
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA case study of the MAC (masked acyl cyanide) oxyhomologation of N,N-dibenzyl-L-phenylalaninal with anti diastereoselectivity: preparation of (2S,3S)-allophenylnorstatin esters†
Xuefeng
He
 ,
Marie
Buchotte
,
Régis
Guillot
,
Sandrine
Deloisy
and
David J.
Aitken
*
,
Marie
Buchotte
,
Régis
Guillot
,
Sandrine
Deloisy
and
David J.
Aitken
*
Université Paris-Saclay, CNRS, ICMMO, CP3A Organic Synthesis Group and Services Communs, 15 rue Georges Clemenceau, 91405 Orsay cedex, France. E-mail: david.aitken@universite-paris-saclay.fr
First published on 7th February 2022
Abstract
The three-component reaction between a protected α-amino aldehyde, an alcohol and an α-silyloxymalononitrile provides an expedient access to protected α-hydroxy-β-amino acid derivatives. The prototypical process, performed on N-Cbz-phenylalaninal, is known to proceed with syn diastereoselectivity. The present study demonstrates that the diastereoselectivity of the reaction can be inverted, using the rationale of a Felkin-Anh interaction model. Reactions performed on N,N-dibenzyl-L-phenylalaninal proceed with a high anti diastereoselectivity, providing a panel of synthetically useful ester derivatives of (2S,3S)-allophenylnorstatin. The procedure is exploited to accomplish one of the most efficient syntheses of the title compound to date, in 3 steps (66% yield) from N,N-dibenzyl-L-phenylalaninal.
Introduction
Since its first advocacy several decades ago,1 the umpolung concept has fructified to provide powerful tools for conducting organic synthesis.2 Particular attention has been paid to the development and application of acyl anion equivalents, such as 1,3-dithianes,3 dialkylhydrazones,4 cyanohydrins5 and α-aminonitriles.6 A significant family of umpolung reagents are constituted by O-functionalized α-hydroxymalononitriles, known as masked acyl cyanide (MAC) reagents, first introduced by Yamamoto and Nemoto.7 The deprotonated form of a MAC reagent is nucleophilic and it reacts with an electrophile; subsequent unmasking reveals an acyl cyanide which can be further transformed into a carboxylic acid derivative by reaction with a nucleophile. This reactivity profile has been widely exploited in multi-step syntheses of biologically active molecules8 and in other synthetic applications.9When the electrophilic partner is a chiral α-amino aldehyde and the MAC reagent is a silyl ether—hereafter referred to generically as H-MAC-[Si]—the one-pot reaction constitutes an oxyhomologation, presumably proceeding via a [1,4]-silyl transfer mechanism, to provide access to an α-hydroxy-β-amino acid derivative in which the alcohol function is protected as a silyl ether (Fig. 1). Despite the importance of α-hydroxy-β-amino acids,10 few applications of MAC methodology have been made thereto and they have, for the most part, been characterized by a syn diastereoselectivity (syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anti ratio around 4:1).11 Recently, however, we discovered that when Garner's aldehyde is used as the electrophilic partner the oxyhomologation gives the corresponding MAC reaction product with an anti diastereoselectivity.12
:anti ratio around 4:1).11 Recently, however, we discovered that when Garner's aldehyde is used as the electrophilic partner the oxyhomologation gives the corresponding MAC reaction product with an anti diastereoselectivity.12
 | ||
| Fig. 1 The proposed mechanism for the reaction of an H-MAC-[Si] reagent with a protected chiral α-amino aldehyde, highlighting two models that may govern the diastereoselectivity in the first step. Once unmasked, the acyl cyanide intermediate reacts with a nucleophile. | ||
One of the appealing features of MAC methodology is that the mild reaction conditions ensure that no erosion of the inherent enantiomeric composition of a chiral substrate is observed;7m,8c this means that the oxyhomologation of a single enantiomer of an α-amino aldehyde substrate will furnish the oxyhomologation product as a single enantiomer.
While the mechanism of the MAC reaction has not been studied in detail, it seems plausible that the syn selectivity observed with unrestricted α-amino aldehydes might arise via an anti-Felkin–Anh model implicating a hydrogen bond between the carbamate-protected amine and the aldehyde during the first step, whereas Garner's aldehyde, devoid of an NH motif, reacts via a Felkin–Anh model (Fig. 1). If this reasoning is valid, it should be possible to perform an anti stereoselective MAC oxyhomologation reaction on an unrestricted α-amino aldehyde on the condition that the protecting group suite does not contain an NH motif. To probe this hypothesis, an N,N-dibenzyl-protected amino aldehyde appeared to us to be an appropriate substrate, since compounds of this type are known to react with nucleophiles13,14 with good Felkin–Anh selectivity and without loss of enantiomeric enrichment. In order to provide direct comparison with Nemoto's reference work using N-Cbz-phenylalaninal,7m the present study focuses on the MAC reaction of N,N-dibenzyl-L-phenylalaninal with alcohols. We describe herein the successful anti oxyhomologation of this substrate to prepare ester derivatives of the 2S,3S stereoisomer of allophenylnorstatin.15
Results and discussion
We began this study with a consideration of H-MAC-[Si] reagents. Almost all of the work described previously in the literature has employed 2-(tert-butyldimethylsilyloxy)malononitrile 1a (H-MAC-TBS);7j–r,8c–g,9a–d the related triisopropylsilyl and tert-butyldiphenylsilyl derivatives 1b (H-MAC-TIPS)7m and 1c (H-MAC-TBDPS)12 have each been alluded to on only one occasion. All three of these reagents are appealing since they possess significant steric bulk, which might have an advantageous impact on the diastereoselectivity of the oxyhomologation reaction. However, an important factor for the synthetic utility of any H-MAC-[Si] reagent is a convenient and reliable access to the reagent itself. No syntheses of the derivatives 1b and 1c have been described to date. The earliest synthesis of 1a required a rather inefficient five-step sequence starting from diethyl 2-bromomalonate.7a Subsequently, a three-step procedure was proposed starting from malononitrile, requiring acetylation, oxidative cleavage, then silyl ether formation;16 this is the basis of the approach that we have adopted for our preparative procedures (Scheme 1). | ||
| Scheme 1 Reaction conditions. Reactions carried out on 4.6 mmol scale unless stated. a: R3SiCl (1.5 equiv.), imidazole (2 equiv.), DMF; R3Si = TBS or TBDPS, 30 min at 0 °C then 30 min at rt; R3Si = TIPS, 16 h, 0 °C slowly to rt. b: R3SiOTf (1.5 equiv.), 2,6-lutidine (2 equiv.), CH2Cl2; R3Si = TBS, 30 min at 0 °C then 30 min at rt; R3Si = TIPS or TBDPS, 16 h, 0 °C slowly to rt. c: Conducted on 20 mmol scale. | ||
Acetylation of malononitrile17 proceeded smoothly to provide 2 essentially in its enol tautomer form in 94% yield. Oxidative cleavage was performed using peracetic acid and, in our experience, the quality of the resulting 2-hydroxymalononitrile 3 was critical; its use immediately after preparation led to the best yields in the subsequent silylation step. For this latter, we compared the efficacy of silyl triflates with that of silyl chlorides on a 4.6 mmol scale. In all three cases the silylation was noticeably more efficient and reproducible with the triflate (yields 78–87%) than with the corresponding chloride (yields 60–65%) (Scheme 1). Using this adaptation and extension of the original procedure, the three MAC reagents 1a-c were prepared conveniently on around gram scale. Gratifyingly, when the H-MAC-TBS 1a synthesis was conducted at a preparative 20 mmol scale, the isolated yield of 1a from 2 improved to 97%.
