
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColour and constitution of conjugate bases of benzodifurantrione, its ring-opened derivatives and benzodifuranone dye analogues†
Michael G.
Hutchings
 *a,
Anthony J.
Lawrence
a and
Alan R.
Kennedy
b
*a,
Anthony J.
Lawrence
a and
Alan R.
Kennedy
b
aDepartment of Chemistry, University of Manchester, Manchester M13 9PL, UK. E-mail: mghutchings@btinternet.com
bDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow G1 1XL, UK
First published on 10th March 2022
Abstract
The observation of ready deprotonation of the phenylogous enol of benzodifurantrione (BDT) to give a bright violet conjugate base has led to two follow up explorations. Extension of BDT enol by insertion of a p-phenylene unit into the enol C–O bond gives the known bright red 4-hydroxylated benzodifuranone dyes. Their deprotonation results in previously unreported near infrared-absorbing conjugate bases. These appear to aggregate in solution, the more so in less polar solvents. Ring-opened derivatives of BDT containing α-dicarbonyl substituents also give coloured conjugate bases, but α-keto-ester and -anilide derivatives differ substantially (ester yellow; anilide intense red). Investigation of this nonintuitive difference leads to the conclusion that while the anilide is essentially planar the ester is nonplanar. The contrast in conformation impacts on the auxochromic effects of the otherwise closely related α-dicarbonyl substituents and thus the variation in colour. The latter observation has potential across colour chemistry in general. In contrast to the readily observed BDT enol, no evidence has been adduced for enol tautomers amongst the ring-opened analogues.
Introduction
While the number of scientific publications worldwide is reported to be doubling every 9 years,1 a recent survey has estimated that the Royal Society of Chemistry's publication of colour-related science and technology has increased by an astonishing 8 times between the two 7-year periods 2003–9 and 2010–16.2a Much of this increase has been fueled by new colour chemistry-derived molecules, materials and phenomena. This paper is a further contribution to this field.A convenient synthetic route to benzodifuranone dyes (1 in Scheme 1; BDF) exploits benzodifurantrione (2; BDT) as a valuable precursor.3 Besides its synthetic value BDT is of interest from another viewpoint. Its novel structure led to the first characterisation of phenylogous enolisation, where keto tautomer 2 is in equilibrium with enol 3, and also with enolate anion 4 (Scheme 1).4 The enol 3 is readily observed as the favoured tautomer in some solvents and the sole tautomer in the crystal form. Tautomers 2 and 3 are specific realisations of the core substructures 5keto and 5enol which characterise rarely encountered enolisation across a benzene ring. The pale yellow colour of enol 3 contrasts with its bright and strong violet enolate conjugate base 4. These observations led to further questions concerning two simple modifications of BDT which we now address. The first part of this paper reports deprotonation of more extensively conjugated phenylogues of 3, the known5 bright red 4-hydroxylated BDF dyes (1; Ar1 = phenyl, Ar2 = 4-hydroxyaryl). The resultant BDF dye conjugate bases absorb in the near infrared (NIR) spectrum, and surprisingly have not previously been reported. The second question follows on from the characterisation of BDT (2), which had been aided by comparison with the properties of the model ring-opened α-ketoester 6 (Scheme 2).4 This and related derivatives were usually near colourless, but in some solvents or the presence of base brightly coloured solutions resulted as readily as the violet of anion 4 from BDT. Significantly, an unexpected marked variation in colour was observed between ostensibly very similar structures that was not readily understood. It therefore became of interest to define the structural differences underlying the colours of the presumed anions of ketoester 6 and analogues, resulting in the colour-structure rationale presented in the second part of this paper. Taken together the results of these two studies are intended to contribute to understanding of the similarities and differences for the quinonoid and benzenoid chromophores encountered amongst these molecules.
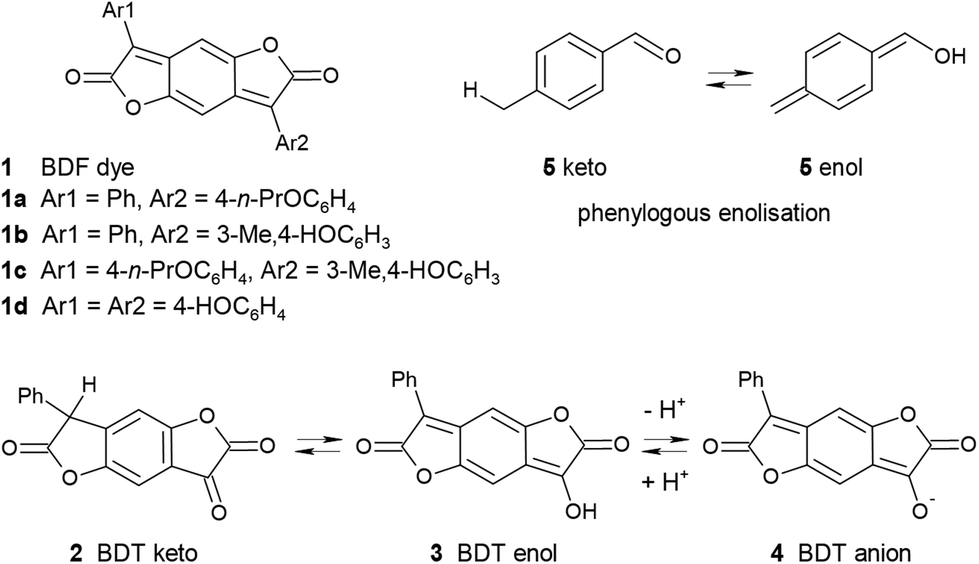 | ||
| Scheme 1 The benzodifuranone chromogen (BDF), and the phenylogous enolisation and ionisation of benzodifurantrione (BDT). | ||
 | ||
| Scheme 2 Reactions of BDT (Ar = 2,5-dimethoxyphenyl) i: ROH (R = Me, 6; R = H, 7); ii: ArNH2, HOAc, 25 °C; iii: o-toluidine, HOAc, H+, Δ; iv: o-toluidine, HCl; v: Me3SiCHN2, toluene, 40 °C; vi: PhCH2Br, K2CO3, DMF, 25 °C. | ||
Results and discussion
Part 1 Colour of the conjugate bases of hydroxylated BDF dyes
