117.2 (CH), 121.2 (C), 124.2 (CH), 124.7 (CH), 126.5 (CH), 127.0 (CH), 128.1 (CH), 128.4 (CH), 128.8 (CH), 129.7 (C), 133.1 (CH), 133.5 (CH), 135.4 (C), 136.6 (C), 157.4 (C); m/z (ESI) 355 (MNa+. 100%).
3-(2′-Bromophenylthio)-4-hydroxybenzaldehyde (4g)
Iron(III) trichloride (4.7 mg, 0.029 mmol) was dissolved in [BMIM]NTf2 (25 μL, 0.087 mmol) and left to stir for 0.5 h at room temperature before being added to a solution of N-(2-bromophenylthio)succinimide (2) (100 mg, 0.35 mmol) in chloroform (0.5 mL). 4-Hydroxybenzaldehyde (1g) (36 mg, 0.29 mmol) and bis(4-methoxyphenyl)sulfane (3) (7.2 mg, 0.029 mmol) was then added and the reaction mixture was left to stir at 85 °C for 24 h. Further N-(2-bromophenylthio)succinimide (2) (100 mg, 0.35 mmol) was added and the reaction mixture was left to stir at 85 °C for a further 24 h. The reaction mixture was concentrated in vacuo and purified using flash column chromatography (hexane/dichloromethane 1:1) to give 3-(2′-bromophenylthio)-4-hydroxybenzaldehyde (4g) (49 mg, 54%) as an off-white solid. Mp 136–138 °C; νmax/cm−1 (neat) 3207 (OH), 1663 (CO), 1593 (CC), 1558, 1488, 1263, 1147, 1018, 745, 719; δH (400 MHz, CDCl3) 6.64 (1H, dd, J 7.9, 1.6 Hz, 6′-H), 7.03–7.08 (2H, m, 4-OH and 4′-H), 7.12–7.16 (1H, m, 5′-H), 7.22 (1H, d, J 8.5 Hz, 5-H), 7.56 (1H, dd, J 7.9, 1.4 Hz, 3′-H), 7.97 (1H, dd, J 8.5, 2.0 Hz, 6-H), 8.08 (1H, d, J 2.0 Hz, 2-H), 9.87 (1H, s, CHO); δC (101 MHz, CDCl3) 116.8 (CH), 117.3 (C), 121.9 (C), 127.5 (CH), 127.9 (CH), 128.4 (CH), 131.0 (C), 133.4 (CH), 134.1 (CH), 136.0 (C), 140.0 (CH), 162.6 (C), 189.9 (CH); m/z (ESI) 330.9397 (MNa+. C13H979BrNaO2S requires 330.9399).
(2-Hydroxy-5-chlorophenyl)(2′-bromophenyl)sulfane (4h)
The reaction was performed as described in general procedure A using 4-chlorophenol (1h) (37 mg, 0.29 mmol). The reaction mixture was stirred at 75 °C for 2 h. Purification by flash column chromatography (hexane/dichloromethane, 4:1) gave (2-hydroxy-5-chlorophenyl)(2′-bromophenyl)sulfane (4h) (50 mg, 54%) as a white solid. Mp 77–79 °C; νmax/cm−1 (neat) 3391 (OH), 3050 (CH), 1464 (CC), 1447, 1188, 1017, 817, 740; δH (400 MHz, CDCl3) 6.36 (1H, s, OH), 6.64 (1H, dd, J 7.9, 1.5 Hz, 6′-H), 7.00–7.08 (2H, m, 3-H and 4′-H), 7.15 (1H, td, J 7.9, 1.4 Hz, 5′-H), 7.38 (1H, dd, J 8.8, 2.6 Hz, 4-H), 7.52 (1H, d, J 2.6 Hz, 6-H), 7.55 (1H, dd, J 7.9, 1.4 Hz, 3′-H); δC (101 MHz, CDCl3) 117.1 (C), 117.2 (CH), 121.6 (C), 125.9 (C), 127.2 (CH), 127.6 (CH), 128.4 (CH), 132.9 (CH), 133.3 (CH), 136.2 (CH), 136.4 (C), 156.3 (C); m/z (ESI) 336.9059 (MNa+. C12H879Br35ClNaOS requires 336.9060).
(2-Hydroxy-5-fluorophenyl)(2′-bromophenyl)sulfane (4i)
The reaction was performed as described in general procedure A using 4-fluorophenol (1i) (33 mg, 0.29 mmol). The reaction mixture was stirred at 75 °C for 3 h. Purification by flash column chromatography (hexane/dichloromethane, 9:1) gave (2-hydroxy-5-fluorophenyl)(2′-bromophenyl)sulfane (4i) (30 mg, 34%) as a colourless oil. νmax/cm−1 (neat) 3437 (OH), 3061 (CH), 1478 (CC), 1445, 1194, 1017, 818, 770, 743; δH (400 MHz, CDCl3) 6.20 (1H, s, OH), 6.66 (1H, dd, J 8.0, 1.5 Hz, 6′-H), 7.01–7.08 (2H, m, 3-H and 4′-H), 7.11–7.17 (2H, m, 6-H and 5′-H), 7.22–7.27 (1H, m, 4-H), 7.55 (1H, dd, J 7.9, 1.4 Hz, 3′-H); δC (101 MHz, CDCl3) 116.3 (d, 3JCF 8.3 Hz, C), 116.8 (d, 3JCF 7.9 Hz, CH), 119.9 (d, 2JCF 23.0 Hz, CH), 121.7 (C), 122.7 (d, 2JCF 23.1 Hz, CH), 127.3 (CH), 127.6 (CH), 128.3 (CH), 133.3 (CH), 136.5 (C), 154.0 (d, 4JCF 2.4 Hz, C), 156.7 (d, 1JCF 242.1 Hz, C); m/z (ESI) 296.9392 ([M − H]−. C12H779BrFOS requires 296.9391).