Each H-MAC-[Si] reagent 1a–c was evaluated in the oxyhomologation of N,N-dibenzyl-L-phenylalaninal using methanol as the standard alcohol component. On the basis of previous work, DMAP was used as the mild base and reactions were run overnight in ether at 0 °C. The number of equivalents of MAC reagent and base were screened, invariably in the presence of a sufficient excess of methanol. Results are presented in Table 1. For H-MAC-TBS 1a, use of a slight excess of the reagent gave good yields of products 4a/4a′ (entries 1 and 2), and this improved when a larger excess of the reagent was employed (entries 3 and 4), reaching 83% in the presence of 2 equivalents of base. Running the reaction at room temperature (entry 5) was slightly deleterious to the yield. For H-MAC-TIPS 1b, a very similar reactivity profile was observed. With one equivalent of reagent and base the yield of 4b/4b′ was moderate 56% (entry 6) but improved when the quantity of either the reagent or the base was increased (entries 7 and 8) and reached a satisfying 80% in the presence of two equivalents of each (entry 9). Running the reaction at room temperature had a marginally negative effect on the yield (entry 10). For H-MAC-TBDPS 1c, a comparable reactivity profile was once again observed, with yields of 4c/4c′ ranging from 51 to 74% depending on the number of reagent equivalents (entries 11–14). Running the reaction at room temperature had no perceptible effect on the yield (entry 15).
|
|
|||||
|---|---|---|---|---|---|
| entry | H-MAC-[Si] (equiv.) | DMAP (equiv.) | Yieldb4/4′ (%) | dr (anti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :syn)c :syn)c |
|
| a Reaction conditions: N,N-dibenzyl-L-phenylalaninal (0.5 mmol; 1 equiv.), stated amounts of H-MAC-[Si] and DMAP, methanol (3 equiv.) in Et2O (5 mL), 16 h, 0 °C, under argon. b Isolated yields are given. c Determined by 1H NMR; see text for details. d Reaction run at room temperature. | |||||
| 1 | 1a | 1.2 | 1 | 68 | 94:6 |
| 2 | 1a | 1.2 | 2 | 77 | 92:8 |
| 3 | 1a | 2.4 | 1 | 80 | 92:8 |
| 4 | 1a | 2.4 | 2 | 83 |
92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :8 :8
|
| 5d | 1a | 2.4 | 2 | 78 | 91:9 |
| 6 | 1b | 1.2 | 1 | 56 | 93:7 |
| 7 | 1b | 1.2 | 2 | 65 | 93:7 |
| 8 | 1b | 2.4 | 1 | 71 | 94:6 |
| 9 | 1b | 2.4 | 2 | 80 |
91:9
|
| 10d | 1b | 2.4 | 2 | 70 | 91:9 |
| 11 | 1c | 1.2 | 1 | 51 | 92:8 |
| 12 | 1c | 1.2 | 2 | 51 | 92:8 |
| 13 | 1c | 2.4 | 1 | 71 | 93:7 |
| 14 | 1c | 2.4 | 2 | 74 |
93:7
|
| 15d | 1c | 2.4 | 2 | 74 | 91:9 |

The diastereomeric ratios (dr) were established by inspecting the 1H NMR spectra of the product mixtures and integrating the signals for protons whose chemical shifts were most conveniently differentiated between diastereomer pairs. In the event, this turned out to be the TBS methyl groups signals for 4a/4a′, the CHOTIPS signals for 4b/4b′ and the methyl ester signals for 4c/4c′. Within the precision limits of this method, the dr was uniformly high in all three cases, regardless of the reaction conditions involved, and was always greater than 10:1. The major diastereomer 4c crystallized and an X-ray diffraction study revealed that it had the 2S,3S configuration (Fig. 2),18 which confirmed that the MAC reaction had indeed proceeded with the desired anti diastereoselectivity. Intuitively we felt that the major diastereomers 4a and 4b should also be anti, and this was confirmed by subjecting each of the three samples 4a/4a′, 4b/4b′ and 4c/4c′ to selective desilylation using TBAF in THF to provide, in good yield after chromatography, a single product 5 (Scheme 2), whose spectroscopic and optical rotation data were the same as those published for the anti 2S,3S stereoisomer.15f In these transformations, the 1H NMR spectra of the crude reaction products indicated the presence of the minor syn diastereomers in silylated form, suggesting that the deprotection process may be diastereoselective, although we did not pursue this matter further.
 | ||
| Fig. 2 X-ray diffraction structure of compound 4c showing the 2S,3S configuration. For clarity, hydrogen atoms have been removed, with the exception of those at the stereogenic centres. | ||
 | ||
| Scheme 2 Reaction conditions. For 4a/4a′ and 4b/4b′: 1 h, 0 °C. For 4c/4c′: 16 h, rt. | ||
The results collected in Table 1 show that all three H-MAC-[Si] reagents performed in a satisfactory manner, with all products 4a/4a′–4c/4c′ being obtained with high anti diastereoselectivity. Comparison of best yields (entries 4, 9 and 14) gave a marginal advantage to 1a over 1b which was in turn better than 1c (83%, 80% and 74% yields, respectively). As mentioned above, H-MAC-TBS is the most studied reagent so far and its synthesis was the most efficient in our hands so we continued our studies with this reagent and retained the conditions of entry 4 as standard.
