
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a synthetic equivalent of α,α-dicationic acetic acid leading to unnatural amino acid derivatives via tetrafunctionalized methanes†
Haruyasu
Asahara
 *ab,
Atsushi
Bonkohara
a,
Masaya
Takagi
a,
Kento
Iwai
ac,
Akitaka
Ito
ac,
Kotaro
Yoshioka
d,
Shinki
Tani
d,
Kazuto
Umezu
d and
Nagatoshi
Nishiwaki
*ac
*ab,
Atsushi
Bonkohara
a,
Masaya
Takagi
a,
Kento
Iwai
ac,
Akitaka
Ito
ac,
Kotaro
Yoshioka
d,
Shinki
Tani
d,
Kazuto
Umezu
d and
Nagatoshi
Nishiwaki
*ac
aSchool of Environmental Science and Engineering, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan. E-mail: nishiwaki.nagatoshi@kochi-tech.ac.jp
bGraduate School of Pharmaceutical Sciences, Osaka University, Yamadaoka 1-6, Suita, Osaka 565-0871, Japan
cResearch Center for Molecular Design, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan
dKumiai Chemical Industry Co. Ltd., Nakanogo, Fuji, Shizuoka 421-3306, Japan
First published on 25th February 2022
Abstract
Diethyl mesoxalate (DEMO) exhibits high electrophilicity and accepts the nucleophilic addition of a less nucleophilic acid amide to afford N,O-hemiacetal. However, our research showed that elimination of the amide moiety proceeded more easily than dehydration upon treatment with a base. This problem was overcome by reacting DEMO with an acid amide in the presence of acetic anhydride to efficiently obtain N,O-acetal. Acetic acid was eliminated leading to the formation of N-acylimine in situ upon treatment with the base. N-Acylimine is also electrophilic, accepting the second nucleophilic addition by pyrrole or indole to form α,α-disubstituted malonates. Subsequent hydrolysis followed by decarboxylation resulted in (α-indolyl-α-acylamino)acetic acid formation; homologs of tryptophan. Through this process, DEMO serves as a synthetic equivalent of α,α-dicationic acetic acid to facilitate nucleophilic introduction of the two substituents.
Introduction
Malonic ester synthesis is an important tool for elaborate organic synthesis. The acidic methylene group readily generates enolate, which reacts with up to two electrophiles, and subsequent hydrolysis and decarboxylation lead to the formation of α,α-disubstituted acetic acids (Scheme 1, upper).1,2 Through this process, diethyl malonate serves as a synthetic equivalent of α,α-dianionic acetic acid (Fig. 1, upper). In contrast, the central carbonyl carbon of DEMO (diethyl mesoxalate, diethyl oxomalonate, diethyl ketomalonate), one of the vicinal tricarbonyl compounds, is highly electrophilic and reacts with versatile nucleophiles to afford hemiacetals (Fig. 2, path a).3,4 High electrophilicity facilitates the nucleophilic addition of acid amide, which is considered a masked amino group but is not generally applied as a nucleophile.4 If α,α-disubstituted malonates can be prepared from DEMO by successive nucleophilic reactions, α,α-disubstituted acetic acid can be obtained upon hydrolysis followed by decarboxylation (Scheme 1, lower). In this process, two types of substituents are introduced nucleophilically, indicating that DEMO acts as a synthetic equivalent of α,α-dicationic acetic acid (Fig. 1, lower).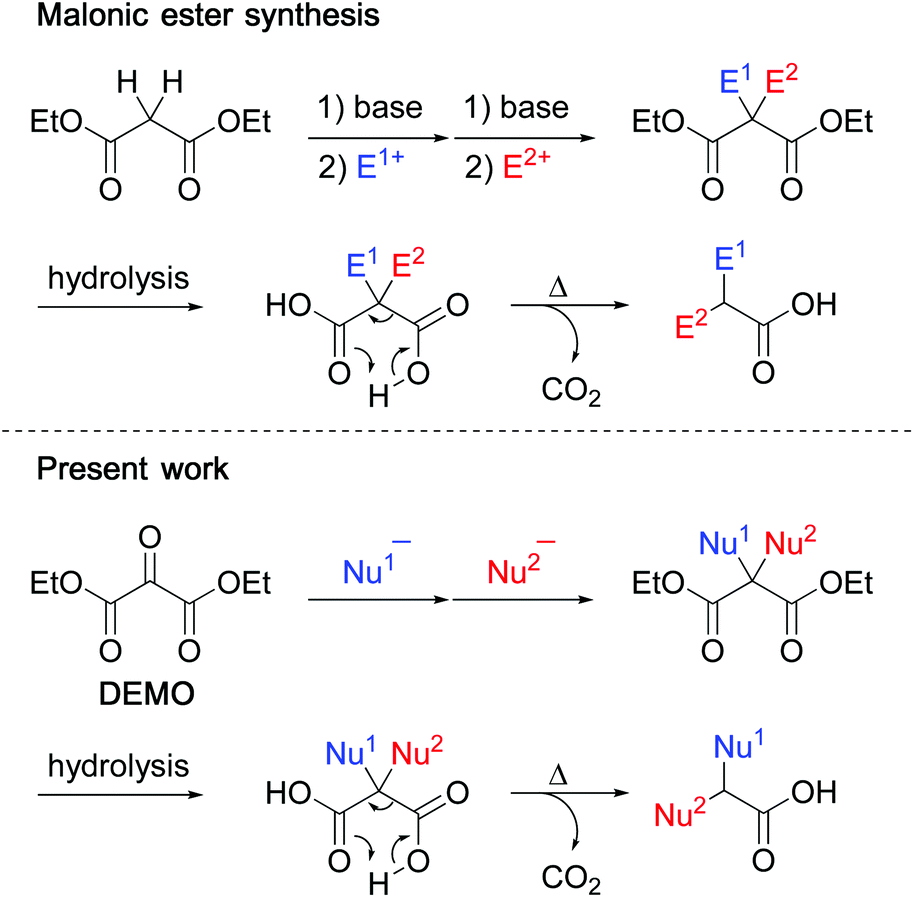 | ||
| Scheme 1 Synthetic schemes of α,α-disubstituted acetic acids using diethyl malonate (upper) and using DEMO (lower). | ||
 | ||
| Fig. 1 Synthetic equivalents of α,α-dianionic and α,α-dicationic acetic acids. | ||
 | ||
| Fig. 2 Synthetic equivalents of α,α-dianionic and α,α-dicationic acetic acids. | ||