Visible absorption data for four oxygen-substituted BDF dyes are recorded in Table 1. Three are OH-substituted and are intended to demonstrate the effect on colour of ionisation at the aryl OH groups. The fourth (1a) is alkoxylated and serves as a nonionising model. It shows the typical strong and bright red colour of such BDF dyes (Table 1), with an absorption maximum at 493 nm in ethyl acetate (EA).5 The dye is weakly solvatochromic and insensitive to acid and base. The closely related 4-OH-substituted analogue 1b (additionally 3-methylated to improve solubility) shows similar behaviour to dye 1a in less polar solvents (e.g. a single absorption near λmax = 509 nm in EA; Table 1). Dye 1b is substantially more solvatochromic due to H-bond donor interaction between its phenol OH group and H-bond acceptor solvents. The relative solvatochromism of these and other BDF dyes is to be reported in detail separately.6 However, in highly dipolar and basic solvents such as DMSO and amides, dye 1b behaves very similarly to BDT enol (3). The absorption at 530 nm in DMSO due to non-ionised dye is now accompanied by a bathochromic absorption in the NIR with maximum at 835 nm (Fig. 1(a)). Addition of base (diazabicycloundecane, DBU) promotes the NIR absorption (εmax 8.4 × 104 M−1 cm−1) at the expense of the former (Fig. 1(b)), to give a near-colourless solution. Conversely, addition of acid removes the NIR absorption and regenerates that in the visible (Fig. 1(c); εmax 5 × 104 M−1 cm−1). These observations are typical of a halochromic molecule and consistent with simple ionisation of the phenolic BDF dye 1b and its re-protonation. | ||
| Fig. 1 Visible-NIR spectra of BDF dye 1b (red arrow) and its conjugate base (green arrow) (a) in DMSO; (b) in DMSO plus a slight excess of DBU; (c) in DMSO plus a slight excess of glacial acetic acid; (d) in dichloromethane with a slight excess of DBU; (e) in ethyl acetate with a slight excess of DBU. | ||
| BDF dye 1 | λ max /nm | |||||
|---|---|---|---|---|---|---|
| Ar1 | Ar2 | Additive | EtOAc | DMSO | DMF | |
| a NMP solvent. b DCM solvent. | ||||||
| 1a | C6H5 | 4-n-PrOC6H4 | None | 493 | 504 | 499 |
| DBU | No change | No change | No change | |||
| HOAc | No change | No change | No change | |||
| 1b | C6H5 | 3-Me,4-HOC6H3 | None | 509 | 530, 835 (770sh) | 530, 834 (770sh)a |
| DBU | 766 (680sh, 820sh) | 834 (770sh) | ||||
| HOAc | 526 | 532 | ||||
| 1c | 4-n-PrOC6H4 | 3-Me,4-HOC6H3 | None | 530 b | 551, 841 (770sh) | 545, 836 (770sh) |
| DBU | 841 (770sh) | 835 (770sh) | ||||
| HOAc | 548 | |||||
| 1d | 4-HOC6H4 | 4-HOC6H4 | None | 530 | 550, 819w | 544, 817 |
| DBU | 710 (550w, sh) | 902 (820sh) | 903 (800sh) | |||
| DBU, HOAc | 550 | 540w, 809 (770sh) | ||||
Blueish-red dye 1c with an additional 4′-propoxy substituent shows similar behaviour to 1b, bathochromically shifted by about 15 nm (Table 1). The same response to base of the visible absorption at ca. 550 nm is observed, resulting in a NIR absorbance maximum at 840 nm that is reversed with acid. Dye 1c is also a model for dye 1d which contains two potentially ionising p-OH substituents. The visible absorption maximum of dye 1d is essentially the same as dye 1c with an NIR absorption in DMSO or DMF somewhat hypsochromically shifted relative to 1c at ca. 820 nm, belonging to the mono-ionised species. Excess DBU added to the solution of 1d now causes a significant further bathochromic shift to about 900 nm assigned to the dianion resulting from ionisation of both the OH groups. Careful addition of glacial acetic acid to the DMF/DBU solution results in a mixture of the non-ionised dye (540 nm) plus the monoanion at about 810 nm. Excess acid quenches the latter with a resultant single absorbance in the visible (see ESI† for the sequence of spectra). Acid also brings the NIR absorption back to the visible in DMSO.
In all cases (1b–1d) the NIR maxima of the monoanions in DMSO and amide solvents are accompanied by blue-shifted shoulders at about 770 nm (Table 1), exemplified by dye 1b in Fig. 1(a) and (b). A further less intense blue-shifted shoulder at about 680 nm is also apparent in the latter spectrum. Similar behaviour is seen in DCM (containing slightly more than an equivalent of DBU) but with a relatively more intense shoulder at 770 nm (Fig. 1(d)). In even less polar EA (plus DBU) the absorption at 770 nm becomes the maximum, with the absorption at 820 nm now relegated to a red-shifted shoulder (Fig. 1(e)). The minor blue-shifted shoulder at 680 nm is still apparent with higher relative absorbance. This behaviour is qualitatively consistent with solvent-dependent molecular aggregation where H-type π-stacking of dyes (“pile of coins”) leads to excitonic coupling and blue-shifted absorbance.7 Anionic phthalocyanines provide well-known precedents.8 In aqueous solution with and without co-dissolved NaCl, absorbances were observed similar to those of the conjugate bases of 1b–d in the NIR. Thus a relatively sharp Q-band centred at about 665 nm was accompanied by a broader less well-defined absorption envelope with λmax about 40 nm blue-shifted. (Compare the ca. 50 nm difference between the maximum and blue-shifted shoulder of the conjugate base of BDF 1b in Fig. 1.) A preliminary quantitative relationship between solvent polarity, as quantified by the Kirkwood–Onsager reaction field dielectric function9g(ε) = (ε − 1)/(ε + 1), and the absorbance ratios of the maxima near 820 and 770 nm for the conjugate base of 1b in different solvents is discussed in the ESI.† In the meantime the conclusion is restricted to the proposal that the mono-anionic conjugate bases of OH-substituted BDF dyes 1b–d aggregate in the solvents studied, with less polar solvents inducing greater degrees of aggregation.