The scope of the MAC oxyhomologation of N,N-dibenzyl-L-phenylalaninal was evaluated using a panel of alcohols. Results are presented in Scheme 3. As before, dr values were determined by integration of the 1H NMR signals of the TBS methyl groups and we considered it plausible that the anti diastereomer predominated in each case. Support for this contention was provided by the 1H NMR data, which invariably showed the diagnostic TBS methyl groups signals at lower field for the major anti isomer than for the minor syn isomer (see ESI†).
 | ||
| Scheme 3 Reaction conditions. N,N-dibenzyl-L-phenylalaninal (0.5 mmol; 1 equiv.), 1a (2.4 equiv.), DMAP (2 equiv.) and ROH (3 equiv.) in Et2O (5 mL), 16 h, 0 °C, under argon. Isolated yields are indicated. The dr was determined by 1H NMR; see text for details. Only the major anti diastereomer is illustrated. | ||
Ethanol and benzyl alcohol gave good yields of products 6/6′ and 7/7′ with high diastereoselectivity. With three other uncongested primary alcohols—allyl alcohol, propargyl alcohol and phenylethanol—the products (8/8′–10/10′ respectively) were likewise obtained in very good yields and high dr, reaching 95:5 for compound 10/10′. When the reaction was carried out with branched chain primary alcohols—isoamyl alcohol, isobutanol and (S)-2-methylbutanol—the yield of the oxyhomologation products (11/11′–13/13′ respectively) was more modest (46–64%), although the dr values remained consistently high. The presence of the chiral center in 2-methylbutanol had no perceptible effect on the dr value. With two representative secondary alcohols—isopropanol and cyclopentanol—the reactions were much less efficient; the desired products 14/14′ and 15/15′ were isolated only in low yields (11% and 14%, respectively), although the dr values were still just as high as those observed for primary alcohol substrates. Notwithstanding the limitations arising from the steric bulk of the alcohol, these oxyhomologation reactions provided rapid access to a selection of ester derivatives of N,O-protected (2S,3S)-allophenylnorstatin, some of which (e.g. allyl and propargyl) appear amenable to subsequent functionalization.
The methyl ester of (2S,3S)-allophenylnorstatin, itself a LTA4 hydrolase inhibitor,19 has been used as a building block for the preparation of BACE1 inhibitors,20 photobiological switches,21 and symmetrical peptidomimetic scaffolds;22 it has also served as an intermediate for the preparation of a variety of other biologically active compounds,23 as has the corresponding ethyl ester.24 These two ester derivatives were prepared readily as follows (Scheme 4). Catalytic hydrogenolysis of the single stereoisomer 5 (obtained viaScheme 2) gave the methyl ester 16 in high yield (88%). In a similar fashion, compound 17 was first obtained as a single anti isomer in 91% yield by selective desilylation of 6/6′ (dr 92:8) using TBAF in THF; taking the diastereomeric composition of the substrate into account, this equates to a near quantitative yield of the available anti component. Compound 17 was subjected to catalytic hydrogenolysis to provide ethyl ester 18 in good yield (73%). These procedures provide a convenient access to esters 16 and 18 as an alternative to the classical approach involving acid-mediated esterification of the parent amino acid.
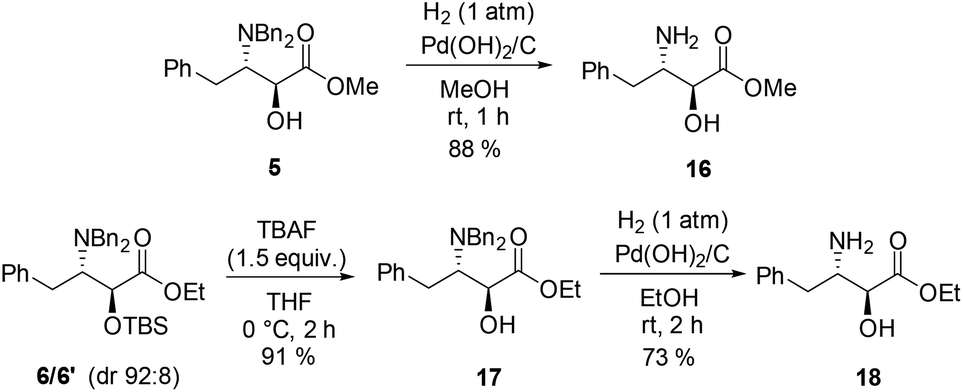 | ||
| Scheme 4 Preparation of (2S,3S)-allophenylnorstatin esters. | ||
To complete this study we prepared (2S,3S)-allophenylnorstatin in its free amino acid form (Scheme 5). The benzyl ester derivative 7/7′ (dr 92:8) was desilylated using TBAF to furnish exclusively the anti derivative 19 in 84% yield; on the basis of the diastereomeric composition of the substrate, this equates to a 91% yield of the available anti component. For the final step, catalytic hydrogenolysis of all three benzyl groups was envisaged in the presence of a palladium catalyst. Initial experiments carried out in methanol were hampered by the formation of mixtures of products resulting from partial N-debenzylation and the unwanted formation of N- and/or O-methylated derivatives.25 To circumvent this problem we performed the hydrogenation in ethyl acetate; mutatits mutandis, complete debenzylation was achieved cleanly to provide the target hydroxy amino acid 20 in 95% yield. Spectroscopic and optical rotation data were in full agreement with the literature data for the (2S,3S) stereoisomer. This 3-step synthesis of (2S,3S)-allophenylnorstatin in 66% overall yield from N,N-dibenzyl-L-phenylalaninal is one of the shortest and most efficient to date.26
 | ||
| Scheme 5 Preparation of (2S,3S)-allophenylnorstatin. | ||
Conclusions
This study reveals that MAC reactions between N,N-dibenzyl-L-phenylalaninal and alcohols proceed with high anti diastereoselectivity. These results contrast significantly with Nemoto's work on MAC reactions involving N-Cbz-phenylalaninal, which gave products with syn diastereoselectivity.7m The combined observations provide the first example of the MAC oxyhomologation of an α-amino aldehyde in a directed diastereoselective manner, through judicious choice of the amine protecting group suite. Using the new anti-directed formulation, a panel of ester derivatives of orthogonally N,O-protected (2S,3S)-allophenylnorstatins has been prepared. While the reactions were sensitive to the steric bulk of the alcohol, a number of synthetically useful esters were prepared in this way and an expedient new synthesis of the title amino acid in single-enantiomer form was achieved in only 3 steps.Experimental