Numerous organic reactions using DEMO have been reported in the literature, and can be categorized into several types (Fig. 2). In the reaction of DEMO with a dinucleophile, a new ring is formed between the central carbon and the adjacent ester functionality in almost all cases (path b).5 Double addition to the central carbonyl group of DEMO, it is limited to intramolecular ring closure (path c);6 however, to the best of our knowledge, the introduction of two substituents via successive double nucleophilic addition to the central carbon has not been performed. The chemical conversion of hemiacetals has been extensively studied. When reagents possess nucleophilic and electrophilic sites, the formed hemiacetal attacks the electrophilic site of the nucleophile via dipolar cycloaddition (path d).7 Conversely, dehydration of the hemiacetal forms an electron-deficient double bond (path e),8–10 which subsequently accepts the addition of less nucleophilic reagents, such as pyrrole.10
We focused on N-acylimines (Y = N, R = acyl), which can accept nucleophilic addition to form α,α-disubstituted malonates. However, the synthesis of N-acylimines involves multi-step reactions: nitrosoation at the α-position of malonate,11 reduction to an amino group,11N-acylation, α-bromination12 and dehydrobromination.12 Although N-acetylimine is directly prepared from DEMOvia the aza-Wittig reaction, the N-substituent is limited to an acetyl group.9 In our previous work, we demonstrated that DEMO efficiently reacted with versatile acid amides to afford the corresponding hemiacetals,4 which serves as precursors of N-acylimines modifiable by the acyl group upon dehydration.
Considering the above-mentioned background and our previous work,4,10 we planned a new synthetic strategy for unnatural amino acid derivatives using double nucleophilic addition to DEMO. The high electrophilicity of DEMO allows the attack of less nucleophilic acid amides, which facilitates the introduction of protected amino groups in a single step.4 Dehydration of the obtained hemiacetals facilitates the formation of N-acylimines possessing versatile acyl groups, which are subjected to reactions with second nucleophiles such as pyrroles and indoles.10 Subsequent hydrolysis, followed by decarboxylation, leads to the formation of N-protected unnatural amino acids.
Oligopeptides are widely used in medicinal chemistry; however, the diversity of the resulting framework is limited when natural amino acids are used as building blocks. Hence, the development of facile methods for the synthesis of unnatural amino acids has gained attention recently. Amino acids are generally synthesized by connecting small molecules via new bond formation (Fig. 3, methods a–c). Method a involves the substitution of α-substituted α-bromoacetic acid with ammonia or ammonium salt.13 Method b, the Strecker reaction,14 provides amino acids by the addition of cyanide to the α-substituted imine, followed by hydrolysis of the cyano group. In these cases, precursors possessing a substituent at the α-position should be prepared beforehand, which is a significant disadvantage when constructing a compound library. Reactions of masked amino acids (hydantoin) with aldehydes or ketones (method c) facilitate the modification of the α-substituents; however, it is necessary to use a strong base and the subsequent hydrogenation limits usable substrates possessing a functional group.15 A combination of methods b and c, referred as the Bucherer–Bergs reaction, also furnishes 5,5-disubstituted hydantoins.16 In contrast, our synthetic method (method d) achieved the synthesis of amino acid derivatives by decarboxylation17 of α,α-disubstituted malonate, in which the α-substituent is easily modified by altering the nucleophile. Because α,α-disubstituted malonate has multiple coordination sites, control of stereochemistry is expected to be easier than methods a–c.