TD-DFT calculations have been reported on BDF dyes to provide understanding of various properties.10–13 The study by Jacquemin and co-workers on substituted BDF dyes in solution is particularly relevant.12para-Donor substituents cause bathochromic shifts closely consistent with experiment, due to a single charge-transfer (CT) S0 → S1 excitation from donor-substituted aryl ring into the tricyclic benzdifuranone core. If both phenyl substituents (Ar1 and Ar2 in 1) are each p-substituted by donor groups as in 1c and 1d, the absorption is modestly red-shifted compared with mono-substituted analogues 1a and 1b (theory and experiment) with electron density more widely distributed over the benzdifuranone tricycle core in S1 (see ESI† for more detail). Our own calculations at the much lower PPP-SCF-CI level of theory14 reach similar conclusions (see ESI†). Higher energy secondary absorbance (S0 → Sn, n > 1) is not predicted by TD-DFT in the NIR and thus is not believed to contribute to the blue-shifted multiple absorbances of the anionic conjugate bases of 1b–d.
The ionisation behaviour of 4-hydroxyphenyl BDF dyes in different solvents and under the influence of base thus follows the same pattern as BDT. The transition energy difference of about 14.9 kcal mole−1 for BDT (λmax enol (3) = 430 nm; enolate (4) = 554 nm) is increased to about 19.6 kcal mol−1 by insertion of the p-phenylene unit to give BDF dye equivalents (λmax 530 nm for 1b; 834 nm for ionised 1b). As far as we are aware NIR-absorbing BDF dyes have only been mentioned twice in the literature. In the only experimental observation a THF solution of the amino BDF dye 10 (Scheme 2) in the presence of tetra-n-butylammonium fluoride absorbed with a maximum at 830 nm (footnote 23 in ref. 15). A strong H-bonding interaction between dye-NH2 (10) and F− was suggested rather than full N–H deprotonation. TD-DFT calculations of a series of hypothetical donor–acceptor substituted BDF dyes for potential DSSC application revealed dyes with calculated λmax above 840 nm.12
Part 2 Colour of conjugate bases of ring-opened analogues of BDT
Ketolactone ring-opened analogues of BDT retain several of its structural features, while relaxing structural constraints enforced within the ketolactone unit of tautomer 2. This ring in BDT is relatively reactive toward simple nucleophiles (Scheme 2). α-Ketoester 6 was readily formed by simply dissolving BDT in dry methanol and allowing to stand overnight.4 The corresponding α-ketoacid 7 resulted from hydrolysis of BDT.4 Reaction between BDT and arylamines is more complex. In acetic acid, ring-opened α-ketoamides are the kinetically favoured products, analogous to formation of 6 and 7. In the current context, reaction between BDT and electron-rich 2,5-dimethoxyaniline gives amide 8. If a few drops of strong acid are added to the heated acetic acid solution, the aniline reacts at the ketone centre of BDT to give arylamino-substituted BDF structures which are evidently thermodynamically favoured (e.g.9 from o-toluidine). The reaction presumably proceeds via a hemiaminal which is likely in equilibrium with amide 8 in the acetic acid solution. Consistent with this, amide 8 and ester 6 both show a clear tendency for reverse ring-closure to BDT (see ESI†). Acid-catalysed dehydration of hemiaminal pulls the equilibrium to the product 9. Under even more strongly acidic conditions, BDT undergoes a type of Friedel–Crafts reaction with anilines, for example o-toluidine, to give the BDF dye 10. Protonated 9 is believed to be a precursor to BDF 10. The remarkable solvatochromism of amino BDF dye 10 has been described16 and will be compared elsewhere to the solvatochromic behaviour of other BDF dyes, including hydroxylated derivative 1b, above.6 Reaction of BDT with trimethylsilyldiazomethane gave the methyl ether 11 of BDT (Scheme 2).4 In contrast attempted methylation of ketoamide 8 under the same conditions leads to isolation of 2-trimethylsilylmethoxybenzofuran 12, where the centre alkylated is the lactone carbonyl oxygen (Scheme 2), and not its ketone. Although the methylation product 13 could be identified as a minor co-product, attempts to convert 12 to 13 failed. Benzylation occurs α to the lactone carbonyl to give 14, characterised as C-linked by IR spectral data, loss of the benzylic proton in 8 by 1H NMR, chemical shifts, and an AB coupling pattern for the diastereotopic benzylic protons in its 1H NMR spectrum. A minor co-product identified as dibenzyl derivative 15 results from further benzylation at the phenolic OH group. No evidence was seen under either of these reaction conditions for alkylation at any other potentially nucleophilic site, especially the putative enolic OH site analogous to that in BDT enol 3 corresponding to the ketone carbonyl oxygen of amide 8 (see below, structure 18 in Fig. 6).| Solvent | BDTa | Ester (6) | Acid (7) | Amide (8) | ||||
|---|---|---|---|---|---|---|---|---|
| Enol 3 | Anion 4 | Parent 6 | Anion 16 | Parent 7 | Anions | Parent 8 | Anion 17 | |
| a Data from ref. 4; keto tautomer 2: λmax = 292–300 nm. b Aqueous carbonate. c TEA added. | ||||||||
| DMSO | 554 | 357 | 468 | 351 | 465 | 340 | 522 | |
| DMF | 552 | 472 | 531 | |||||
| Acetone | 432 | 355 | 470c | 357 | 442c | 374 | 532c | |
| DCM | 427 | 543c | 369 | 451c | 382 | 424c | 390 | 496c |
| PhMe | 438 | 530c | 371 | 443c | 375 | 421c | 398 | 498c |
| H2O | 347 | 424b | ||||||
BDT. In the earlier paper4 the structures of the tautomers of BDT and its derived anion 4 were fully characterised. The latter was generated in DMSO or DMF spontaneously without the addition of base, and absorbed in the visible spectrum at λmax 554 and 552 nm respectively, giving a bright violet solution (εmax 3.5 × 104 M−1 cm−1). Subsequent measurements have shown the anion is generated in other solvents in the presence of base (Table 2). Triethylamine (TEA) has been commonly applied, but it has also been observed that moisture present in some laboratory solvents (e.g. non-rigorously dried THF) or even merely absorbed from the atmosphere by dry solvent solutions on standing, results in generation of the distinctive violet colouration of the anion (see ESI†). In fact, pale yellow solutions of non-ionised BDT cause immediate violet colouration of paper, wood, human skin, and the surface of some glass, inter alia, due to either adventitious moisture or basic sites within the material. In iso-propanol at least, a deprotonation–reprotonation cycle can be demonstrated by subsequent addition of methanesulphonic acid (MSA) to the anion to give yellow enol, and then a second addition of TEA regenerates the violet anion. The visible absorption is slightly solvatochromic, ranging 24 nm upward from 530 nm, comparable to the solvatochromism of the parent enol of BDT (3; 14 nm range for the same solvents).4
Ring-opened analogues. Solutions of 6, 7, and 8 in less polar solvents such as ethyl acetate or toluene are almost colourless. Dilute DMSO solutions are brightly coloured; those from ester 6 and acid 7 are yellow, but that of amide 8 differs as a bright bluish-red. The reason for the difference in colour between two ostensibly very similar chromogens was initially unclear, and instigated detailed investigation of these ring-opened analogs of BDT. All compounds 6–8 have double absorbance maxima in DMSO, one each in the UV and visible regions. Addition of a trace of MSA leads to complete removal of the absorbances in the visible region, leaving only single UV absorbances due to the benzenoid parent compounds. The observed λmax values are between 350–400 nm consistent with an H-bonding 2-substituted aryl ketone chromophore.17 Alternatively, addition of TEA removes the absorptions in the UV region to give correspondingly stronger single absorbances at visible wavelengths (Table 2). The effect of acid (base) is again reversed by subsequent addition of base (acid), consistent with the bathochromic species being due to ionised forms. Addition of TEA to DCM, EA or toluene solutions also generates coloured solutions in the same way.