General experimental methods
N,N-Dibenzyl-L-phenylalaninal {[α]24D = −91.2 (c 1.0, CHCl3), lit.27 [α]20D = −90.0 (c 1.0, CHCl3)} was prepared from commercial (S)-phenylalaninol according to literature procedure.28 Peracetic acid solution (35 wt% in acetic acid) was purchased from Acros. Solvents and reagents were purified under argon as follows: Et2O and THF were distilled from Na/benzophenone; DMF and 2,6-lutidine were distilled from CaH2; CH2Cl2 was passed through an activated alumina column immediately before use; methanol and ethanol were distilled from Mg/I2; benzyl alcohol, isopropanol, isobutanol, isoamyl alcohol and (S)-2-methylbutan-1-ol were distilled from CaO; allyl alcohol, propargyl alcohol and phenylethyl alcohol were dried over CaSO4, K2CO3 and Na2SO4 respectively, followed by distillation. All other reagents were obtained commercially and were used directly as supplied. Preparative flash chromatography was performed using columns packed with SDS (35–70 μm) or Macherey–Nagel (40–63 μm) silica gel. Analytical thin-layer chromatography, used to monitor preparative flash chromatography and to provide characteristic retention factors (Rf), was performed on 0.25 mm commercial silica gel plates (Merck 60F-254); plates were visualized by UV fluorescence at 254 nm and then revealed by heating after dipping in a ninhydrin solution (1.5% in n-BuOH) or a potassium permanganate solution (7.5% in water). 1H and 13C NMR spectra were recorded on Bruker DPX250 (250 and 62.9 MHz, respectively), Bruker AV300 (300 and 75.5 MHz, respectively), Bruker AV360 (360 and 90.6 MHz, respectively), Bruker Avance I 400 and Bruker Avance III 400 spectrometers (400 and 100.6 MHz, respectively). Chemical shifts (δ) are given in parts per million, using solvent signals as internal standards (CDCl3: δH = 7.26 ppm, δC = 77.0 ppm; DMSO-d6: δH = 2.50 ppm, δC = 39.5 ppm; CD3OD: δH = 3.31 ppm, δC = 49.0 ppm; D2O: δH = 4.79 ppm). Assignments were aided by JMOD pulse sequences and 2D experiments (HSQC, HMBC, COSY). Splitting patterns for 1H signals are designated as s (singlet), d (doublet), t (triplet), q (quartet), br s (broad singlet), or m (multiplet). Coupling constants (J) are reported in Hz. Positive and negative electrospray (ES+, ES−) high resolution mass spectra (HRMS) were recorded with a Bruker Daltonics micrOTOF-Q instrument. Infrared spectroscopy (IR) analyses were recorded on a FT-IR PerkinElmer Spectrum Two spectrophotometer using an ATR diamond accessory; maximum absorbances (ν) are given in cm−1. Elemental analyses were performed by the Microanalysis Service of the I.C.S.N. (Gif-sur-Yvette, France). Melting points (Mp) were determined with a Büchi M-560 apparatus in open capillary tubes and are uncorrected. Optical rotations were measured on a Jasco P-1010 polarimeter using a 10 cm quartz cell; values for [α]TD were obtained with the D-line of sodium at the indicated temperature T, using solutions of concentration (c) in units of g 100 mL−1.Preparation of MAC reagents: H-MAC-[Si]
:1). IR (ATR): ν 3045, 2610, 2238, 2225, 1600, 1574, 1507, 1402, 1359, 1227 cm−1. HRMS (ES−): calcd for C5H3N2O [M − H]− 107.0251; found 107.0250. 1H NMR (360 MHz, DMSO-d6), δ 2.21 (s, 3H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH3), 12.1 (br s, 1H, C–OH). 13C NMR (62.9 MHz, DMSO-d6), δ 21.2 (C–CH3), 59.2 (C(CN)2), 113.8 (C(CN)CN), 115.7 (C(CN)CN), 189.3 (C–OH). Anal. calcd for C5H4N2O: C, 55.56; H, 3.73; N, 25.91. Found: C, 55.54; H, 3.53; N, 25.97.
C–CH3), 12.1 (br s, 1H, C–OH). 13C NMR (62.9 MHz, DMSO-d6), δ 21.2 (C–CH3), 59.2 (C(CN)2), 113.8 (C(CN)CN), 115.7 (C(CN)CN), 189.3 (C–OH). Anal. calcd for C5H4N2O: C, 55.56; H, 3.73; N, 25.91. Found: C, 55.54; H, 3.53; N, 25.97.
General procedure for 2-hydroxymalononitrile silylation
:1; then concentration of fractions on a rotary evaporator with a water bath at 40 °C until 450 mbar) to give H-MAC-TBS 1a (583 mg, 65% over 2 steps) as a colorless liquid. According to method B: silylation of 2-hydroxymalononitrile 3 (380 mg, 4.63 mmol) was carried out with tert-butyldimethylsilyl triflate (1.6 mL, 6.97 mmol) and 2,6-lutidine in CH2Cl2 for 30 min at 0 °C followed by 30 min at room temperature. After work-up and concentration under reduced pressure (until 450 mbar), the pale brown liquid residue was purified by flash chromatography (pentane then pentane/Et2O, 20:1; then concentration of fractions on a rotary evaporator with a water bath at 40 °C until 450 mbar) to furnish compound 1a (790 mg, 87% over 2 steps) as a colorless liquid. H-MAC-TBS 1a can be stored under argon for several months at −18 °C. Rf 0.18 (pentane/Et2O, 20:1). IR (ATR): ν 2957, 2934, 2862, 2252, 1473, 1259, 1115 cm−1. 1H NMR (300 MHz, CDCl3), δ 0.28 (s, 6H, Si(CH3)2), 0.94 (s, 9H, SiC(CH3)3), 5.33 (s, 1H, CH(CN)2). 13C NMR (75.5 MHz, CDCl3), δ −5.5 (Si(CH3)2), 17.9 (SiC(CH3)3), 25.0 (SiC(CH3)3), 50.8 (CH(CN)2), 112.3 (CH(CN)2). Anal. calcd for C9H16N2OSi: C, 55.06; H, 8.21; N, 14.27. Found: C, 54.81; H, 8.12; N, 14.13. 1H and 13C NMR data were in agreement with those described in literature.7a
:1; then concentration of fractions on a rotary evaporator with a water bath at 40 °C until 450 mbar) to furnish compound 1a (3.79 g, 97% over 2 steps) as a colorless liquid.