1,4-Bis-[(2′-bromophenyl)sulfane]-2,5-dihydroxybenzene (4j)
Iron(III) trichloride (5.2 mg, 0.032 mmol) was dissolved in [BMIM]NTf2 (28 μL, 0.095 mmol) and left to stir for 0.5 h at room temperature before being added to a solution of N-(2-bromophenylthio)succinimide (2) (200 mg, 0.70 mmol) in chloroform (0.6 mL). Hydroquinone (1j) (35 mg, 0.32 mmol) and bis(4-methoxyphenyl)sulfane (3) (7.8 mg, 0.032 mmol) was then added and the reaction mixture was left to stir at 75 °C for 1 h. The reaction mixture was concentrated in vacuo and purified using flash column chromatography (hexane/dichloromethane 3:2) to give 1,4-bis-[(2′-bromophenyl)sulfane]-2,5-dihydroxybenzene (4j) (121 mg, 79%) as a white solid. Mp 189–190 °C; νmax/cm−1 (neat) 3414 (OH), 3056 (CH), 1458 (CC), 1443, 1302, 1189, 1018, 798, 739; δH (400 MHz, CDCl3) 5.98 (2H, s, 2 × OH), 6.81 (2H, dd, J 7.8, 1.5 Hz, 2 × 6′-H), 7.07 (2H, td, J 7.8, 1.5 Hz, 2 × 4′-H), 7.19 (2H, td, J 7.8, 1.5 Hz, 2 × 5′-H), 7.24 (2H, s, 3-H and 6-H), 7.57 (2H, dd, J 7.8, 1.5 Hz, 2 × 3′-H); δC (101 MHz, CDCl3) 120.6 (2 × C), 122.3 (2 × C), 122.5 (2 × CH), 127.9 (2 × CH), 128.1 (2 × CH), 128.4 (2 × CH), 133.4 (2 × CH), 136.1 (2 × C), 151.1 (2 × C); m/z (ESI) 482.8698 (MH+. C18H1379Br2O2S2 requires 482.8718).
General procedure B: preparation of phenoxathiins 5
To a solution of biaryl sulfide (1.0 equiv.) in N,N-dimethylacetamide (0.03 M in arene) was added copper(I) thiophene-2-carboxylate (1.0 equiv.). The reaction mixture was stirred at 100 °C for 18 h. The reaction mixture was extracted with ethyl acetate (30 mL) and washed with 5% aqueous lithium chloride (2 × 30 mL), dried over MgSO4, filtered and concentrated in vacuo. The resulting material was purified by flash column chromatography.2-Methylphenoxathiin (5a)10a
The reaction was performed as described in general procedure B using (2-hydroxy-5-methylphenyl)(2′-bromophenyl)sulfane (4a) (60 mg, 0.46 mmol). Purification by flash column chromatography (hexane) gave 2-methylphenoxathiin (5a) (38 mg, 86%) as a colourless oil. Spectroscopic data was consistent with the literature.10aδH (500 MHz, CDCl3) 2.26 (3H, s, CH3), 6.87–6.93 (3H, m, 1-H, 3-H and 4-H), 6.97–7.02 (2H, m, 6-H and 8-H), 7.07–7.14 (2H, m, 7-H and 9-H); δC (126 MHz, CDCl3) 20.7 (CH3), 117.5 (CH), 117.9 (CH), 119.7 (C), 120.3 (C), 124.4 (CH), 126.9 (CH), 127.2 (CH), 127.7 (CH), 128.4 (CH), 134.3 (C), 150.0 (C), 152.5 (C); m/z (ESI) 214 (M+. 100%).2,3-Dimethylphenoxathiin (5b)
The reaction was performed as described in general procedure B using (2-hydroxy-4,5-dimethylphenyl)(2′-bromophenyl)sulfane (4b) (50 mg, 0.16 mmol). Purification by flash column chromatography (hexane) gave 2,3-dimethylphenoxathiin (5b) (30 mg, 81%) as a white solid. Mp 117–119 °C; νmax/cm−1 (neat) 2984 (CH), 1449 (CC), 1441, 1218, 1023, 874, 756; δH (400 MHz, CDCl3) 2.17 (3H, s, CH3), 2.20 (3H, s, CH3), 6.81 (1H, s, 4-H), 6.85 (1H, s, 1-H), 6.95–7.01 (2H, m, 6-H and 8-H), 7.06–7.13 (2H, m, 7-H and 9-H); δC (101 MHz, CDCl3) 19.0 (CH3), 19.6 (CH3), 116.2 (C), 117.8 (CH), 118.9 (CH), 120.6 (C), 124.3 (CH), 126.9 (CH), 127.4 (CH), 127.6 (CH), 132.9 (C), 136.4 (C), 150.1 (C), 152.5 (C); m/z (ESI) 251.0502 (MNa+. C14H12NaOS requires 251.0501).
2-t-Butylphenoxathiin (5c)
The reaction was performed as described in general procedure B using (2-hydroxy-5-t-butylphenyl)(2′-bromophenyl)sulfane (4c) (55 mg, 0.16 mmol). Purification by flash column chromatography (hexane) gave 2-t-butylphenoxathiin (5c) (35 mg, 83%) as a colourless oil. νmax/cm−1 (neat) 2960 (CH), 1462 (CC), 1442, 1262, 1227, 853, 750; δH (400 MHz, CDCl3) 1.29 (9H, s, 3 × CH3), 6.95 (1H, d, J 8.4 Hz, 4-H), 6.97–7.02 (2H, m, 6-H and 8-H), 7.09–7.16 (4H, m, 1-H, 3-H, 7-H and 9-H); δC (101 MHz, CDCl3) 31.5 (3 × CH3), 34.5 (C), 117.3 (CH), 117.9 (CH), 119.4 (C), 120.4 (C), 123.8 (CH), 124.5 (CH), 124.9 (CH), 126.9 (CH), 127.7 (CH), 147.8 (C), 150.0 (C), 152.5 (C); m/z (ESI) 279.0815 (MNa+. C16H16NaOS requires 279.0814).
2,3-(Methylenedioxy)phenoxathiin (5d)
The reaction was performed as described in general procedure B using [2-hydroxy-4,5-(methylenedioxy)phenyl](2′-bromophenyl)sulfane (4d) (54 mg, 0.17 mmol). Purification by flash column chromatography (hexane/dichloromethane 4:1) gave 2,3-(methylenedioxy)phenoxathiin (5d) (38 mg, 93%) as a colourless oil. νmax/cm−1 (neat) 2888 (CH), 1459 (CC), 1219, 1118, 1032, 933, 850, 746; δH (400 MHz, CDCl3) 5.94 (2H, s, CH2), 6.57 (1H, s, 4-H), 6.62 (1H, s, 1-H), 6.98–7.04 (2H, m, 6-H and 8-H), 7.10–7.16 (2H, m, 7-H and 9-H); δC (101 MHz, CDCl3) 100.6 (CH), 101.8 (CH2), 106.2 (CH), 111.4 (C), 117.8 (CH), 121.0 (C), 124.7 (CH), 126.9 (CH), 127.9 (CH), 144.6 (C), 147.4 (C), 147.5 (C), 152.9 (C); m/z (ESI) 267.0087 (MNa+. C13H8NaO3S requires 267.0086).