A single crystal X-ray structure determination of carboxylic acid 7 confirms its ketone tautomer structure in the solid state and reveals an intramolecular H-bond between phenol OH and ketone oxygen (Fig. 2). The proton of the carboxylic OH is disordered over two sites. One of these sites also gives intramolecular H-bonding to the α-keto group. Intermolecular hydrogen bonding interactions are between CO2H donors and both CO2H and the adjacent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O acceptors. This gives 1-dimensional hydrogen bonded chains that propagate parallel to the crystallographic b direction. The α-dicarbonyl unit adopts a planar anti relationship, and is almost coplanar with its attached aryl ring. This results in a close separation (2.27 Å) between the carboxyl carbonyl oxygen and the ortho-H of the attached ring. The plane of the pendant phenyl group is orthogonal to the least squares plane of its attached lactone (angle between least squares planes 89.97(18)°). Non-heteroatom intermolecular features of note are the π-interactions between antiparallel neighbour molecules that give dimers (closest C⋯C distance 3.189(7) Å between phenol ring and COCOOH units) and a longer range edge-to-face interaction between the phenyl and phenol rings that leads these dimers to stack parallel to the a direction.
O acceptors. This gives 1-dimensional hydrogen bonded chains that propagate parallel to the crystallographic b direction. The α-dicarbonyl unit adopts a planar anti relationship, and is almost coplanar with its attached aryl ring. This results in a close separation (2.27 Å) between the carboxyl carbonyl oxygen and the ortho-H of the attached ring. The plane of the pendant phenyl group is orthogonal to the least squares plane of its attached lactone (angle between least squares planes 89.97(18)°). Non-heteroatom intermolecular features of note are the π-interactions between antiparallel neighbour molecules that give dimers (closest C⋯C distance 3.189(7) Å between phenol ring and COCOOH units) and a longer range edge-to-face interaction between the phenyl and phenol rings that leads these dimers to stack parallel to the a direction.
 | ||
| Fig. 2 (a) Molecular structure of ketoacid 7 from X-ray diffraction. Displacement ellipsoids are shown at the 50% probability level. Intramolecular hydrogen bonds are present between the OH groups of O3 and O5 and the ketone atom O4 (H⋯O 1.85 and 2.14 Å respectively). There is also a relatively short interaction between O6 and the aromatic C–H of the phenol ring (H⋯O 2.27 Å). The essentially coplanar nature of the phenol ring and the C2HO3 substituent is shown by the torsion angles C(O4)–C–C–O3, C–C–C–O4, and O4–C–C–O6 (4.2(7), −0.5(7) and −175.2(5)° respectively). (b) Part of the packing structure of 7 viewed down the a axis and highlighting the 1-dimensional hydrogen bonded chain that propagates parallel to the b direction. | ||
The following discussion defines the solution structures of ester 6 and amide 8, and then their respective conjugate bases 16 and 17. In particular, an understanding of the difference in the colours of the latter pair follows from conformational differences. Fig. 3 summarises significant spectroscopic and other properties of these four species. Full data which lead to unambiguous nmr assignments are reported in the ESI.†
 | ||
| Fig. 3 Selected spectroscopic and other data for ester 6 and amide 8, and derived conjugate bases 16 and 17. The negative charges in the latter are arbitrarily shown localised on the exocyclic oxybenzfuran oxygen, although they are inevitably delocalised across other centres, see text. Nmr shift data in red (CDCl3 for 6 and 8; DMSO-d6 for 16 and 17); important NOE interactions in green; IR data in blue; pKa estimates in DMSO (cyan) and water (violet). | ||
The downfield phenolic proton nmr shifts near δH 12 ppm (CDCl3) in both 6 and 8 indicate intramolecular H-bonds in a 6-membered ring, thus in both cases to ketone oxygen.18 Alternative 7-ring H-bonds to ester or amide carbonyl oxygen are feasible in principle and characterised in reported example molecules,19 but proton nmr shifts of phenol OH in these larger H-bonded rings are typically more than 2 ppm upfield near δH 10 ppm relative to 6-ring analogues, and thus are inconsistent with the current observed values. The IR stretch frequencies of the ketones in both cases are equally consistent with intramolecular H-bonding, at 1644 and 1628 cm−1 for 6 and 8 respectively. For comparison the non-H-bonded ketone IR frequency in the model compound ethyl phenylglyoxalate (PhCOCO2Et) is blue-shifted at 1689 cm−1.20 Unless hindered, anilide conformations are normally close to planar (torsion about C–N near 0/180°), and this along with the NOE between NH and the 2-methoxy group in amide 8 implies the ortho-H at δH 8.2 is deshielded by the anilide carbonyl.21 Significantly the abnormal downfield shift of the proton Hc ortho to ketone in amide 8 at δH 8.7 is also concluded to be deshielded by the same nearby amide carbonyl. The inference is that the ketoamide plus attached benzlactone and dimethoxyphenyl units in 8 are tending toward overall planarity, with a dihedral angle between the two carbonyl groups near 180°. In contrast the conventional shift of the corresponding proton Hc in ketoester 6 (Fig. 3) implies little