:1; pentane then pentane/Et2O, 20:1) to separate H-MAC-TIPS from triisopropylsilanol. H-MAC-TIPS 1b (665 mg, 60% over 2 steps) was isolated as a colorless viscous liquid. According to method B: silylation of 2-hydroxymalononitrile 3 (380 mg, 4.63 mmol) was carried out with triisopropylsilyl triflate (1.9 mL, 7.05 mmol) and 2,6-lutidine in CH2Cl2 overnight from 0 °C to room temperature. After work-up and concentration under reduced pressure, the brown liquid residue was purified by two successive flash chromatographies (pentane then pentane/Et2O, 50:1; pentane then pentane/Et2O, 20:1) to separate H-MAC-TIPS from triisopropylsilanol. H-MAC-TIPS 1b (862 mg, 78% over 2 steps) was obtained as a colorless liquid. H-MAC-TIPS 1b can be stored under argon for several months at −18 °C. Rf 0.23 (pentane/Et2O, 20:1). IR (ATR): ν 2948, 2870, 2252, 1463, 1114 cm−1. 1H NMR (300 MHz, CDCl3), δ 1.07–1.14 (m, 18H, SiCH(CH3)2), 1.16–1.24 (m, 3H, SiCH(CH3)2), 5.40 (s, 1H, CH(CN)2). 13C NMR (75.5 MHz, CDCl3), δ 11.5 (SiCH(CH3)2), 17.3 (SiCH(CH3)2), 51.3 (CH(CN)2), 112.3 (CH(CN)2). Anal. Calcd for C12H22N2OSi: C, 60.46; H, 9.30; N, 11.75. Found: C, 60.20; H, 9.08; N, 11.67.
:1) to give H-MAC-TBDPS 1c (965 mg, 65% over 2 steps) as a colorless viscous oil, which crystallized as a white solid after standing for 24 h at −18 °C. According to method B: silylation of 2-hydroxymalononitrile 3 (380 mg, 4.63 mmol) was carried out with tert-butyldiphenylsilyl triflate29 (1.43 M in CHCl3, 5.0 mL, 7.15 mmol) and 2,6-lutidine in CH2Cl2 overnight from 0 °C to room temperature. After work-up and concentration under reduced pressure, the brown liquid residue was purified by flash chromatography (pentane then pentane/Et2O: 20:1) to furnish H-MAC-TBDPS 1c (1.18 g, 80% over 2 steps) as a colorless viscous oil, which crystallized as a white solid after standing for 16 h under high vacuum (10−2 mbar). H-MAC-TBDPS 1c can be stored under argon for several months at −18 °C. Mp 48 °C. Rf 0.25 (pentane/Et2O, 20:1). IR (ATR): ν 3074, 2962, 2933, 2890, 2860, 2253, 1590, 1472, 1428, 1112 cm−1. HRMS (ES+): calcd for C19H20N2NaOSi [M + Na]+ 343.1237; found 343.1228. 1H NMR (300 MHz, CDCl3), δ 1.15 (s, 9H, SiC(CH3)3), 5.08 (s, 1H, CH(CN)2), 7.46–7.59 (m, 6H, SiPh), 7.69–7.73 (m, 4H, SiPh). 13C NMR (75.5 MHz, CDCl3), δ 19.3 (SiC(CH3)3), 26.3 (SiC(CH3)3), 51.5 (CH(CN)2), 111.9 (CH(CN)2), 128.5 (CHPh), 129.3 (CPh), 131.2 (CHPh), 135.5 (CHPh). Anal. calcd for C19H20N2OSi: C, 71.21; H, 6.29; N, 8.74. Found: C, 71.22; H, 6.33; N, 8.55.
MAC reactions with alcohols as nucleophiles
:1) to give the corresponding ester MAC reaction products.
The diastereoisomeric ratio (dr) was determined by 1H NMR analysis (CDCl3 at 293 K) on crude products either before or (in the cases of 14/14′, 15/15′, 4b/4b′ and 4c/4c′) after chromatography. For product samples obtained from H-MAC-TBS, dr was determined by integration of the TBS methyl groups signals. For product samples obtained from H-MAC-TIPS, dr was determined by integration of the CHOTIPS signals. For product samples obtained from H-MAC-TBDPS, dr was determined by integration of the OMe signals.
:8, 196 mg, 83%), as a colorless oil. Rf 0.39 (pentane/Et2O, 20:1). IR (ATR): ν 3022, 2951, 2925, 2858, 1752, 1738, 1605, 1494, 1454, 1250, 1125 cm−1. HRMS (ES+): calcd for C31H42NO3Si [M + H]+ 504.2928; found 504.2904. Spectroscopic data for the major anti (2S,3S)-diastereomer (4a): 1H NMR (360 MHz, CDCl3), δ 0.08 (s, 3H, SiCH3), 0.13 (s, 3H, SiCH3), 0.96 (s, 9H, SiC(CH3)3), 2.91–3.05 (m, 2H, PhCH2CH), 3.37–3.44 (m, 1H, CHN), 3.53 (s, 3H, OCH3), 3.61 (d, J = 13.9 Hz, 2H) and 3.78 (d, J = 13.9 Hz, 2H) (AB syst., N(CH2Ph)2), 4.60 (d, J = 4.3 Hz, 1H, CHOTBS), 7.08–7.14 (m, 6H, Ph), 7.17–7.31 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.8 (Si(CH3)2), 18.2 (SiC(CH3)3), 25.8 (SiC(CH3)3), 32.6 (PhCH2CH), 51.5 (OCH3), 54.4 (N(CH2Ph)2), 62.7 (CHN), 71.9 (CHOTBS), 125.8 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.7 (CHPh), 129.6 (CHPh), 139.5 (CPh), 140.3 (CPh), 173.4 (COOCH3).