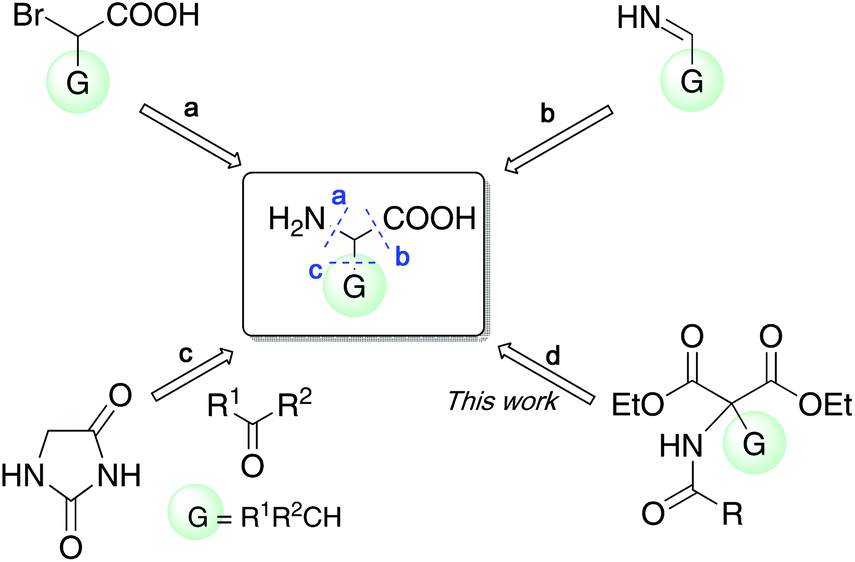 | ||
| Fig. 3 Commonly used synthetic methods for α-substituted amino acids. | ||
Results and discussion
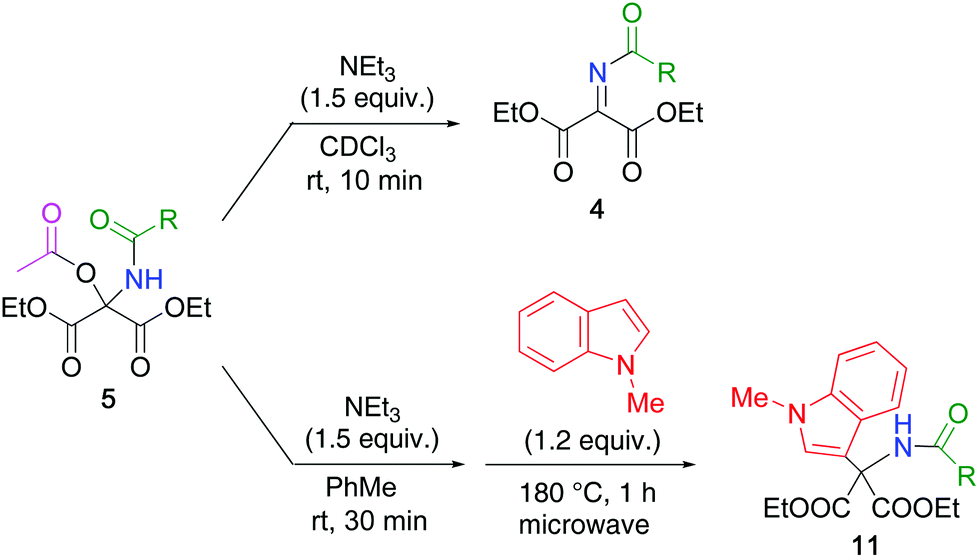
4-Methylbenzamide 1a (Scheme 1, R = 4-MeC6H4) was selected as a model substrate for the first nucleophile. Upon heating at 60 °C for 18 h in acetonitrile, DEMO reacted with 1a to afford N,O-hemiacetal 2a with 92% yield. When 2a was subjected to the reaction with the second nucleophile, pyrrole 3, under the same conditions, no reaction proceeded, presumably due to the congested structure and low electrophilicity of 2a. Thus, the dehydration of 2a leading to N-acylimine 4a was studied. Although microwave heating (at 140 °C) and treatment with acids (BF3·OEt2, H2SO4), bases (NEt3, K2CO3, tert-BuOK), or molecular sieves 3 Å were attempted, the recovery of 2a or elimination of amide 1a yielding DEMO was observed without any detectable 4a. The low reactivity of 2a was due to the reduced elimination ability and high acidity of the hydroxy group. This disadvantage was addressed by using the reaction between DEMO and 1a in the presence of acetic anhydride. Although the less nucleophilic 1a cannot attack acetic anhydride, the formed N,O-hemiacetal 2a can attack it by mooring the amide functionality to afford N,O-acetal 5a (Scheme 2). However, only trace amounts of 5a were detected, which was due to the competitive hydration of DEMO leading to the formation of gem-diol (Table 1, entry 1). The addition of molecular sieves 3 Å considerably increased the yield, and 5a was quantitatively obtained when two equivalents of acetic anhydride were utilized (entries 2 and 3). | ||
| Scheme 2 Synthesis of N,O-acetal 5 by nucleophilic addition of amide 1 to DEMO in the presence of acetic anhydride. | ||
|
|
|||
|---|---|---|---|
| Ac2O/equiv. | Additive | Yield of 5a/% | |
| 1 | 1 | — | 4 |
| 2 | 1 | Molecular sieves 3 Å | 72 |
| 3 | 2 | Molecular sieves 3 Å | Quant. |
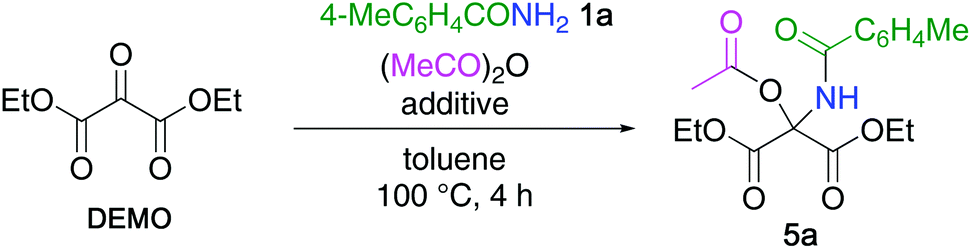
The preparative method was applied to other acid amides 1b–j (R = Ph 1b, C6F51c, Me 1d, Et 1e, Pr 1f, Me(CH2)101g, tert-Bu 1h, CF31i, tert-BuO 1j) to quantitatively afford the corresponding N,O-acetals 5b–j, except for 5i (94% yield). It is noteworthy that the reaction proceeded efficiently even in the case of highly electron-deficient amides such as pentafluorobenzamide 1c and trifluoroacetamide 1i, and bulky pivalamide 1h. In addition, this protocol enabled the direct introduction of Boc-protected amino group by using urethane 1j.