or no effect due to nearby carbonyl anisotropy. Consequently a nonplanar α-dicarbonyl conformation is deduced for the ketoester. The most acidic proton in each of 6 and 8 is the doubly benzylic proton α to carbonyl, with an estimated pKa in DMSO at 9.6, about 5 orders of magnitude more acidic than the phenol OH.22 TEA (pKBH+ = 9 in DMSO23) readily deprotonates 6 and 8 at this position in DMSO and DCM, clearly confirmed by the absence of a benzylic proton in the 1H NMR spectra of 16 and 17 and the expected shift for the corresponding carbon in the 13C NMR spectrum (DMSO-d6). The lactone carbonyl IR resonances in 6 and 8 (1819, 1818 cm−1 respectively, in DCM) are replaced by red-shifted enolate absorbances below 1700 cm−1 (DCM + TEA) for 16 and 17. The phenol 1H NMR resonances of the resultant conjugate bases 16 and 17 similarly remain at low field (12 and 13.5 ppm) consistent with continuing intramolecular H-bonding as in their parent molecules (6 and 8), although now appreciably broadened presumably due to proton-exchange reactions. Consistent with this, the ketone stretches in 16 and 17 remain at H-bonded values (ca. 1619 cm−1 in DCM/TEA). The benzenoid protons Hc in each also remain with the same relative shifts: in amide anion 17 deshielded by the nearby amide carbonyl, and in ester anion 16 unaffected by any carbonyl group. Overall this implies the same conformations for the conjugate bases as the parent molecules, with ketoamide (near) planarity and ketoester nonplanarity. Table 3 records 1H NMR shift values for proton Hc in related molecules, in some cases in a variety of solvents. In all of amide 8, its conjugate base 17, O-alkylated 12 and 13, and C-benzylated 14 it resonates well downfield in all cases, consistent with amide carbonyl anisotropic effects due to (near) planar conformations for all these species. However, O-benzylation of the phenolic OH in 15 removes intramolecular H-bonding to carbonyl and consequently eliminates one of the sources driving molecular planarity in these amides. Thus the 1H resonance of Hc in 15 returns upfield to a value similar to that in nonplanar ester 6 and its conjugate base 16. The shifts are essentially independent of solvent for 6 and 8.
| NMR solvent | Solvent dependent δH (Hc) /ppm | |||||||
|---|---|---|---|---|---|---|---|---|
| BDT 2 | ester 6 | acid 7 | amide 8 | 12 | 13 | 14 | 15 | |
| a Data unavailable because of unsatisfactory 1H NMR spectra for 7 in DMSO-d6 under all conditions. b Average shift for the parent molecule across all nmr solvents studied unless otherwise noted. c Difference between the average shift of neutral species and the shift in DMSO-d6 for the conjugate base. d CDCl3 value excluded; see text. | ||||||||
| MeCN-d3 | 7.55 | 7.61 | 7.74 | 8.33 | ||||
| CDCl3 | 7.53 | 7.57 | 8.25 | 8.68 | 9.02 | 9.05 | 8.34 | 7.33 |
| Acetone-d6 | 7.61 | 7.62 | 7.68 | 8.34 | ||||
| Acetic acid-d4 | 7.56 | 7.63 | 7.77 | 8.58 | ||||
| o-Dichlorobenzene-d4 | 7.59 | |||||||
| Dioxan-d8 | 7.66 | |||||||
| THF-d8 | 7.55 | |||||||
| Anion 4 | Anion 16 | Anion | Anion 17 | |||||
| DMSO-d6 | 6.81 | 6.87 | 7.89 | |||||
| Averageb | 7.58 | 7.61 | 7.73d | 8.48 | ||||
| Differencec | 0.77 | 0.74 | 0.59 | |||||
The ionisation equilibria of acid 7 are potentially more complex than ester 6 because of the comparable value of its CO2H pKa to that of the doubly benzylic C–H. Estimates based on an aqueous pKa of 2.15 for PhCOCO2H and relevant correlations give pKa values 7.6 (DMSO; 2 units lower than the doubly benzylic proton) and 8.9 (DMF) inter alia for CO2H in 7.24 The consequence is potential equilibria between the parent 7, two monoanions, and a dianion (neglecting ionisation of the less acidic phenolic OH). While many of the spectroscopic properties of acid 7 are similar to ester 6, including its colour and λmax values, there is one notable exception. Although its Hc nmr shifts in several solvents are constant (δHca. 7.7 ppm, Table 3), in CDCl3 a marked downfield value (δH 8.25) suggests deshielding by the carboxyl carbonyl, analogous to that seen for the amides. The inference is that the conformation in CDCl3 is as found in the crystal and as shown in Fig. 2, whereas in the other solvents an intramolecular 5-ring H-bond between CO2H and α-ketone is absent. A conclusion is that energy differences between various conformations are likely to be low. Two detailed published studies support this generalisation. In the first, the energetics of conformations and conversion barriers for the simplest model compound glyoxal were determined spectroscopically (single vibronic level fluorescence spectra of jet-cooled glyoxal).25 The s-cis conformation (ϕ = 0°) of glyoxal was about 4.8 kcal mol−1 less stable than s-trans (ϕ = 180°), with a s-cis to s-trans conversion energy barrier of only about 1.1 kcal mol−1. Elsewhere theoretical calculations of the conformation and isomerisation energetics of PhCOCO2Et revealed a ground state where the ketone lay in the same plane as the Ph ring, but with the ester group out of plane due to torsion by 50° about the CO–CO2Et bond.20 The energy barrier for interconversion of the ground state nonplanar enantiomers via the planar s-trans conformation was low (ca. 1.2 kcal mol−1), while that for interconversion via the sterically more hindered planar s-cis conformation was higher (ca. 4.5 kcal mol−1).