:9, 217 mg, 80%), as a colorless oil. Rf 0.36 (pentane/Et2O, 20:1). IR (ATR): ν 3028, 2946, 2864, 2804, 1755, 1738, 1604, 1495, 1455, 1256, 1122 cm−1. HRMS (ES+): calcd for C34H48NO3Si [M + H]+ 546.3398; found 546.3375. Spectroscopic data for the major anti (2S,3S)-diastereomer (4b): 1H NMR (250 MHz, CDCl3), δ 0.96–1.10 (m, 21H, Si(CH(CH3)2)3), 2.99 (dd, J = 14.8, 9.5 Hz, 1H) and 3.08 (dd, J = 14.8, 5.0 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.35–3.44 (m, 1H, CHN), 3.55 (s, 3H, OCH3), 3.60 (d, J = 13.8 Hz, 2H) and 3.69 (d, J = 13.8 Hz, 2H) (AB syst., N(CH2Ph)2), 4.62 (d, J = 5.8 Hz, 1H, CHOTIPS), 7.04–7.10 (m, 4H, Ph), 7.14–7.29 (m, 11H, Ph). 13C NMR (62.9 MHz, CDCl3), δ 12.7 (Si(CH(CH3)2)3), 18.1 (Si(CH(CH3)2)3), 32.8 (PhCH2CH), 51.4 (OCH3), 54.6 (N(CH2Ph)2), 63.8 (CHN), 73.2 (CHOTIPS), 125.9 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.1 (CHPh), 128.9 (CHPh), 129.6 (CHPh), 139.5 (CPh), 140.8 (CPh), 173.4 (COOCH3).
:7, 203 mg, 74%), as a colorless oil. Rf 0.20 (pentane/Et2O, 20:1). IR (ATR): ν 3027, 2950, 2929, 2855, 2800, 1748, 1601, 1496, 1451, 1428, 1360, 1257, 1106 cm−1. HRMS (ES+): calcd for C41H46NO3Si [M + H]+ 628.3241; found 628.3210. Spectroscopic data for the major anti (2S,3S)-diastereomer (4c): 1H NMR (360 MHz, CDCl3), δ 1.09 (s, 9H, SiC(CH3)3), 2.98 (dd, J = 14.8, 9.7 Hz, 1H) and 3.14 (dd, J = 14.8, 4.0 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.19 (s, 3H, OCH3), 3.38–3.44 (m, 1H, CHN), 3.45 (d, J = 13.7 Hz, 2H) and 3.62 (d, J = 13.7 Hz, 2H) (AB syst., N(CH2Ph)2), 4.54 (d, J = 5.4 Hz, 1H, CHOTBDPS), 6.96–7.02 (m, 4H, Ph), 7.11–7.17 (m, 7H, Ph), 7.20–7.27 (m, 4H, Ph), 7.32–7.43 (m, 6H, Ph), 7.62–7.67 (m, 4H, Ph). 13C NMR (100.6 MHz, CDCl3), δ 19.5 (SiC(CH3)3), 27.1 (SiC(CH3)3), 33.1 (PhCH2CH), 51.1 (OCH3), 54.4 (N(CH2Ph)2), 63.4 (CHN), 73.2 (CHOTBDPS), 125.9 (CHPh), 126.6 (CHPh), 127.4 (CHPh), 127.6 (CHPh), 127.8 (CHPh), 128.1 (CHPh), 128.8 (CHPh), 129.6 (CHPh), 129.7 (CHPh), 129.8 (CHPh), 132.9 (CPh), 133.0 (CPh), 136.0 (CHPh), 136.1 (CHPh), 139.4 (CPh), 140.6 (CPh), 172.5 (COOCH3).
:8; 161 mg, 0.32 mmol) in dry THF (10 mL) at 0 °C under argon, was added dropwise tetrabutylammonium fluoride (1 M in THF, 480 μL, 0.48 mmol). After stirring for 1 h at 0 °C, the reaction mixture was quenched by addition of a saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine (20 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash chromatography (pentane/EtOAc, 5:1) to furnish the anti alcohol 5 (100 mg, 80%), as a colorless oil. From4b/4b′: Following the above procedure using (2SR,3S)-3-(dibenzylamino)-2-(triisopropylsilyloxy)-4-phenylbutanoate 4b/4b′ (dr 91:9; 169 mg, 0.31 mmol), anti alcohol 5 (104 mg, 86%) was obtained after flash chromatography (pentane/EtOAc, 3:1). From4c/4c′: Following a minor modification of the above procedure (reaction performed at rt for 16 h) using methyl (2SR,3S)-2-(tert-butyldiphenylsilyloxy)-3-(dibenzylamino)-4-phenylbutanoate 4c/4c′ (dr 93:7; 200 mg, 0.32 mmol), anti alcohol 5 (88 mg, 71%) was obtained after flash chromatography (pentane/EtOAc, 5:1). Rf 0.20 (pentane/EtOAc, 5:1). [α]22D = +35.9 (c 1.0, CHCl3) [lit.14e [α]20D = + 35.8 (c 1.0, CHCl3)]. IR (ATR): ν 3513, 3062, 3026, 2952, 2803, 1729, 1602, 1494, 1453, 1252, 1220, 1105 cm−1. HRMS (ES+): calcd for C25H28NO3 [M + H]+ 390.2064; found 390.2051. 1H NMR (360 MHz, CDCl3), δ 2.82 (dd, J = 14.0, 7.6 Hz, 1H) and 3.04 (dd, J = 14.0, 7.2 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.14 (br s, 1H, CHOH), 3.43 (ddd, J = 7.6, 7.2, 1.8 Hz, 1H, CHN), 3.53 (s, 3H, OCH3), 3.67 (d, J = 13.7 Hz, 2H) and 3.83 (d, J = 13.7 Hz, 2H) (AB syst., N(CH2Ph)2), 4.50 (br s, 1H, CHOH), 7.04–7.10 (m, 2H, Ph), 7.19–7.32 (m, 13H, Ph). 13C NMR (90.6 MHz, CDCl3), δ 31.9 (PhCH2CH), 52.3 (OCH3), 54.5 (N(CH2Ph)2), 62.1 (CHN), 69.6 (CHOH), 126.1 (CHPh), 126.9 (CHPh), 128.0 (CHPh), 128.1 (CHPh), 128.8 (CHPh), 129.5 (CHPh), 139.0 (CPh), 139.5 (CPh), 174.9 (COOCH3).