Next, the formation of N-acylimine 4 was investigated (Scheme 3). When N,O-acetal 5a was treated with triethylamine at room temperature in chloroform-d, acetic acid was easily eliminated, leading to N-acylimine 4ain situ, which was confirmed by the 1H and 13C NMR spectra. Other N-acylimines, 4i and 4j, were similarly obtained from 5i and 5j, respectively.
 | ||
| Scheme 3 Conversion of N,O-acetal 5 to N-acylimine 4. | ||
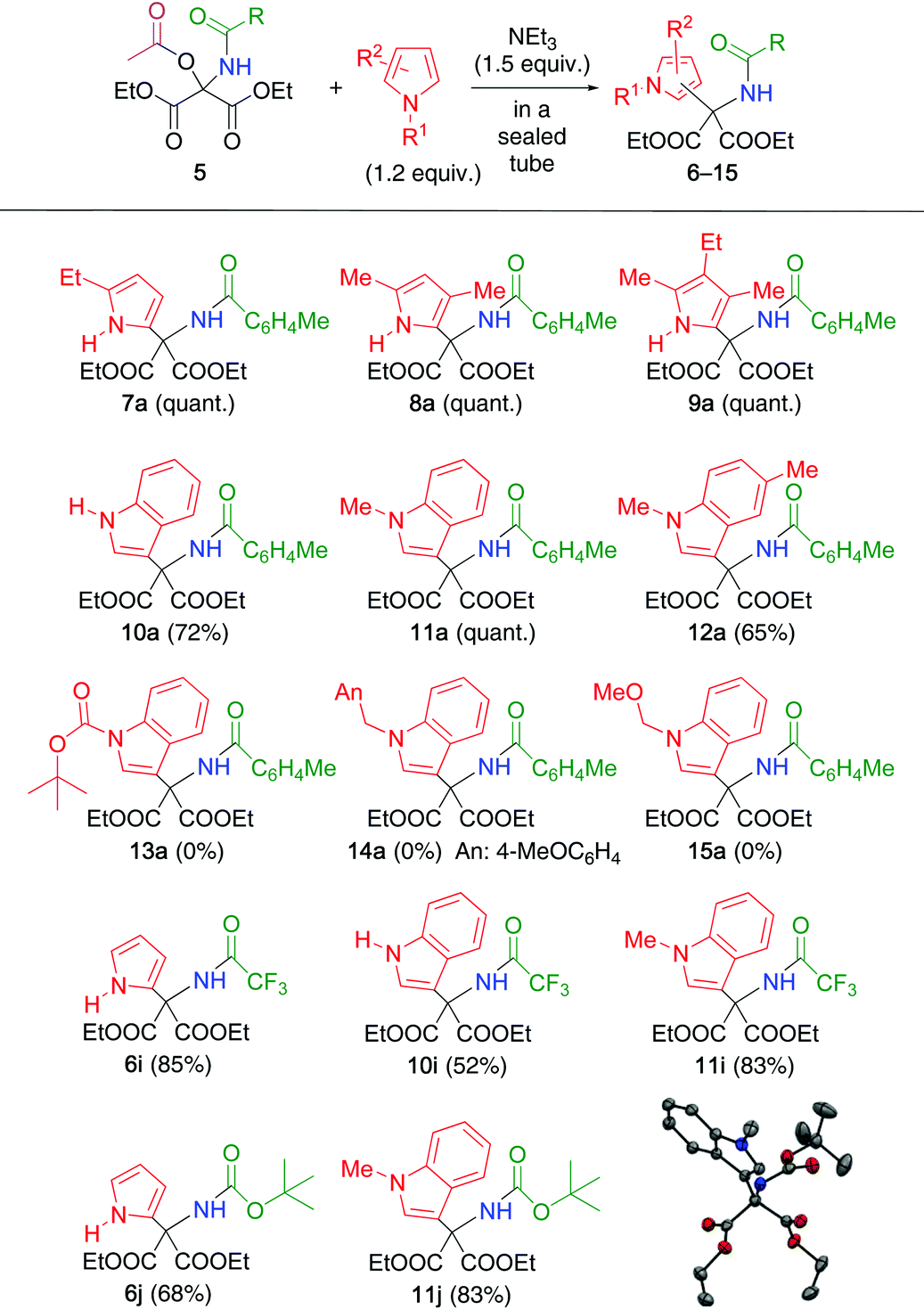
The generated N-acylimines 4 were subjected to a second nucleophilic addition in one-pot without the isolation of 4 because they gradually hydrolyze leading to DEMO and amide 1. To a solution of N,O-acetal 5a in toluene, were added triethylamine and pyrrole 3, and the resulting mixture was heated at 50 °C for 1 d in a sealed tube to produce the double adduct 6a with 26% yield (Table 2, entry 1). Elevating the temperature and using 1.2 equiv. of 3 increased reaction efficiency (entries 2–5). Among the solvents tested, toluene was the most suitable (entries 6–8). Consequently, the reaction conditions in entry 4 were determined to be optimal.

In the series of reactions listed in Table 2, the reaction mixture was washed with hydrochloric acid to remove the base, during which adduct 6a was obtained without decomposition. Approximately half of the 6a decomposed to DEMO and amide 1a upon treatment with column chromatography on silica gel. This was presumably because of the difference in time under acidic conditions. A plausible mechanism for this phenomenon is shown in Scheme 3. After prototropy from 1-position to 2-position of the pyrrole ring, pyrrole 3 was eliminated to afford DEMO and benzamide 1avia the hydrolysis of the intermediately formed N-acylimine 4a (Scheme 4).