Further support for the conclusions on conformations in the above molecules derives from a survey of structures of analogues in the CSD.26 We have carried out substructure searches for acyclic C*–CO–CO–R, where C* is any 3-coordinate C atom (sp2), and R is any nitrogen- or oxygen-linked substituent (see ESI†). Pyruvate derivatives and similar (C* sp3) are intentionally excluded. The basic statistics from the survey are revealing and are presented as histograms in Fig. 4. Primary and secondary amides with potential for intramolecular 5-ring H-bonding show a strong preference for s-trans conformations, tending toward planarity (ϕ = 180°) (Fig. 4(a)). This is exactly what we conclude above for amide 8 and its analogues from the spectroscopic data. Tertiary amides provide no H-bonding option, and additionally are subject to steric hindrance in planar conformations. They thus tend toward orthogonal geometries about the CO–CO bond (ϕ ca. 90°) (Fig. 4(b)). The fewer α-ketocarboxylic acid derivatives in the CSD demonstrate the same preference for s-trans planarity (Fig. 4(c)) as the primary and secondary amides, as well as the structure of acid 7 in the crystal (Fig. 2). Corresponding α-ketoesters tend toward CO–CO orthogonality although the trend is weaker and all conformations are represented amongst reported crystal structures (Fig. 4(d)). This is at least consistent with the conclusions for nonplanarity in ester 6, and variable conformations for acid 7 in different solvents and the crystalline phase.
 | ||
| Fig. 4 Histograms of OC–CO dihedral angles observed amongst structures reported in the CSD (see ESI†). Bins are 15° wide and cover conformations from planar s-cis (ϕ = 0°) to planar s-trans (ϕ = 180°) (x-axes). The y-axes represent the relative number of hits per bin. The molecules are divided into 4 substructure types defined by the functionalities shown in (a–d), where C* is any 3-coordinate carbon. Carboxylate anion termini are omitted. | ||
There is now a clearly delineated difference between ester 6 and amide 8, as well as their conjugate bases 16 and 17, which we believe is germane to the primary question of colour difference between the latter pair. A few basic electronic properties have been reported for various CO–COR functionalities in the ground state (S0),27 but nothing of its auxochromic influence in excited states. While the theoretical level of PPP-SCF-CI calculations14 is fully accepted to be much below state-of-the-art, they have nevertheless been applied to help explain the auxochromic properties of CO-COR substituents (see further justification in ESI†). Key results are summarised in Fig. 5. Initial calculations were run on model structures based on idealised planar benzofuran plus coplanar α-dicarbonyl substituents for both anions 16 (shown as 16p) and 17, each substituted by a phenyl group out of plane by 20°. The calculated absorption maximum for 17 is gratifyingly close to experiment. Three other aspects are significant. First, the terminal methoxy unit of ester anion 16p and dimethoxyaniline unit of 17 play no part in their S0 → S1 excitations. Second, the idealised planar α-ketoester functionality of 16p has the same λmax (calc.) as amide 17. This is no surprise given the absence of any calculated auxochromic effect by substituents beyond the terminal CO unit (OMe or NHAr). Third, the S0 → S1 excitations of 17 and planar 16p involve net π-electron transfer from the anionic oxyfuran portion of the molecules to both carbonyl units of the α-dicarbonyl functionality. This is perhaps unexpected, but provides the source of a possible explanation for the hypsochromic nature of the conjugate base 16 of ester 6 (coloured yellow) relative to that (17) of amide 8 (red). If torsion about the CO–CO bond – as has been concluded above – is now applied to the ketoester substructure of the molecular input to the calculation, it would be expected to diminish the π-electron accepting ability of the terminal (ester) carbonyl in S1, and thus its contribution to the overall CO–COR auxochromic effect. Indeed, this is what is observed: deplanarisation by 50° out-of-plane torsion about the CO–CO bond in the nonplanar input structure 16np results in a significant reduction in electron transfer into the terminal ester group, and corresponding reduction in λmax (calc.), reproducing the experimental λmax in DMSO solution (Fig. 5). While this nonplanarity appears to be sufficient alone to explain the relatively hypsochromic effect of the ketoester substituent, an additional possibility cannot be excluded. In the planar conformation the amide NH could be helping magnify the acceptor effect of ketone in amide 17 through weak (5-ring) stabilising H-bonding.
 | ||
| Fig. 5 Net π-charge transfer on S0 → S1 excitation according to PPP-SCF-CI calculation. Experimental λmax values are recorded in basic DMSO. Molecule skeletons shown represent atoms contributing to π-electron bonding. Hetero atoms are not distinguished in this graphic; see structures in Fig. 3 for their identities. Circle diameters signify relative degree of charge difference on excitation: red circles are atom sites of π-electron donation; blue circles are π-electron acceptor atoms. | ||
Conclusions – the nature of the chromogens
The known trend of increasing bathochromicity covering essentially the whole of the visible spectrum from the yellow-to-orange unsubstituted BDF chromogen successively through dyes (1) 4-substituted by hydroxy or alkoxy (scarlet through to bluish-red), amino (red through to navy), and mono-, then dialkylamino (bright blue, near cyan), is now further extended by the effect of the even stronger phenoxide anion π-electron-donor to give new NIR-absorbing BDF dyes. This ordering is consistent with the well-known trend in (ground state) π-electron donor strengths reflected by their Hammett σR constants: OH/OMe −0.43; NH2 −0.5; MeNH −0.52; Me2N −0.53; Et2N −0.57; O− −0.59.27 The effect of oxide anion is thus hardly surprising apart perhaps from the fact that in BDF dyes the resultant absorption reaches as far as the NIR. More surprising is that it has gone unreported through more than 40 years of BDF research. The main characteristic of these BDF dyes is their donor–acceptor excitation where the tricyclic quinonoid dilactone unit plays the role of a novel delocalised electron-acceptor. New NIR absorbers are of increasing interest and exploitation in the wider world of applied colour chemistry.2bThe difference between εmax values for anions 16 and 17 (0.48 × 104 and 1.82 × 104 M−1 cm−1) mirrors the trend amongst published εmax values for a series of analogous anions derived by benzylic deprotonation of aryl acetic acid esters p-XC6H4CH2CO2Et.28 In these, εmax values of anions with electron-withdrawing substituents varied from 0.84 × 104 (X = CO2Et) to 3.3 × 104 M−1 cm−1 (X = NO2). All the evidence points to chromogens 16 and 17 as donor–acceptor types, with the deprotonated lactone acting as an electron donor into α-dicarbonyl acceptor units, across a benzenoid conjugation link. Spectroscopic and reactivity evidence suggests the anion is more delocalised on the enolate from the lactone ring ((a) and (b) Fig. 6) rather than extended into the conjugated ketone unit ((c) in Fig. 6). This contrasts with the reactivity of the anionic lactone conjugate base of BDT (4), whose unique properties have been discussed elsewhere.4,29 Thus methylation and acetylation of BDT's conjugate base 4 occurs exclusively at the exocyclic enol-derived oxygen of the quinonoid structure rather than at the lactone enolate at the opposite side of the molecule. This situation is more closely analogous to that of the strictly quinonoid BDF dyes 1. The location of the delocalised charge of the conjugate bases is biased by the presence or absence of the oxylactone ring structure versus its alternative ring-opened version. The underlying reasons for this rather subtle effect remains opaque, but is at least further defined by the results for the ring-opened analogues presented in this paper. Probably related is the contrasting relative thermodynamic stability for the easily observed4 phenylogous enolisation of ring-closed BDT 2 ⇆ 3, compared with the failure to adduce any evidence for the corresponding enol of ring-opened analogues 6–8 (e.g.18, Fig. 6) from amongst the spectroscopic and reactivity properties now reported. The benzo-fused ketolactone ring of BDT retains its unique position in enabling easily observable phenylogous enolisation.