:9, 174 mg, 73%), as a viscous colorless oil. Rf 0.29 (pentane/Et2O, 20:1). IR (ATR): ν 3027, 2956, 2929, 2854, 2796, 1747, 1730, 1601, 1494, 1454, 1365, 1254, 1130 cm−1. HRMS (ES+): calcd for C32H44NO3Si [M + H]+ 518.3085; found 518.3062. Spectroscopic data for the major anti (2S,3S)-diastereomer (6): 1H NMR (360 MHz, CDCl3), δ 0.09 (s, 3H, SiCH3), 0.13 (s, 3H, SiCH3), 0.96 (s, 9H, SiC(CH3)3), 1.10 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.90 (dd, J = 14.4, 5.0 Hz, 1H) and 2.98 (dd, J = 14.4, 8.8 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.34–3.41 (m, 1H, CHN), 3.57 (d, J = 13.7 Hz, 2H) and 3.83 (d, J = 13.7 Hz, 2H) (AB syst., N(CH2Ph)2), 3.87–4.05 (m, 2H, OCH2CH3), 4.62 (d, J = 3.6 Hz, 1H, CHOTBS), 7.02–7.07 (m, 2H, Ph), 7.08–7.13 (m, 4H, Ph), 7.15–7.23 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.7 (SiCH3), −4.6 (SiCH3), 14.0 (OCH2CH3), 18.2 (SiC(CH3)3), 25.8 (SiC(CH3)3), 32.6 (PhCH2CH), 54.4 (N(CH2Ph)2), 60.6 (OCH2CH3), 62.7 (CHN), 71.4 (CHOTBS), 125.7 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.8 (CHPh), 129.6 (CHPh), 139.6 (CPh), 140.2 (CPh), 173.1 (COOEt).
:8, 215 mg, 83%), as a colorless oil. Rf 0.34 (pentane/Et2O, 20:1). IR (ATR): ν 3031, 2956, 2929, 2858, 2801, 1752, 1730, 1601, 1494, 1454, 1363, 1253, 1123 cm−1. HRMS (ES+): calcd for C37H46NO3Si [M + H]+ 580.3241; found 580.3217. Spectroscopic data for the major anti (2S,3S)-diastereomer (7): 1H NMR (360 MHz, CDCl3), δ 0.05 (s, 3H, SiCH3), 0.13 (s, 3H, SiCH3), 0.95 (s, 9H, SiC(CH3)3), 2.80 (dd, J = 14.4, 4.3 Hz, 1H) and 2.95 (dd, J = 14.4, 9.7 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.31–3.38 (m, 1H, CHN), 3.55 (d, J = 14.6 Hz, 2H) and 3.87 (d, J = 14.6 Hz, 2H) (AB syst., N(CH2Ph)2), 4.71 (d, J = 2.9 Hz, 1H, CHOTBS), 4.85 (d, J = 12.2 Hz, 1H) and 5.03 (d, J = 12.2 Hz, 1H) (AB syst., OCH2Ph), 6.82–6.91 (m, 2H, Ph), 7.03–7.11 (m, 6H, Ph), 7.12–7.19 (m, 9H, Ph), 7.27–7.31 (m, 3H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.7 (SiCH3), −4.6 (SiCH3), 18.2 (SiC(CH3)3), 25.8 (SiC(CH3)3), 32.4 (PhCH2CH), 54.4 (N(CH2Ph)2), 62.6 (CHN), 66.3 (OCH2Ph), 70.9 (CHOTBS), 125.7 (CHPh), 126.6 (CHPh), 127.8 (CHPh), 128.0 (CHPh), 128.1 (CHPh), 128.4 (CHPh), 128.6 (CHPh), 129.7 (CHPh), 135.4 (CPh), 139.6 (CPh), 140.1 (CPh), 172.9 (COOBn).
:9, 188 mg, 82%), as a colorless oil. Rf 0.35 (pentane/Et2O, 20:1). IR (ATR): ν 3027, 2956, 2929, 2858, 2800, 1751, 1730, 1602, 1494 1453, 1362, 1252, 1126 cm−1. HRMS (ES+): calcd for C33H44NO3Si [M + H]+ 530.3085; found 530.3060. Spectroscopic data for the major anti (2S,3S)-diastereomer (8): 1H NMR (360 MHz, CDCl3), δ 0.08 (s, 3H, SiCH3), 0.13 (s, 3H, SiCH3), 0.96 (s, 9H, SiC(CH3)3), 2.91 (dd, J = 14.4, 5.0 Hz, 1H) and 2.99 (dd, J = 14.4, 9.0 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.37–3.45 (m, 1H, CHN), 3.58 (d, J = 14.0 Hz, 2H) and 3.83 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 4.35 (ddt, J = 13.0, 5.8, 1.1 Hz, 1H) and 4.43 (ddt, J = 13.0, 6.1, 1.1 Hz, 1H) (AB part of ABXM syst., OCH2CHCH2), 4.66 (d, J = 3.6 Hz, 1H, CHOTBS), 5.11–5.21 (m, 2H, OCH2CHCH2), 5.74 (dddd, J = 16.9, 10.4, 6.1, 5.8 Hz, 1H, OCH2CHCH2), 7.01–7.08 (m, 2H, Ph), 7.08–7.13 (m, 4H, Ph), 7.15–7.23 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.7 (SiCH3), −4.6 (SiCH3), 18.2 (SiC(CH3)3), 25.9 (SiC(CH3)3), 32.5 (PhCH2CH), 54.5 (N(CH2Ph)2), 62.7 (CHN), 65.3 (OCH2CHCH2), 71.4 (CHOTBS), 118.6 (OCH2CHCH2), 125.8 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.7 (CHPh), 129.7 (CHPh), 131.7 (OCH2CHCH2), 139.5 (CPh), 140.1 (CPh), 172.7 (COOAll).
:7, 217 mg, 95%), as a colorless oil. Rf 0.15 (pentane/Et2O, 20:1). IR (ATR): ν 3311, 3027, 2951, 2929, 2856, 2802, 2134, 1757, 1740, 1602, 1493, 1452, 1363, 1250, 1124 cm−1. HRMS (ES+): calcd for C33H42NO3Si [M + H]+ 528.2928; found 528.2906. Spectroscopic data for the major anti (2S,3S)-diastereomer (9): 1H NMR (300 MHz, CDCl3), δ 0.09 (s, 3H, SiCH3), 0.13 (s, 3H, SiCH3), 0.95 (s, 9H, SiC(CH3)3), 2.42 (t, J = 2.7 Hz, 1H, OCH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 2.89–3.03 (m, 2H, PhCH2CH), 3.36–3.45 (m, 1H, CHN), 3.58 (d, J = 14.0 Hz, 2H) and 3.79 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 4.40 (dd, J = 15.6, 2.7 Hz, 1H) and 4.56 (dd, J = 15.6, 2.7 Hz, 1H) (AB syst., OCH2CCH), 4.63 (d, J = 3.9 Hz, 1H, CHOTBS), 7.05–7.13 (m, 6H, Ph), 7.16–7.25 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.8 (SiCH3), −4.7 (SiCH3), 18.1 (SiC(CH3)3), 25.8 (SiC(CH3)3), 32.6 (PhCH2CH), 51.9 (OCH2CCH), 54.4 (N(CH2Ph)2), 62.6 (CHN), 71.5 (CHOTBS), 75.0 (OCH2CCH), 77.3 (OCH2CCH), 125.8 (CHPh), 126.7 (CHPh), 128.0 (CHPh), 128.8 (CHPh), 129.6 (CHPh), 139.4 (CPh), 140.1 (CPh), 172.2 (COOCH2CCH).