2-Phenylphenoxathiin (5e)
The reaction was performed as described in general procedure B using (2-hydroxy-5-biphenyl)(2′-bromophenyl)sulfane (4e) (65 mg, 0.18 mmol). Purification by flash column chromatography (hexane) gave 2-phenylphenoxathiin (5e) (42 mg, 84%) as a white solid. Mp 85–86 °C; νmax/cm−1 (neat) 3056 (CH), 1456 (CC), 1439 (CC), 1262, 1210, 1077, 938, 745; δH (400 MHz, CDCl3) 6.98–7.05 (2H, m, 6-H and 8-H), 7.07 (1H, d, J 8.3 Hz, 4-H), 7.10–7.18 (2H, m, 7-H and 9-H), 7.29–7.39 (3H, m, 1-H, 3-H and 4′-H), 7.40–7.47 (2H, m, 3′-H and 5′-H), 7.50–7.56 (2H, m, 2′-H and 6′-H); δC (101 MHz, CDCl3) 117.9 (CH), 118.1 (CH), 119.9 (C), 120.5 (C), 124.7 (CH), 125.4 (CH), 126.6 (CH), 126.9 (2 × CH), 127.0 (CH), 127.5 (CH), 127.9 (CH), 129.0 (2 × CH), 138.0 (C), 139.9 (C), 151.6 (C), 152.2 (C); m/z (ESI) 299.0501 (MNa+. C18H12NaOS requires 299.0501).
Benzo[a]phenoxathiin (5f)23
The reaction was performed as described in general procedure B using (2-hydroxynaphthalen-1-yl)(2′-bromophenyl)sulfane (4f) (80 mg, 0.24 mmol). Purification by flash column chromatography (hexane) gave 2-methylphenoxathiin (5f) (50 mg, 83%) as a white solid. Mp 63–64 °C (lit.23 63 °C); δH (400 MHz, CDCl3) 7.02–7.08 (2H, m, 11-H and 13-H), 7.13–7.24 (3H, m, 1-H, 10-H and 12-H), 7.44 (1H, ddd, J 8.1, 6.9, 1.0 Hz, 4-H), 7.55 (1H, ddd, J 8.4, 6.9, 1.3 Hz, 5-H), 7.65 (1H, d, J 8.8 Hz, 2-H), 7.79 (1H, dd, J 8.1, 1.3 Hz, 3-H), 7.92 (1H, dd, J 8.4, 1.0 Hz, 6-H); δC (101 MHz, CDCl3) 113.6 (C), 117.8 (CH), 118.5 (CH), 119.7 (C), 123.1 (CH), 124.8 (CH), 125.1 (CH), 127.0 (CH), 127.2 (CH), 127.8 (CH), 128.1 (CH), 128.5 (CH), 130.3 (C), 131.1 (C), 149.8 (C), 152.5 (C); m/z (ESI) 273 (MNa+. 100%).2-Formylphenoxathiin (5g)
The reaction was performed as described in general procedure B using 3-(2′-bromophenylthio)-4-hydroxybenzaldehyde (4g) (53 mg, 0.17 mmol). Purification by flash column chromatography (hexane/dichloromethane 7:3) gave 2-formylphenoxathiin (5g) (36 mg, 92%) as a yellow solid. Mp 82–83 °C; νmax/cm−1 (neat) 2727 (CH), 1683 (CO), 1592 (CC), 1466, 1238, 1198, 1080, 810, 752; δH (400 MHz, CDCl3) 6.97–7.08 (4H, m, 4-H, 6-H, 8-H and 9-H), 7.14 (1H, ddd, J 8.0, 6.9, 2.1 Hz, 7-H), 7.59 (1H, d, J 2.0 Hz, 1-H), 7.62 (1H, dd, J 8.3, 2.0 Hz, 3-H), 9.84 (1H, s, CHO); δC (101 MHz, CDCl3) 118.0 (CH), 118.3 (CH), 118.6 (C), 121.4 (C), 125.4 (CH), 126.9 (CH), 128.1 (CH), 128.2 (CH), 130.2 (CH), 133.3 (C), 150.9 (C), 156.6 (C), 190.1 (CH); m/z (ESI) 251.0136 (MNa+. C13H8NaO2S requires 251.0137).
2-Chlorophenoxathiin (5h)
The reaction was performed as described in general procedure B using (2-hydroxy-5-chlorophenyl)(2′-bromophenyl)sulfane (4h) (40 mg, 0.13 mmol). Purification by flash column chromatography (hexane) gave 2-chlorophenoxathiin (5h) (25 mg, 83%) as a white solid. Mp 88–90 °C; νmax/cm−1 (neat) 3059 (CH), 1462 (CC), 1439, 1377, 1285, 1261, 1099, 799, 737; δH (400 MHz, CDCl3) 6.89–6.94 (1H, m, 6-H), 6.96–7.10 (5H, m, 1-H, 3-H, 4-H, 8-H and 9-H), 7.14 (1H, ddd, J 7.9, 7.4, 1.8 Hz, 7-H); δC (101 MHz, CDCl3) 118.0 (CH), 118.8 (CH), 119.2 (C), 122.2 (C), 124.9 (CH), 126.4 (CH), 126.9 (CH), 127.7 (CH), 128.1 (CH), 129.5 (C), 150.9 (C), 152.0 (C); m/z (ESI) 234.9958 (MH+. C12H835ClOS requires 234.9979).
2-Fluorophenoxathiin (5i)
The reaction was performed as described in general procedure B using (2-hydroxy-5-fluorophenyl)(2′-bromophenyl)sulfane (4i) (45 mg, 0.15 mmol). Purification by flash column chromatography (hexane) gave 2-fluorophenoxathiin (5i) (30 mg, 91%) as a white solid. Mp 58–60 °C; νmax/cm−1 (neat) 3062 (CH), 1458 (CC), 1442, 1245, 1183, 853, 753; δH (400 MHz, CDCl3) 6.78–6.85 (2H, m, 1-H and 3-H), 6.95 (1H, ddd, J 8.5, 4.8, 0.7 Hz, 4-H), 6.99–7.04 (2H, m, 6-H and 8-H), 7.09 (1H, dd, J 7.7, 1.7 Hz, 9-H), 7.12–7.17 (1H, m, 7-H); δC (101 MHz, CDCl3) 113.5 (d, 2JCF 26.0 Hz, CH), 114.3 (d, 2JCF 23.3 Hz, CH), 118.0 (CH), 118.7 (d, 3JCF 8.6 Hz, CH), 119.3 (C), 122.1 (d, 3JCF 8.9 Hz, C), 124.8 (CH), 126.9 (CH), 128.1 (CH), 148.4 (d, 4JCF 2.7 Hz, C), 152.3 (C), 159.3 (d, 1JCF 243.9 Hz, C); m/z (ESI) 236.0529 (MNH4+. C12H11FNOS requires 236.0540).