 | ||
| Scheme 4 A plausible mechanism for the decomposition of double adduct 6a. | ||
The optimized conditions were applied to other N,O-acetals 5 and pyrroles/indoles (Table 3). Although double adducts 6–11 were purified by extraction or recrystallization, it was sometimes difficult, depending on the structure of the substrates 1 and pyrroles/indoles. Alkyl-substituted pyrroles efficiently underwent nucleophilic addition to 5a, leading to adducts 7a–9a because of the high electron density on the pyrrole ring. Indole (benzo[b]pyrrole) reacted with 5a to produce adduct 10a, which was also unstable under acidic conditions. In this case, prototropy from the 1-position to the 3-position of the indole ring was a key step. Adducts 11a and 12a obtained from the corresponding N-methylindoles, respectively, were stable for purification by using column chromatography on silica gel. Hence, other N-protected indoles were employed; however, adducts 13a–15a were not detected because of the electron-withdrawing properties and steric hindrance of the N-substituent. N-Trifluoroacetyl and N-Boc N,O-acetals 5i and 5j, respectively, exhibited similar reactivities to furnish the corresponding double adducts 6 and 11.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| a A toluene solution of N,O-acetal 5 and pyrrole/indole (1.2 equiv.), and triethylamine (1.5 equiv.) was heated at 120 °C for 3 h in a sealed tube. The yields were determined by 1H NMR. |
|---|
|
Surprisingly, the reaction of N-acetyl-N,O-acetal 5d with N-methylindole resulted in only trace amounts of adduct 11d, even though severe reaction conditions were utilized (Table 4, entry 1). When the reaction was monitored by 1H NMR, only a small amount of N-acylimine 4d was detected, which indicates that the yield of 11d has a parallel relationship with the efficiency generation of 4d. Interestingly, the longer the alkyl group, the higher the yield of 11 (entries 1–4), and the bulkiness of the alkyl group was found to increase the reactivity of N,O-acetal 5. N,O-Acetal 5h derived from pivalamide 1h exhibited considerably higher reactivity to afford 11h with 57% yield (entry 5).
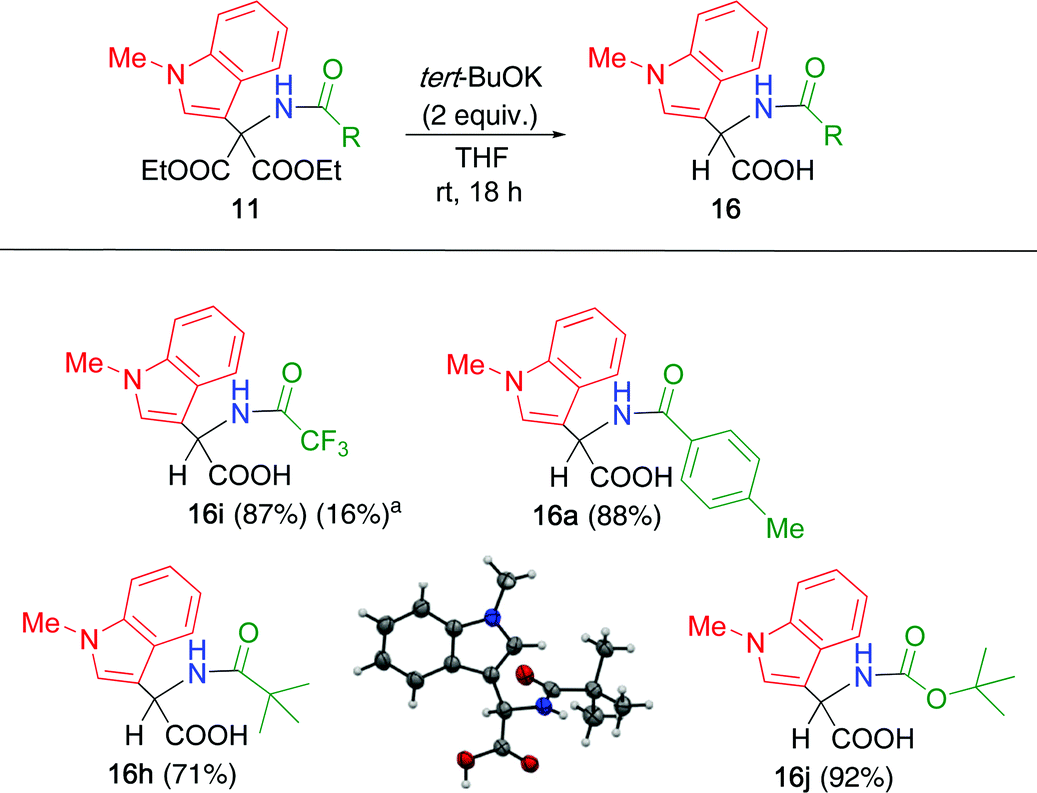
To gain insight into this steric effect, DFT calculations were conducted for 5d (R = Me), 5h (R = tert-Bu), and 5k (R = Bu); however, notable differences in bond lengths and bond angles between them were not confirmed. Although no clear evidence was obtained, one possible reason is as follows: when the bulky N-acyl group repels with ethoxycarbonyl groups, the N–H and acetyl groups are closed together, which accelerates the elimination of acetic acid (Fig. 4).
 | ||
| Fig. 4 Elimination of acetic acid leading to N-acylimine 4. | ||
The synthesized α,α-disubstituted malonates 11 were subjected to hydrolysis under various conditions. In the cases of 6a and 10a, all attempts failed because the presence of the N–H group caused prototropy followed by elimination of the heteroaromatic moiety, as shown in Scheme 3. However, such decomposition was not observed for 11a derived from N-methylindole. Hence, the reaction conditions were investigated using 11 (Table 5). When the double adduct 11a was treated with sodium hydroxide in ethanol, hydrolysis and subsequent decarboxylation proceeded quantitatively to afford α,α-disubstituted acetic acid 16a (entry 1). Boc-substituted adduct 11j also supplied only decarboxylated product 16j without any detectable malonic acid (entry 2). In the case of trifluoroacetyl adduct 11i, only a complex mixture was obtained under both the reaction conditions (entries 3 and 4).