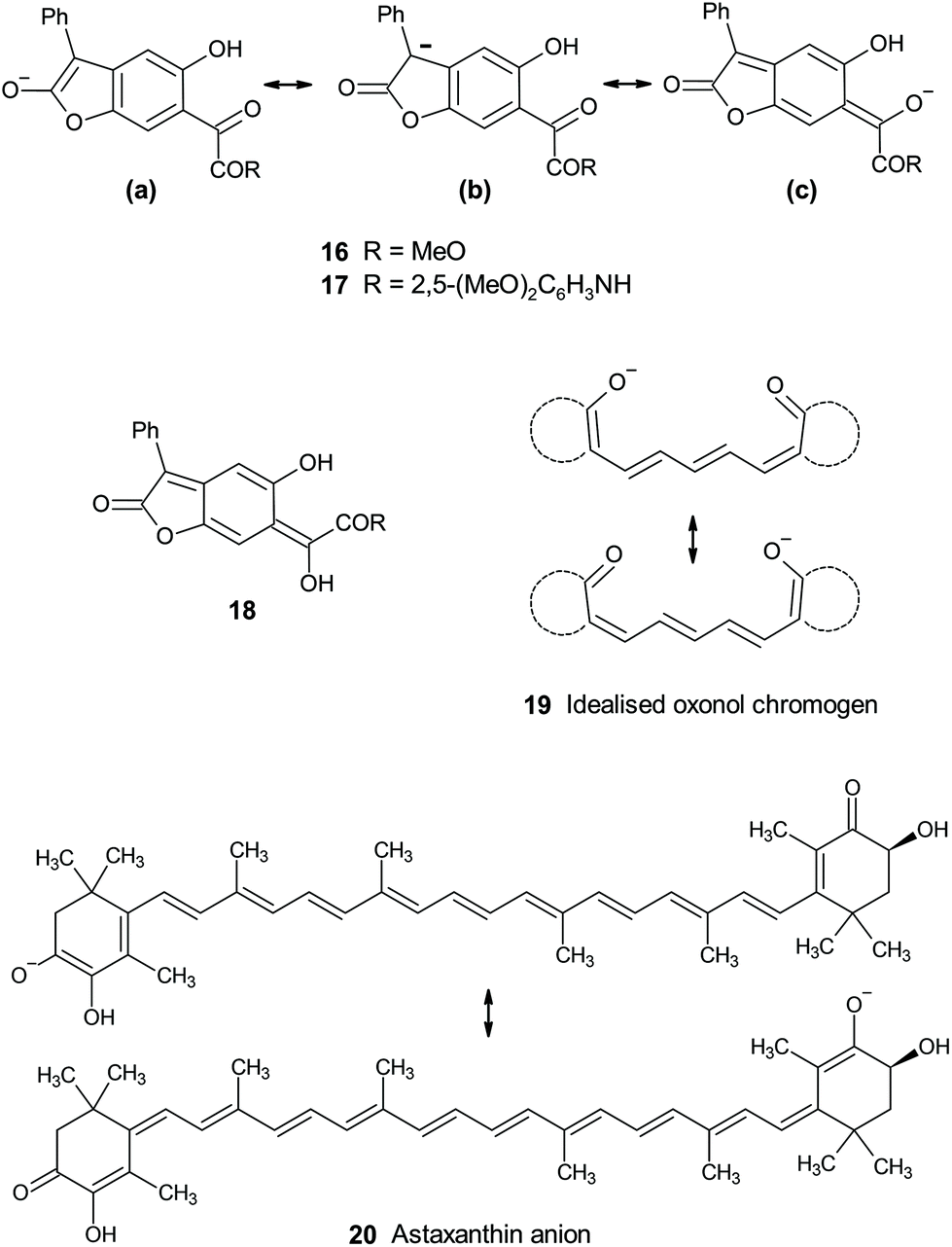 | ||
| Fig. 6 Delocalised anionic chromogens, and an elusive enol tautomer (18) of ring-opened BDT. | ||
From amongst related conjugated anionic chromogens, the limiting case of full charge delocalisation in the ground state is exemplified by symmetrical charge distribution in some oxonol anions (e.g. idealised 19, Fig. 6), which are anionic polymethine analogues of better known cationic cyanine dyes.30 The oxonol chromogen is usually generated by deprotonation analogous to that described for 6 and 8 above, but is brought to lower pH by acidification of the relevant proton by inclusion of further conjugating groups in the ring structures implied in 19. Oxonols have historical application as indicators of cell wall potentials (voltage sensitive dyes, VSD, thus electrochromes). Chromogens 16 and 17 can be viewed as unsymmetric analogues. These are more akin to types such as the delocalised astaxanthin anion chromogen 20 responsible for the blue-grey colour of uncooked lobsters and other crustaceans.31 Astaxanthin itself is not particularly activated toward deprotonation, but in vivo the anionic chromogen is stabilised at a basic site within a protein complex (crustacyanin). This suggests that halochromic species such as 1b, 2 ⇆ 3, 6 and 8 might respond to basic sites within cells and thus be viable indicators in biological environments. NIR-absorbing conjugate bases of BDF dyes such as 1b are particularly attractive as potential bio-indicators, absorbing in the first biological transparency window (NIR-I; 700–950 nm).