5,12-Dioxa-7,14-dithiapentacene (5j)
To a solution of 1,4-bis-[(2′-bromophenyl)sulfane]-2,5-dihydroxybenzene (4j) (90 mg, 0.19 mmol) in N,N-dimethylacetamide (6.4 mL) was added copper(I) thiophene-2-carboxylate (71 mg, 0.37 mmol). The reaction mixture was stirred at 100 °C for 18 h. The reaction mixture was extracted with ethyl acetate (30 mL) and washed with 5% aqueous lithium chloride (2 × 30 mL), dried over MgSO4, filtered and concentrated in vacuo. Purification by flash column chromatography (hexane/dichloromethane 9:1) gave 5,12-dioxa-7,14-dithiapentacene (5j) (49 mg, 82%) as a white solid. Mp 216–218 °C; νmax/cm−1 (neat) 3070 (CH), 1450 (CC), 1439, 1370, 1261, 1205, 1169, 874, 747; δH (400 MHz, CDCl3) 6.77 (2H, s, 6-H and 13-H), 6.95–7.03 (4H, m, 2 × 2-H and 2 × 4-H), 7.07–7.15 (4H, m, 2 × 1-H and 2 × 3-H); δC (101 MHz, CDCl3) 115.5 (2 × CH), 118.0 (2 × CH), 119.3 (2 × C), 119.5 (2 × C), 124.8 (2 × CH), 126.9 (2 × CH), 128.1 (2 × CH), 148.7 (2 × C), 152.1 (2 × C); m/z (ESI) 340.0465 ([MNH4]+. C18H14NO2S2 requires 340.0460).
N-(Benzyloxycarbonyl)-[3′-(2′′-bromophenylthio)]-L-tyrosine methyl ester (7)
The reaction was performed as described in general procedure A using N-(benzyloxycarbonyl)-L-tyrosine methyl ester (6) (192 mg, 0.582 mmol). The reaction mixture was stirred at 75 °C for 20 h. Purification by flash column chromatography (dichloromethane) gave N-(benzyloxycarbonyl)-[3′-(2′′-bromophenylthio)]-L-tyrosine methyl ester (7) (240 mg, 80%) as a colourless oil. νmax/cm−1 (neat) 3379 (OH), 3318 (NH), 2948 (CH), 1705 (CO), 1507 (CC), 1446, 1216, 1019, 757; [α]D25 +42.7 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.01 (1H, dd, J 14.0, 5.8 Hz, 3-HH), 3.11 (1H, dd, J 14.0, 5.5 Hz, 3-HH), 3.67 (3H, s, CO2CH3), 4.63 (1H, ddd, J 8.0, 5.8, 5.5 Hz, 2-H), 5.07 (1H, d, J 12.2 Hz, PhCHH), 5.10 (1H, d, J 12.2 Hz, PhCHH), 5.29 (1H, d, J 8.0 Hz, NH), 6.36 (1H, s, OH), 6.53 (1H, dd, J 8.0, 1.5 Hz, 6′′-H), 6.95–7.03 (2H, m, 5′-H and 4′′-H), 7.05–7.12 (1H, m, 5′′-H), 7.15 (1H, dd, J 8.4, 2.2 Hz, 6′-H), 7.28–7.39 (6H, m, 2′-H and Ph), 7.52 (1H, dd, J 7.9, 1.3 Hz, 3′′-H); δC (101 MHz, CDCl3) 37.4 (CH2), 52.6 (CH3), 55.0 (CH), 67.2 (CH2), 115.4 (C), 116.2 (CH), 121.1 (C), 126.8 (CH), 127.1 (CH), 128.2 (2 × CH), 128.3 (CH), 128.4 (CH), 128.7 (2 × CH), 129.2 (C), 133.1 (CH), 133.9 (CH), 136.3 (C), 137.3 (C), 137.9 (CH) 155.6 (C), 156.8 (C), 171.8 (C); m/z (ESI) 538.0288 (MNa+. C24H2279BrNNaO5S requires 538.0294).
Methyl (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-2′-yl)propanoate (8)
The reaction was performed as described in general procedure B using N-(benzyloxycarbonyl)-[3′-(2′′-bromophenylthio)]-L-tyrosine methyl ester (7) (100 mg, 0.194 mmol). Purification by flash column chromatography (dichloromethane) gave methyl (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-2′-yl)propanoate (8) (78.0 mg, 93%) as a colourless oil. νmax/cm−1 (neat) 3342 (NH), 2950 (CH), 1699 (CO), 1511 (CC), 1464, 1442, 1230, 1206, 1056, 747; [α]D25 +48.7 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 2.98 (1H, dd, J 14.0, 6.0 Hz, 3-HH), 3.06 (1H, dd, J 14.0, 5.6 Hz, 3-HH), 3.73 (3H, s, CO2CH3), 4.62 (1H, ddd, J 8.0, 6.0, 5.6 Hz, 2-H), 5.08 (1H, d, J 12.2 Hz, PhCHH), 5.13 (1H, d, J 12.2 Hz, PhCHH), 5.23 (1H, d, J 8.0 Hz, NH), 6.81–6.85 (2H, m, 1′-H and 4′-H), 6.87–6.91 (1H, m, 3′-H), 6.97–7.02 (2H, m, 6′-H and 8′-H), 7.08 (1H, dd, J 7.7, 1.5 Hz, 9′-H), 7.10–7.14 (1H, m, 7′-H), 7.29–7.38 (5H, m, Ph); δC (101 MHz, CDCl3) 37.5 (CH2), 52.6 (CH3), 54.9 (CH), 67.2 (CH2), 117.9 (CH), 118.0 (CH), 119.9 (C), 120.5 (C), 124.7 (CH), 126.9 (CH), 127.5 (CH), 127.9 (CH), 128.2 (2 × CH), 128.4 (CH), 128.7 (CH), 128.7 (2 × CH), 132.3 (C), 136.3 (C), 151.4 (C), 152.2 (C), 155.7 (C) 171.9 (C); m/z (ESI) 458.1031 (MNa+. C24H21NNaO5S requires 458.1033).