The reaction conditions were tested again using 11i, which is more reactive than 11a and 11j. Several bases and solvents were evaluated, among which a combination of potassium tert-butoxide and THF was effective for hydrolysis, followed by decarboxylation, leading to 16i with 87% yield (Table 6). In this reaction, the water contained in THF acted as a source of hydroxide. The yield considerably decreased when the same reaction was conducted under the same conditions in an argon atmosphere using dry THF as the solvent. Optimized conditions were applied to the other adducts 11. Toluoyl-substituted adduct 11a was efficiently converted to 16a. Adducts possessing a bulky acyl group, such as pivaloyl and tert-butoxycarbonyl (Boc) groups, underwent hydrolysis and decarboxylation to produce the corresponding α,α-disubstituted acetic acids 16h and 16j, respectively. The α-amino acid derivatives 16 are homologs of tryptophan, which is expected to be useful for identifying new biologically active compounds.
| a Dry THF was used in an argon atmosphere. |
|---|
|
This protocol facilitates modification of the acyl group by altering amide 1. Synthesis of dipeptide was attempted using this feature (Scheme 5). When DEMO was reacted with L-prolineamide 1l in the presence of acetic anhydride, protection of not only the hydroxy group but also the ring nitrogen of the pyrrolidine was achieved. This is because amine can attack anhydride while amide cannot, by which doubly acetylated N,O-acetal 5l was obtained with 88% yield. After the generation of N-acylimine 4l by triethylamine in situ, a second nucleophilic addition by N-methylindole formed 11l with 75% yield. Subsequent treatment with potassium tert-butoxide in THF resulted in the hydrolysis of ester functions, followed by decarboxylation, and resulted in the dipeptide 16l with 84% yield. In the 1H NMR of 16l, signals of four kinds of isomers were observed. When this compound was subjected to the measurement of temperature-variable NMR spectra, each type of signals coalesced (see ESI†). Hence, these isomers are not diastereomers caused by two chiral centers but rotamers caused by two amide functions.
 | ||
| Scheme 5 Synthesis of amino acid derivative 7Lf using prolineamide 1L. | ||
Conclusions
A new approach for unnatural amino acid derivatives was demonstrated using DEMO. The high electrophilicity of DEMO facilitates the acceptance of the double nucleophilic addition via N-acylimine 4. In this protocol, an even less nucleophilic acid amide can be used as the first nucleophile, and pyrroles and indoles 3 are used as the second nucleophile, which produced α,α-disubstituted malonates 6–12. Subsequent hydrolysis followed by decarboxylation furnished (α-indolyl-α-acylamino)acetic acids 11, which are homologs of tryptophan. Through this process, DEMO served as a synthetic equivalent of α,α-dicationic acetic acid to nucleophilically introduce two substituents. The application of this method by using other nucleophiles is currently being studied, and the results will be shown in subsequent papers.Experimental section
General
All reagents were purchased from commercial sources and used without further purification. Dry acetonitrile was also purchased from commercial source and used as received. 1H and 13C NMR spectra were recorded on Bruker DPX-400 and JEOL JMN-ECZ400S spectrometers (400 MHz and 100 MHz, respectively) using TMS as an internal standard. The assignments of the 13C NMR were performed by DEPT experiments. IR spectra were recorded on a JASCO FT/IR-4200 spectrometer equipped with an ATM detector. High-resolution mass spectra were obtained on an AB SCEIX Triplet TOF 4600 mass spectrometer. Melting points were recorded on an SRS-Optimelt automated melting point system and were uncorrected. HPLC analysis was directly performed with chiral stationary phase column, DAICEL CHIRALPAK or CHIRALCEL. Diffraction data were collected at 93 K under a cold N2-gas stream on a Rigaku XtaLAB Synergy-S/Mo system (λ = 0.71073 Å (Mo-Kα)). The integrated data were analyzed by using an Olex2 crystallographic software package.18 The structures were solved with the ShelXT structure solution program19 using Intrinsic Phasing and refined with the ShelXL refinement package20 using the least-squares minimization. Anisotropic refinement was performed for all non-hydrogen atoms, and all the hydrogen atoms were put at calculated positions.18–20 The geometrical optimization was carried out for at the B3LYP/6-31 g(d,p) level of theory implemented on Gaussian 09 package.21General procedure for synthesis of N,O-acetal 5
To a solution of DEMO (0.87 g, 5.0 mmol) in toluene (20 mL), 4-methylbenzamide 1a (0.81 g, 6.0 mmol), molecular sieves 3A (1.7 g) and acetic anhydride (1.1 g, 10 mmol) were added, and the resultant mixture was heated at 100 °C for 4 h. After filtration of molecular sieves, the filtrate was washed with water (30 mL × 2), dried over magnesium sulfate, and concentrated in vacuo to afford N,O-acetal 5a (1.36 g, 4.94 mmol, yield 99%) as a white solid.For the synthesis of other N,O-acetals 5b–j, the same experiments were conducted.
General procedure for synthesis of double adduct 6
In a screw capped test-tube, a toluene solution (3 mL) of N,O-acetal 5a (140 mg, 0.4 mmol), triethylamine (84 μL, 0.6 mmol), and 1-methylindole (63 mg, 0.48 mmol) was heated at 120 °C for 3 h. The resultant solution was poured into chloroform (50 mL), and 12 M hydrochloric acid (1 mL, 12 mmol) was added, then, washed with water (50 mL × 2). The organic layer was dried over magnesium sulfate and concentrated. The residue was washed with hexane (100 mL) to afford double adduct 11a (169 mg, 0.4 mmol, yield quant.) as a white solid. Further purification was performed by recrystallization from mixed solvent (Et2O–hexane).The experimental procedure were conducted in the same way for the synthesis of other double adducts 6–12.