A further unanticipated and significant new feature from this study is the nature of the α-dicarbonyl auxochromic electron acceptor of planar 17 where both carbonyl units act as acceptor centres on photoexcitation. The resultant relatively bathochromic shades achievable suggest this auxochrome could be a useful new addition to conventional acceptors typified by ester, nitrile, sulphone and nitro, all routinely applied across many dye types. It has the additional benefit of potential for structural modification via amide N-substituents. These could be varied to tailor physical properties beyond colour (e.g. solubility, hydrophobicity/philicity, etc.), as well as to introduce further remote reaction sites to enable cross-linking or other covalent functionalisation.
Experimental
See ESI.†Author contributions
MGH conceived the study, carried out spectroscopic measurements and interpretation, and wrote the paper. AJL carried out all synthesis and characterisation, plus spectroscopic measurements and their interpretation. ARK carried out CSD searches and X-ray structure determination and wrote the corresponding section. All authors reviewed and agreed the final draft of the ms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Nigel Hall shared unpublished results on the mechanism of acid-catalysed arylation of BDT by anilines. M.Chem. students Collette Turner and Khalid Ali initiated early synthesis work. Dr Sally Bloodworth (University of Southampton) carried out valuable spectroscopic measurements. Dr Rudolf Naef (University of Basel) provided gratis the latest version of his PiSystems PPP-SCF-CI software. We thank all of these for their contributions.References
- http://blogs.nature.com/news/2014/05/global-scientific-output-doubles-every-nine-years.html (accessed April 2021).
-
(a)
P. Bamfield and M. G. Hutchings, Chromic Phenomena: Technological Applications of Colour Chemistry, Royal Society of Chemistry, London, UK, 3rd edn, 2018, Appendix 2, p. 765 Search PubMed
; (b) ibid, p. 432 ff Search PubMed
-
(a)
N. Hughes, D. F. Newton, D. J. Milner and G. A. De Boos, World PatentWO 94/12501, 1994 Search PubMed
- A. J. Lawrence, M. G. Hutchings, A. R. Kennedy and J. J. W. McDouall, J. Org. Chem., 2010, 75, 690–701 CrossRef CAS PubMed
- C. W. Greenhalgh, J. L. Carey and D. F. Newton, Dyes Pigm., 1980, 1, 103–120 CrossRef CAS
- M. G. Hutchings , unpublished work.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed
- P. J. Camp, A. C. Jones, R. K. Neely and N. M. Speirs, J. Phys. Chem. A, 2002, 106, 10725–10732 CrossRef CAS
- C. Laurence, P. Nicolet, M. T. Dalati, J.-L. M. Abboud and R. Notario, J. Phys. Chem., 1994, 98, 5807–5816 CrossRef CAS
- W. Bartkowiak and P. Lipkowski, J. Mol. Model., 2005, 11, 317–322 CrossRef CAS PubMed
- S. Lunák Jr., J. Vyňuchal and R. Hrdina, J. Mol. Struct., 2009, 935, 82–91 CrossRef
- J. P. Cerón-Carrasco, A. Ripoche, F. Odobel and D. Jacquemin, Dyes Pigm., 2012, 92, 1144–1152 CrossRef
- Y.-Z. Zhan, X. Zhao and W. Wang, Color. Technol., 2016, 132, 1–7 Search PubMed
-
(a)
R. Naef, PiSystem98, Im Budler 6, CH-4419 Lupsingen, Switzerland Search PubMed
- A. Oehlke, K. Hofmann and S. Spange, New J. Chem., 2006, 30, 533–536 RSC
- A. A. Gorman, M. G. Hutchings and P. D. Wood, J. Am. Chem. Soc., 1996, 118, 8497–8498 CrossRef CAS
- C. H. Eugster and P. Bosshard, Helv. Chim. Acta, 1963, 46, 815–851 CrossRef CAS
- P. E. Hansen and J. Spanget-Larsen, Molecules, 2017, 22, 552–572 CrossRef CAS PubMed
- R. K. Castellano, Y. Li, E. A. Homan, A. J. Lampkins, I. V. Marín and K. A. Abboud, Eur. J. Org. Chem., 2012, 4483–4492 CrossRef CAS
- D. Ferri, T. Bürgi and A. Baiker, J. Chem. Soc., Perkin Trans. 2, 2000, 221–228 RSC
- G. W. Gribble and F. P. Bousquet, Tetrahedron, 1971, 27, 3785–3794 CrossRef CAS
-
(a) D. M. Heathcote, G. A. De Boos, J. H. Atherton and M. I. Page, J. Chem. Soc., Perkin Trans. 2, 1998, 535–540 RSC
- I. M. Kolthoff, M. K. Chantooni and S. Bhowmik, J. Am. Chem. Soc., 1968, 90, 23–28 CrossRef CAS
-
B. G. Cox, Acids and bases, solvent effects on acid-base strength, Oxford University Press, 2013, ch. 9 Search PubMed
- K. W. Butz, D. J. Krajnovich and C. S. Parmenter, J. Chem. Phys., 1990, 93, 1557–1567 CrossRef CAS
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed
-
C. Hansch, A. Leo and D. Hoekman, Exploring QSAR: Hydrophobic, Electronic, and Steric Constants, American Chemical Society, Washington DC, 1995 Search PubMed
- S. Kiyooka, Y. Ueda and K. Suzuki, Bull. Chem. Soc. Jpn., 1980, 53, 1656–1660 CrossRef CAS
- E. Kleinpeter, U. Bölke and A. Koch, J. Phys. Chem. A, 2010, 114, 7616–7623 CrossRef CAS PubMed
-
H. Zollinger, Color Chemistry; Synthesis, Properties and Applications of Organic Dyes and Pigments, 3rd edition, Wiley VCH, Weinheim, 2003, Ch. 3 Search PubMed
- S. Begum, M. Cianci, B. Durbeej, O. Falklöf, A. Hädener, J. R. Helliwell, M. Helliwell, A. C. Regan and C. I. F. Watt, Phys. Chem. Chem. Phys., 2015, 17, 16723–16732 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and characterisation data; visible/NIR spectra; X-ray structure determination; search of the Cambridge Crystal Structure Database; theoretical calculations. CCDC 1994670. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob02442f |
| This journal is © The Royal Society of Chemistry 2022 |