(2S)-2-[(Benzyloxycarbonyl)amino]-3-(phenoxathiin-2′-yl)propanoic acid
To a stirred solution of methyl (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-2′-yl)propanoate (8) (130 mg, 0.299 mmol) in methanol (6 mL), 1,4-dioxane (3 mL) and water (3 mL) was added cesium carbonate (126 mg, 0.388 mmol). The resulting reaction mixture was left to stir at room temperature for 20 h. The reaction mixture was concentrated in vacuo, diluted with water (20 mL) and acidified to pH 1 with 1 M aqueous hydrochloric acid. The aqueous layer was extracted with dichloromethane (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure to give (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-2′-yl)propanoic acid as a colourless oil (103 mg, 82%). νmax/cm−1 (neat) 3306 (OH), 2920 (CH), 1697 (CO), 1462 (CC), 1265, 1231, 1053, 748; [α]D20 +7.1 (c 0.1, MeOH); δH (400 MHz, CD3OD) 2.84 (1H, dd, J 14.0, 9.4 Hz, 3-HH), 3.13 (1H, dd, J 14.0, 4.8 Hz, 3-HH), 4.39 (1H, dd, J 9.4, 4.8 Hz, 2-H), 4.98 (1H, d, J 12.5 Hz, PhCHH), 5.06 (1H, d, J 12.5 Hz, PhCHH), 6.88 (1H, d, J 8.2 Hz, 6′-H), 6.96–7.06 (4H, m, 1′-H, 4′-H, 8′-H and 9′-H), 7.10 (1H, dd, J 7.7, 1.5 Hz, 3′-H), 7.12–7.31 (5H, m, 7′-H and Ph); δC (101 MHz, CD3OD) 37.8 (CH2), 56.7 (CH), 67.5 (CH2), 118.5 (CH), 118.7 (CH), 121.1 (C), 121.3 (C), 125.7 (CH), 127.8 (CH), 128.5 (CH), 128.6 (2 × CH), 128.9 (CH), 129.0 (CH), 129.4 (2 × CH), 129.9 (CH), 135.4 (C), 138.2 (C), 152.3 (C), 153.5 (C), 158.4 (C), 175.0 (C); m/z (ESI) 422.1060 (MH+. C23H20NO5S requires 422.1057).
(2S)-2-Amino-3-(phenoxathiin-2′-yl)propanoic acid hydrochloride (9)
A solution of (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-2′-yl)propanoic acid (75 mg, 0.16 mmol) in 6 M aqueous hydrochloric acid (7.5 mL) and 1,4-dioxane (1.5 mL) was heated under reflux for 4 h. After cooling to room temperature, the reaction mixture was concentrated in vacuo and the resulting residue recrystallised from methanol and diethyl ether to afford (2S)-2-amino-3-(phenoxathiin-2′-yl)propanoic acid hydrochloride (9) as a white solid (49 mg, 95%). Mp 234–236 °C; νmax/cm−1 (neat) 3387 (NH), 2889 (OH), 1728 (CO), 1470 (CC), 1439, 1265, 1200, 1123, 741; [α]D21 −3.1 (c 0.1, MeOH); δH (400 MHz, CD3OD) 3.09 (1H, dd, J 14.6, 7.7 Hz, 3-HH), 3.25 (1H, dd, J 14.6, 5.5 Hz, 3-HH), 4.22 (1H, dd, J 7.7, 5.5 Hz, 2-H), 6.99–7.20 (7H, m, 1′-H, 3′-H, 4′-H, 6′-H, 7′-H, 8′-H and 9′-H); δC (101 MHz, CD3OD) 36.4 (CH2), 55.0 (CH), 118.7 (CH), 119.2 (CH), 120.9 (C), 122.2 (C), 126.0 (CH), 127.9 (CH), 128.8 (CH), 129.2 (CH), 130.1 (CH), 132.3 (C), 153.2 (C), 153.4 (C), 171.2 (C); m/z (ESI) 288.0690 (MH+. C15H14NO3S requires 288.0689).
Methyl (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-10′,10′-dioxide-2′-yl)propanoate (10)
Methyl (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-2′-yl)propanoate (8) (60.0 mg, 0.138 mmol) was dissolved in dichloromethane (1.30 mL) with stirring under argon. Glacial acetic acid (217 μL, 3.79 mmol) was then added to the solution followed by 30% H2O2 (78.0 μL). The reaction mixture was stirred at 50 °C for 20 h. The reaction mixture was cooled to room temperature and diluted with dichloromethane (20 mL). The organic layer was washed with water (20 mL) and the aqueous layer was further extracted with dichloromethane (2 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over MgSO4, filtered, and concentrated in vacuo. Purification by flash chromatography (dichloromethane/methanol 199:1) gave methyl (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-10′,10′-dioxide-2′-yl)propanoate (10) (62.0 mg, 75%) as a white solid. Mp 122–124 °C; νmax/cm−1 (neat) 3348 (NH), 2954 (CH), 1714 (CO), 1520 (CC), 1469, 1273, 1152, 1063, 757; [α]D25 +65.9 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.15 (1H, dd, J 14.0, 6.2 Hz, 3-HH), 3.26 (1H, dd, J 14.0, 5.4 Hz, 3-HH), 3.75 (3H, s, CO2CH3), 4.67 (1H, ddd, J 7.6, 6.2, 5.4 Hz, 2-H), 5.10 (2H, s, PhCH2), 5.42 (1H, d, J 7.6 Hz, NH), 7.26–7.43 (9H, m, 3′-H, 4′-H, 6′-H, 8′-H and Ph), 7.61–7.67 (1H, m, 7′-H), 7.79 (1H, d, J 1.9 Hz, 1′-H), 8.04 (1H, dd, J 7.9, 1.2 Hz, 9′-H); δC (101 MHz, CDCl3) 37.7 (CH2), 52.8 (CH3), 54.8 (CH), 67.2 (CH2), 119.0 (CH), 119.2 (CH), 123.5 (CH), 123.8 (CH), 124.9 (C), 124.9 (C), 125.0 (CH), 128.2 (2 × CH), 128.3 (CH), 128.6 (2 × CH), 133.4 (C), 134.3 (CH), 135.2 (CH), 136.2 (C), 150.6 (C), 151.6 (C), 155.7 (C), 171.4 (C); m/z (ESI) 490.0931 (MNa+. C24H21NNaO7S requires 490.0931).