General procedure for hydrolysis and decarboxylation of 11a
To a solution of double adduct 11a (21 mg, 0.05 mmol) in THF (2 mL), potassium tert-butoxide (11 mg, 0.10 mmol) was added, and the resultant mixture was stirred at room temperature overnight. After evaporation, the residue was dissolved in chloroform (20 mL), and 12 M hydrochloric acid (1 mL, 12 mmol) was added, then, washed with water (10 mL). The organic layer was dried over magnesium sulfate and concentrated. The residue was washed with hexane (100 mL) to afford amino acid derivative 16a (14 mg, 0.044 mmol, yield 88%) as a white solid.The decarboxylation of other double adducts were also performed by the same experimental procedure.
Author contributions
Asahara and Nishiwaki came up with the idea for this study and were the main driving force behind it. Bonkohara, Takagi, and Iwai performed experiments. Iwai and Ito conducted X-ray crystallography and DFT calculations. Yoshioka, Tani, and Umezu supplied DEMO and discussed on the Experimental results.Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Sawada, T. Nakayama, N. Esaki, H. Tanaka, K. Soda and R. K. Hill, J. Org. Chem., 1986, 51, 3384 CrossRef CAS; H. R. Snyder and C. W. Smith, J. Am. Chem. Soc., 1944, 66, 350 CrossRef; R. Adams and C. S. Marvel, J. Am. Chem. Soc., 1920, 42, 310 CrossRef.
- Recent papers: I. Banerjee, K. C. Ghosh, E. Oheix, M. Jean, J.-V. Naubron, M. Réglier, O. Iranzo and S. Sinha, J. Org. Chem., 2021, 86, 2210 CrossRef CAS PubMed; Q.-K. Zhao, X. Wu, L.-P. Li, F. Yang, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2021, 23, 1675 CrossRef PubMed; R. Kucera, F. W. Goetzke and S. P. Fletcher, Org. Lett., 2020, 22, 2991 CrossRef PubMed.
- Recent papers: Y. Chen, H.-L. Wan, Y. Huang, S. Liu, F. Wang, C. Lu, J. Nie, Z. Chen, Y. Chen, G. Yang and C. Mao, Org. Lett., 2020, 22, 7797 CrossRef CAS PubMed; K. Hirano, K. Yoshioka, K. Umezu, T. Kagawa, Y. Sumi and N. Shibata, Chem. Lett., 2020, 49, 330 CrossRef; P. Mandal, D. Patra and R. Shunmugam, Polym. Chem., 2020, 11, 3340 RSC; G. Ragusa, M. Gomez-Canas, P. Morales, C. Rodriguez-Cueto, M. R. Pazos, B. Asproni, E. Cichero, P. Fossa, G. A. Pinna, N. Jagerovic, J. Fernandez-Ruiz and G. Murineddu, Eur. J. Med. Chem., 2017, 127, 398 CrossRef PubMed; K. Balaraman, R. Ding and C. Wolf, Adv. Synth. Catal., 2017, 359, 4165 CrossRef PubMed; L. J. Patalag, J. A. Ulrichs, P. G. Jones and D. B. Werz, Org. Lett., 2017, 19, 2090 CrossRef PubMed.
- H. Asahara, K. Inoue, S. Tani, K. Umezu and N. Nishiwaki, Adv. Synth. Catal., 2016, 358, 2817 CrossRef CAS.
- Recent papers: X. Wu, Z.-H. Zhu, H. He, L. Ren, C.-F. Zhu and Y.-G. Li, J. Org. Chem., 2020, 85, 6216 CrossRef CAS PubMed; Z. Wang, J. Shi, X. Zhu, W. Zhao, Y. Gong, X. Hao, Y. Hou, Y. Liu, S. Ding, J. Liu and Y. Chen, Bioorg. Chem., 2020, 105, 104371 CrossRef PubMed; B. Oyallon, M. Brachet-Botineau, C. Loge, P. Bonnet, M. Souab, T. Robert, S. Ruchaud, S. Bach, P. Berthelot, F. Gouilleux, M.-C. Viaud-Massuard and C. Denevault-Sabourin, Eur. J. Med. Chem., 2018, 154, 101 CrossRef PubMed; A. P. Dhondge, P.-C. Tsai, C.-Y. Nien, W.-Y. Xu, P.-M. Chen, Y.-H. Hsu, K.-W. Li, F.-M. Yen, S.-L. Tseng, Y.-C. Chang, H. J. H. Chen and M.-Y. Kuo, Org. Lett., 2018, 20, 2538 CrossRef PubMed; C. I. Briguglio, S. Piras, P. Corona, M. A. Pirisi, L. Burrai, G. Boatto, E. Gavini and G. Rassu, J. Heterocycl. Chem., 2016, 53, 1721 CrossRef.
- Recent papers: M. V. Campbell, A. V. Iretskii and R. A. Mosey, J. Org. Chem., 2020, 85, 11211 CrossRef CAS PubMed; S. Kimura and N. Saito, Tetrahedron, 2018, 74, 4504 CrossRef; P. A. Wender, M. S. Jeffreys and A. G. Raub, J. Am. Chem. Soc., 2015, 137, 9088 CrossRef PubMed; J. Tian, Q. Du, R. Guo, Y. Li, B. Cheng and H. Zhai, Org. Lett., 2014, 16, 3173 CrossRef PubMed; V. F. Mironov, G. A. Ivkova, L. M. Abdrakhmanova, L. M. Burnaeva, R. Z. Musin, S. V. Kharlamov and I. V. Konovalova, Russ. J. Org. Chem., 2012, 48, 306 CrossRef; R. R. Cesati III and J. A. Katzenellenbogen, Org. Lett., 2000, 2, 3635 CrossRef PubMed.