(2S)-2-[(Benzyloxycarbonyl)amino]-3-(phenoxathiin-10′,10′-dioxide-2′-yl)propanoic acid
To a stirred solution of methyl (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-10′,10′-dioxide-2′-yl)propanoate (10) (45.0 mg, 0.0963 mmol) in methanol (2 mL), 1,4-dioxane (1 mL) and water (1 mL) was added cesium carbonate (40.7 mg, 0.125 mmol). The resulting reaction mixture was left to stir at room temperature for 20 h. The reaction mixture was concentrated in vacuo, diluted with water (10 mL) and acidified to pH 1 with 1 M aqueous hydrochloric acid. The aqueous layer was extracted with dichloromethane (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure to give (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-10′,10′-dioxide-2′-yl)propanoic acid as a white solid (37.0 mg, 85%). Mp 231–234 °C; νmax/cm−1 (neat) 3013 (CH), 2672 (OH), 1698 (CO), 1469 (CC), 1273, 1150, 1062, 750; [α]D24 +13.8 (c 0.1, MeOH); δH (500 MHz, CD3OD) 3.05 (1H, dd, J 14.0, 9.6 Hz, 3-HH), 3.34 (1H, dd, J 14.0, 4.4 Hz, 3-HH), 4.48 (1H, dd, J 9.6, 4.4 Hz, 2-H), 4.97 (1H, d, J 12.5 Hz, PhCHH), 5.02 (1H, d, J 12.5 Hz, PhCHH), 7.13–7.29 (5H, m, Ph), 7.34 (1H, d, J 8.5 Hz, 4′-H), 7.43–7.48 (2H, m, 6′-H and 8′-H), 7.59 (1H, dd, J 8.5, 1.9 Hz, 3′-H), 7.72 (1H, t, J 7.8 Hz, 7′-H), 7.89 (1H, d, J 1.9 Hz, 1′-H), 8.00 (1H, d, J 8.1 Hz, 9′-H); δC (126 MHz, CD3OD) 37.8 (CH2), 56.5 (CH), 67.6 (CH2), 120.1 (CH), 120.2 (CH), 124.1 (CH), 124.4 (CH), 125.9 (C), 126.2 (CH), 126.3 (C), 128.7 (2 × CH), 128.8 (CH), 129.4 (2 × CH), 135.7 (CH), 136.5 (C), 136.7 (CH), 138.1 (C), 151.8 (C), 153.0 (C), 158.4 (C) 174.6 (C); m/z (ESI) 476.0776 (MNa+. C23H19NNaO7S requires 476.0774).
(2S)-2-Amino-3-(phenoxathiin-10′,10′-dioxide-2′-yl)propanoic acid hydrochloride (11)
A solution of (2S)-2-[(benzyloxycarbonyl)amino]-3-(phenoxathiin-10′,10′-dioxide-2′-yl)propanoic acid (25 mg, 0.055 mmol) in 6 M aqueous hydrochloric acid (2.5 mL) and 1,4-dioxane (0.5 mL) was heated under reflux for 4 h. After cooling to room temperature, the reaction mixture was concentrated in vacuo and the resulting residue recrystallised from methanol and diethyl ether to afford (2S)-2-amino-3-(phenoxathiin-10′,10′-dioxide-2′-yl)propanoic acid hydrochloride (11) as a white solid (18 mg, 92%). Mp 235–236 °C; νmax/cm−1 (neat) 3075 (NH), 2948 (CH), 2831 (OH), 1732 (CO), 1592 (CC), 1471, 1272, 1199, 1148, 758; [α]D24 +30.3 (c 0.1, MeOH); δH (500 MHz, CD3OD) 3.34 (1H, dd, J 14.7, 7.4 Hz, 3-HH), 3.45 (1H, dd, J 14.7, 5.9 Hz, 3-HH), 4.37 (1H, dd, J 7.4, 5.9 Hz, 2-H), 7.49–7.55 (3H, m, 4′-H, 6′-H and 8′-H), 7.72 (1H, dd, J 8.6, 1.5 Hz, 3′-H), 7.78 (1H, t, J 7.9 Hz, 7′-H), 7.99 (1H, d, J 1.5 Hz, 1′-H), 8.03 (1H, d, J 7.9 Hz, 9′-H); δC (126 MHz, CD3OD) 36.4 (CH2), 54.7 (CH), 120.2 (CH), 120.9 (CH), 124.1 (CH), 125.0 (CH), 126.3 (C), 126.4 (CH), 126.5 (C) 133.4 (C), 135.9 (CH), 136.9 (CH), 152.6 (C), 152.9 (C), 170.9 (C); m/z (ESI) 342.0405 (MNa+. C15H13NNaO5S requires 342.0407).
2-(2′-Bromophenylthio)-17-β-estradiol-17-acetate (13)
The reaction was performed as described in general procedure A using 17-β-estradiol-17-acetate (12) (91.5 mg, 0.291 mmol). The reaction mixture was stirred at 40 °C for 4 h. Purification by flash column chromatography (hexane/dichloromethane, 3:2) gave 2-(2′-bromophenylthio)-17-β-estradiol-17-acetate (13) (107 mg, 73%) as a white solid. Mp 172–175 °C; νmax/cm−1 (neat) 3406 (OH), 2924 (CH), 1724 (CO), 1481 (CC), 1427, 1346, 1015, 891, 745; [α]D20 +48.7 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 0.84 (3H, s, 13-CH3), 1.21–1.61 (7H, m, 7-H2, 8-H, 11-HH, 15-H2 and 16-HH), 1.70–1.81 (1H, m, 9-H), 1.84–1.94 (2H, m, 11-HH and 14-H), 2.05 (3H, s, COCH3), 2.16–2.28 (3H, m, 12-H2 and 16-HH), 2.86–2.92 (2H, m, 6-H2), 4.68 (1H, dd, J 9.0, 8.0 Hz, 17-H), 6.13 (1H, s, OH), 6.59 (1H, dd, J 7.8, 1.5 Hz, 6′-H), 6.82 (1H, s, 4-H), 6.99 (1H, td, J 7.8, 1.5 Hz, 4′-H), 7.11 (1H, td, J 7.8, 1.3 Hz, 5′-H), 7.41 (1H, s, 1-H), 7.52 (1H, dd, J 7.8, 1.3 Hz, 3′-H); δC (101 MHz, CDCl3) 12.2 (CH3), 21.3 (CH3), 23.4 (CH2), 26.3 (CH2), 27.1 (CH2), 27.7 (CH2), 29.8 (CH2), 36.9 (CH2), 38.4 (CH), 43.0 (C), 43.8 (CH), 49.9 (CH), 82.8 (CH), 112.0 (C), 115.7 (CH), 120.8 (C), 126.6 (CH), 126.8 (CH), 128.1 (CH), 133.0 (CH), 134.2 (CH), 134.3 (C), 138.0 (C), 142.6 (C), 155.3 (C), 171.4 (C); m/z (ESI) 501.1091 (MH+. C26H3079BrO3S requires 501.1090).
(15S,18S,19S,22S,23R)-18-Methyl-4-oxa-11-thiahexacyclo[12.11.0.03,12.05,10.015,23.018,22]pentacosa-1(14),2,5(10),6,8,12-hexaen-19-yl acetate (14)
The reaction was performed as described in general procedure B using 2-(2′-bromophenylthio)-17-β-estradiol-17-acetate (13) (50 mg, 0.17 mmol). Purification by flash column chromatography (hexane/dichloromethane 7:3) gave (15S,18S,19S,22S,23R)-18-methyl-4-oxa-11-thiahexacyclo[12.11.0.03,12.05,10.015,23.018,22]pentacosa-1(14),2,5(10),6,8,12-hexaen-19-yl acetate (14) (32 mg, 76%) as a white solid. Mp 185–187 °C; νmax/cm−1 (neat) 2928 (CH), 1724 (CO), 1462 (CC), 1288, 1037, 879, 752; [α]D21 +44.9 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 0.82 (3H, s, 18-CH3), 1.21–1.56 (7H, m, 16-HH, 20-HH, 21-H2, 23-H and 24-H2), 1.69–1.77 (1H, m, 15-H), 1.82–1.91 (2H, m, 16-HH and 22-H), 2.06 (3H, s, COCH3), 2.09–2.28 (3H, m, 17-H2 and 20-HH), 2.76–2.82 (2H, m, 25-H2), 4.68 (1H, dd, J 9.1, 7.9 Hz, 19-H), 6.73 (1H, s, 2-H), 6.87–7.00 (3H, m, 6-H, 8-H and 13-H), 7.05–7.13 (2H, m, 7-H and 9-H); δC (101 MHz, CDCl3) 12.2 (CH3), 21.3 (CH3), 23.4 (CH2), 26.3 (CH2) 27.1 (CH2), 27.7 (CH2), 29.3 (CH2), 36.9 (CH2), 38.4 (CH), 43.0 (C), 43.9 (CH), 49.9 (CH), 82.8 (CH), 116.6 (C), 117.7 (CH), 117.8 (CH), 120.6 (C), 123.7 (CH), 124.4 (CH), 126.9 (CH), 127.6 (CH), 136.8 (C), 136.9 (C), 150.0 (C), 152.4 (C), 171.3 (C); m/z (ESI) 443.1651 (MNa+. C26H28NaO3S requires 443.1651).
(15S,18S,19S,22S,23R)-18-Methyl-4-oxa-11-thiahexacyclo[12.11.0.03,12.05,10.015,23.018,22]pentacosa-1(14),2,5(10),6,8,12-hexaen-19-ol (15)
A solution of (15S,18S,19S,22S,23R)-18-methyl-4-oxa-11-thiahexacyclo[12.11.0.03,12.05,10.015,23.018,22]pentacosa-1(14),2,5(10),6,8,12-hexaen-19-yl acetate (14) (17 mg, 40 μmol) in tetrahydrofuran (0.5 mL) and methanol (0.5 mL) was treated with potassium carbonate (7.0 mg, 49 μmol). The resulting reaction mixture was left to stir at room temperature for 18 h. The reaction mixture was concentrated in vacuo and the residue was partitioned between a saturated aqueous solution of ammonium chloride (10 mL) and ethyl acetate (10 mL). The aqueous layer was extracted with ethyl acetate (2 × 10 mL) and the combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (hexane/ethyl acetate 7:3) gave (15S,18S,19S,22S,23R)-18-methyl-4-oxa-11-thiahexacyclo[12.11.0.03,12.05,10.015,23.018,22]pentacosa-1(14),2,5(10),6,8,12-hexaen-19-ol (15) (14 mg, 93%) as a white solid. Mp 197–199 °C; νmax/cm−1 (neat), 3565 (OH), 2905 (CH), 1466 (CC), 1443, 1215, 1065, 860, 745; [α]D21 +89.1 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 0.77 (3H, s, 18-CH3), 1.10–1.55 (8H, m, OH, 16-HH, 20-HH, 21-H2, 23-H and 24-H2), 1.65 (1H, m, 15-H), 1.83–1.90 (1H, m, 16-HH), 1.92–1.97 (1H, m, 22-H), 2.06–2.17 (2H, m, 17-H2), 2.22–2.29 (1H, m, 20-HH), 2.76–2.82 (2H, m, 25-H2), 3.72 (1H, t, J 8.4 Hz, 19-H), 6.74 (1H, s, 2-H), 6.93–7.01 (3H, m, 6-H, 8-H and 13-H), 7.07–7.12 (2H, m, 7-H and 9-H); δC (101 MHz, CDCl3) 11.2 (CH3), 23.3 (CH2), 26.4 (CH2), 27.2 (CH2), 29.3 (CH2), 30.7 (CH2), 36.7 (CH2), 38.7 (CH), 43.3 (C), 44.0 (CH), 50.1 (CH), 82.0 (CH), 116.6 (C), 117.7 (CH), 117.8 (CH), 120.6 (C), 123.7 (CH), 124.4 (CH), 126.9 (CH), 127.6 (CH), 136.9 (C), 137.1 (C), 150.0 (C), 152.4 (C); m/z (ESI) 401.1532 (MNa+. C24H26NaO2S requires 401.1546).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from The Carnegie Trust for the Universities of Scotland (Ph.D. studentship to A. C. D.) and the University of Glasgow is gratefully acknowledged.Notes and references
- (a) N. N. Makhova, L. I. Belen'kii, G. A. Gazieva, I. L. Dalinger, L. S. Konstantinova, V. V. Kuznetsov, A. N. Kravchenko, M. M. Krayushkin, O. A. Rakitin, A. M. Starosotnikov, L. L. Fershtat, S. A. Shevelev, V. Z. Shirinian and V. N. Yarovenko, Russ. Chem. Rev., 2020, 89, 55 CrossRef CAS; (b) V. Jaiswal, B. Mondal and J. Saha, Asian J. Org. Chem., 2020, 9, 1466 CrossRef CAS; (c) S. Liu, G.-J. Deng and H. Huang, Synlett, 2020, 32, 142 Search PubMed.