- Recent papers: J. R. Hwu, A. Panja, N. K. Gupta, Y.-C. Hu, K.-T. Tan, C.-C. Lin, K.-C. Hwang, M.-H. Hsu, W.-C. Huang and S.-C. Tsay, Eur. J. Org. Chem., 2021, 683 CrossRef CAS; L.-J. Chen, J.-H. Yu, B. He, J.-W. Xie, Y.-X. Liu and W.-D. Zhu, J. Org. Chem., 2020, 85, 7793 CrossRef PubMed; Y.-H. Deng, W.-D. Chu, Y.-H. Shang, K.-Y. Yu, Z.-L. Jia and C.-A. Fan, Org. Lett., 2020, 22, 8376 CrossRef PubMed; Y.-L. Zhang, R.-T. Guo, H. Luo, X.-S. Liang and X.-C. Wang, Org. Lett., 2020, 22, 5627 CrossRef PubMed; H. Asahara, S. Kikuchi, Y. Unno, S. Yokoyama, K. Yoshioka, S. Tani, K. Umezu and N. Nishiwaki, Curr. Org. Chem., 2019, 23, 97 CrossRef; C.-C. Xie, R. Tan and Y.-K. Liu, Org. Chem. Front., 2019, 6, 919 RSC; X.-B. Ding, D. P. Furkert and M. A. Brimble, Angew. Chem., Int. Ed., 2019, 58, 11830 CrossRef PubMed.
- Recent papers: Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216 CrossRef CAS PubMed; L. Zhou, X.-D. An, S. Yang, X.-J. Li, C.-L. Shao, Q. Liu and J. Xiao, Org. Lett., 2020, 22, 776 CrossRef PubMed; B. Li, J. Liu, F. Gao, M. Sun, Y. Guo, Y. Zhou, D. Wen, Y. Deng, H. Chen, K. Wang and W. Yan, Org. Biomol. Chem., 2019, 17, 2892 RSC; A. Kondoh and M. Terada, Org. Lett., 2018, 20, 5309 CrossRef PubMed.
- S. Sano, H. Shimizu, K. Kim, W. S. Lee, M. Shiro and Y. Nagao, Chem. Pharm. Bull., 2006, 54, 196 CrossRef CAS PubMed.
- H. Asahara, S. Kawakami, K. Yoshioka, S. Tani, K. Umezu and N. Nishiwaki, Bull. Chem. Soc. Jpn., 2018, 91, 1715 CrossRef CAS.
- J. Liu, Y. Li, M. Ke, M. Liu, P. Zhan, Y.-C. Xiao and F. Chen, J. Org. Chem., 2020, 85, 15360 CrossRef CAS PubMed.
- A. V. Prosyanik, D. V. Fedoseenko and V. I. Markov, Zh. Org. Khim., 1985, 21, 1637 CAS.
- Recent papers: N. Sakai, K. Sasaki, H. Suzuki and Y. Ogiwara, Synthesis, 2020, 1823 CrossRef CAS; N. Kaplaneris, C. Spyropoulos, M. G. Kokotou and C. G. Kokotos, Org. Lett., 2016, 18, 5800 CrossRef PubMed; H. Biava and N. Budisa, J. Fluorine Chem., 2013, 156, 372 CrossRef.
- Reviews: W. Masamba, Molecules, 2021, 26, 1707 CrossRef CAS PubMed; P. Galatsis, Strecker amino acid synthesis” in “Name Reactions for Functional Group Transformations, ed. J. J. Li and E. J. Corey, John Wiley & Sons, Hoboken, US, 2007, pp. 477–499 Search PubMed; Y. Larry, Recent developments in catalytic asymmetric Strcker-type reactions, in Organic Synthesis Highlights V, ed. H.-G. Schmaltz and T. Wirth, John Wiley & Sons, Hoboken, US, 2003 Search PubMed.
- Recent papers: Z. Imani, V. R. Mundlapati, G. Goldsztejn, V. Brenner, E. Gloaguen, R. Guillot, J.-P. Baltaze, K. Le Barbu-Debus, S. Robin, A. Zehnacker, M. Mons and D. J. Aitken, Chem. Sci., 2020, 11, 9191 RSC; L. A. Hardegger, F. Mallet, B. Bianchi, C. Cai, A. G.-G. Perrenoud, R. Humair, R. Kaehny, S. Lanz, C. Li, J. Li, F. Rampf, L. Shi, C. Spoendlin, J. Stauble, S. Teng, X. Tian, B. Wietfeld, Y. Yang, B. Yu, C. Zepperitz, X. Zhang and Y. Zhang, Org. Process Res. Dev., 2020, 24, 1743 CrossRef CAS; J. Yu, J. Li, S. Cao, T. Wu, S. Zeng, H. Zhang, J. Liu and Q. Jiao, Catal. Commun., 2019, 120, 28 CrossRef.
- Recent papers: M. Kalník, P. Gabko, M. Bella and M. Koós, Molecules, 2021, 26, 4024 CrossRef CAS PubMed; O. A. Kolyamshin, Y. N. Mitrasov, V. A. Danilov and A. N. Vasil'ev, Russ. J. Gen. Chem., 2021, 91, 1459 CrossRef.
- H. Fu, P. Li, Z. Wang, X. Li, Q. Dai and C. Hu, Org. Biomol. Chem., 2020, 18, 4439 RSC; T. Schneider, V. Kubyshkin and N. Budisa, Eur. J. Org. Chem., 2018, 2053 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data and copies of NMR spectra of compounds 5–12 and 16, crystallographic data of 11j and 16h, and computational study for 5. CCDC 2130060 and 2130061. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob02482e |
| This journal is © The Royal Society of Chemistry 2022 |