- (a) N. Svenstrup and J. Becher, Synthesis, 1995, 215 CrossRef CAS; (b) S. Pathania, R. K. Narang and R. K. Rawal, Eur. J. Med. Chem., 2019, 180, 486 CrossRef CAS PubMed; (c) K. Laxmikeshav, P. Kumari and N. Shankaraiah, Med. Res. Rev., 2022, 42, 513 CrossRef CAS PubMed.
- M. Li, X. Cai, Z. Qiao, K. Liu, W. Xie, L. Wang, N. Zheng and S.-J. Su, Chem. Commun., 2019, 55, 7215 RSC.
- S. Ionescu, D. Gavriliu, O. Maior and M. Hillebrand, J. Photochem. Photobiol., A, 1999, 124, 67 CrossRef CAS.
- R. Kikumoto, Y. Tamao, K. Ohkubo, T. Tezuka and S. Tonomura, J. Med. Chem., 1980, 23, 1293 CrossRef CAS PubMed.
- (a) M. Harfenist, D. M. Joseph, S. C. Spence, D. P. C. McGee, M. D. Reeves and H. L. White, J. Med. Chem., 1997, 40, 2466 CrossRef CAS PubMed; (b) M. Harfenist, D. P. C. McGee, M. D. Reeves and H. L. White, J. Med. Chem., 1998, 41, 2118 CrossRef CAS PubMed.
- C. T. Supuran, A. Scozzafava, F. Briganti, G. Loloiu and O. Maior, Eur. J. Med. Chem., 1998, 33, 821 CrossRef CAS.
- (a) J. Wu, Z. Wang, X.-Y. Chen, Y. Wu, D. Wang, Q. Peng and P. Wang, Sci. China: Chem., 2020, 63, 336 CrossRef CAS; (b) F. Juliá, Q. Shao, M. Duan, M. B. Plutschack, F. Berger, J. Mateos, C. Lu, X.-S. Xue, K. N. Houk and T. Ritter, J. Am. Chem. Soc., 2021, 143, 16041 CrossRef PubMed.
- G. M. Bennett, M. S. Lesslie and E. E. Turner, J. Chem. Soc., 1937, 444 RSC.
- (a) K. Komeyama, K. Aihara, T. Kashihara and K. Takaki, Chem. Lett., 2011, 40, 1254 CrossRef CAS; (b) P. Saravanan and P. Anbarasan, Org. Lett., 2014, 16, 848 CrossRef CAS PubMed; (c) S. Yang, B. Feng and Y. Yang, J. Org. Chem., 2017, 82, 12430 CrossRef CAS PubMed; (d) N. Liu, F. Chao, Y. Huang, Y. Wang, X. Meng, L. Wang and X. Liu, Tetrahedron Lett., 2019, 60, 151259 CrossRef CAS.
- F. Hu, X. Zhao, Y. Li, Y. Feng and C. Ma, Synthesis, 2013, 45, 966 CrossRef CAS.
- B. Luo, Q. Cui, H. Luo, Y. Hu, P. Huang and S. Wen, Adv. Synth. Catal., 2016, 358, 2733 CrossRef CAS.
- K. Kanemoto, Y. Sakata, T. Hosoya and S. Yoshida, Chem. Lett., 2020, 49, 593 CrossRef CAS.
- (a) M. C. Henry, H. M. Senn and A. Sutherland, J. Org. Chem., 2019, 84, 346 CrossRef CAS PubMed; (b) M. C. Henry and A. Sutherland, Org. Lett., 2020, 22, 2766 CrossRef CAS PubMed; (c) M. C. Henry, V. M. Abbinante and A. Sutherland, Eur. J. Org. Chem., 2020, 2819 CrossRef CAS.
- A. C. Dodds and A. Sutherland, J. Org. Chem., 2021, 86, 5922 CrossRef CAS PubMed.
- For other methods of phenol thioarylation, see: (a) C. D. Prasad, S. J. Balkrishna, A. Kumar, B. S. Bhakuni, K. Shrimali, S. Biswas and S. Kumar, J. Org. Chem., 2013, 78, 1434 CrossRef CAS PubMed; (b) H. Tian, C. Zhu, H. Yang and H. Fu, Chem. Commun., 2014, 50, 8875 RSC; (c) S. Alazet and T. Billard, Synlett, 2015, 26, 76 CrossRef CAS; (d) T. Hostier, V. Ferey, G. Ricci, D. G. Pardo and J. Cossy, Org. Lett., 2015, 17, 3898 CrossRef CAS PubMed.
- The Gustafson group have reported Lewis base catalysed thioarylation reactions using a thiourea and a diarylselenide: (a) C. J. Nalbandian, E. M. Miller, S. T. Toenjes and J. L. Gustafson, Chem. Commun., 2017, 53, 1494 RSC; (b) C. J. Nalbandian, Z. E. Brown, E. Alvarez and J. L. Gustafson, Org. Lett., 2018, 20, 3211 CrossRef CAS PubMed.
- While the copper-mediated cyclisation step would be faster using iodide rather than bromide intermediates, the iodobenzenethiol required to generate the iodinated biaryl sulfide is not readily available and is non-trivial to synthesise. Cyclisation of bromide intermediates 4 does require 18 hours reaction times, but the 2-bromobenzenethiol is inexpensive and readily available, and the cyclisations with these intermediates are high yielding.
- C. A. Horiuchi, A. Haga and J. Y. Satoh, Bull. Chem. Soc. Jpn., 1986, 59, 2459 CrossRef CAS.
- (a) D. T. Racys, C. E. Warrilow, S. L. Pimlott and A. Sutherland, Org. Lett., 2015, 17, 4782 CrossRef CAS PubMed; (b) D. T. Racys, S. A. I. Sharif, S. L. Pimlott and A. Sutherland, J. Org. Chem., 2016, 81, 772 CrossRef CAS PubMed.
- (a) S. Antoniotti, V. Dalla and E. Duñach, Angew. Chem., Int. Ed., 2010, 49, 7860 CrossRef CAS PubMed; (b) M. J. Earle, U. Hakala, B. J. McAuley, M. Nieuwenhuyzen, A. Ramani and K. R. Seddon, Chem. Commun., 2004, 1368 RSC.
- W.-C. Gao, T. Liu, B. Zhang, X. Li, W.-L. Wei, Q. Liu, J. Tian and H.-H. Chang, J. Org. Chem., 2016, 81, 11297 CrossRef CAS PubMed.
- H. A. Stevenson and S. Smiles, J. Chem. Soc., 1931, 718 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all compounds. See DOI: 10.1039/d2ob00022a |
| This journal is © The Royal Society of Chemistry 2022 |