:5, 210 mg, 80%), as a colorless oil. Rf 0.05 (pentane/Et2O, 20:1). IR (ATR): ν 3027, 2953, 2929, 2857, 2801, 1751, 1729, 1601, 1495, 1453, 1361, 1254, 1130 cm−1. HRMS (ES+): calcd for C38H48NO3Si [M + H]+ 594.3398; found 594.3372. Spectroscopic data for the major anti (2S,3S)-diastereomer (10): 1H NMR (300 MHz, CDCl3), δ 0.05 (s, 3H, SiCH3), 0.11 (s, 3H, SiCH3), 0.95 (s, 9H, SiC(CH3)3), 2.75 (t, J = 7.2 Hz, 2H, OCH2CH2Ph), 2.84 (dd, J = 14.4, 4.8 Hz, 1H) and 2.98 (dd, J = 14.4, 9.0 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.32–3.43 (m, 1H, CHN), 3.57 (d, J = 14.2 Hz, 2H) and 3.83 (d, J = 14.2 Hz, 2H) (AB syst., N(CH2Ph)2), 4.12 (t, J = 7.2 Hz, 2H, OCH2CH2Ph), 4.63 (d, J = 3.3 Hz, 1H, CHOTBS), 6.97–7.04 (m, 2H, Ph), 7.06–7.13 (m, 6H, Ph), 7.16–7.35 (m, 12H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.8 (SiCH3), −4.6 (SiCH3), 18.2 (SiC(CH3)3), 25.9 (SiC(CH3)3), 32.5 (PhCH2CH), 34.8 (OCH2CH2Ph), 54.4 (N(CH2Ph)2), 62.5 (CHN), 65.1 (OCH2CH2Ph), 71.3 (CHOTBS), 125.7 (CHPh), 126.5 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.4 (CHPh), 128.6 (CHPh), 128.7 (CHPh), 129.7 (CHPh), 137.4 (CPh), 139.6 (CPh), 140.0 (CPh), 173.0 (COOCH2CH2Ph).
:9, 154 mg, 64%), as a colorless oil. Rf 0.44 (pentane/Et2O, 20:1). IR (ATR): ν 3031, 2956, 2928, 2857, 2801, 1750, 1726, 1603, 1494, 1455, 1362, 1253, 1126 cm−1. HRMS (ES+): calcd for C35H50NO3Si [M + H]+ 560.3554; found 560.3531. Spectroscopic data for the major anti (2S,3S)-diastereomer (11): 1H NMR (400 MHz, CDCl3), δ 0.10 (s, 3H, SiCH3), 0.14 (s, 3H, SiCH3), 0.83 (d, J = 6.8 Hz, 6H, OCH2CH2CH(CH3)2), 0.97 (s, 9H, SiC(CH3)3), 1.34 (q, J = 6.8 Hz, 2H, OCH2CH2CH(CH3)2), 1.44–1.52 (m, 1H, OCH2CH2CH(CH3)2), 2.88 (dd, J = 14.8, 4.8 Hz, 1H) and 3.00 (dd, J = 14.8, 9.2 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.34–3.41 (m, 1H, CHN), 3.57 (d, J = 13.6 Hz, 2H) and 3.87 (d, J = 13.6 Hz, 2H) (AB syst., N(CH2Ph)2), 3.90–4.02 (m, 2H, OCH2CH2CH(CH3)2), 4.66 (d, J = 2.8 Hz, 1H, CHOTBS), 7.01–7.05 (m, 2H, Ph), 7.06–7.11 (m, 4H, Ph), 7.14–7.23 (m, 9H, Ph). 13C NMR (100.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.5 (SiCH3), 18.2 (SiC(CH3)3), 22.3 (OCH2CH2CHCH3), 22.4 (OCH2CH2CHCH3), 24.7 (OCH2CH2CH(CH3)2), 25.9 (SiC(CH3)3), 32.5 (PhCH2CH), 37.1 (OCH2CH2CH(CH3)2), 54.4 (N(CH2Ph)2), 62.6 (CHN), 63.2 (OCH2CH2CH(CH3)2), 71.1 (CHOTBS), 125.8 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.7 (CHPh), 129.7 (CHPh), 139.6 (CPh), 140.1 (CPh), 173.2 (COOCH2).
:9, 105 mg, 46%), as a colorless oil. Rf 0.39 (pentane/Et2O, 20:1). IR (ATR): ν 3026, 2957, 2931, 2857, 2800, 1750, 1730, 1603, 1495, 1453, 1363, 1251, 1129 cm−1. HRMS (ES+): calcd for C34H48NO3Si [M + H]+ 546.3398; found 546.3375. Spectroscopic data for the major anti (2S,3S)-diastereomer (12): 1H NMR (250 MHz, CDCl3), δ 0.10 (s, 3H, SiCH3), 0.15 (s, 3H, SiCH3), 0.76 (d, J = 6.7 Hz, 3H, OCH2CHCH3), 0.79 (d, J = 6.7 Hz, 3H, OCH2CHCH3), 0.98 (s, 9H, SiC(CH3)3), 1.73 (nonuplet, J = 6.7 Hz, 1H, OCH2CH(CH3)2), 2.87 (dd, J = 14.5, 5.0 Hz, 1H) and 3.01 (dd, J = 14.5, 9.0 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.34–3.44 (m, 1H, CHN), 3.57 (d, J = 14.0 Hz, 2H) and 3.89 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 3.65 (dd, J = 10.5, 6.7 Hz, 1H) and 3.70 (dd, J = 10.5, 6.7 Hz, 1H) (AB part of ABX syst., OCH2CH(CH3)2), 4.70 (d, J = 2.8 Hz, 1H, CHOTBS), 6.97–7.06 (m, 2H, Ph), 7.06–7.12 (m, 4H, Ph), 7.14–7.24 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.5 (SiCH3), 18.2 (SiC(CH3)3), 19.1 (OCH2CH(CH3)2), 25.9 (SiC(CH3)3), 27.5 (OCH2CH(CH3)2), 32.4 (PhCH2CH), 54.4 (N(CH2Ph)2), 62.5 (CHN), 70.9 (OCH2CH(CH3)2), 71.0 (CHOTBS), 125.8 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.6 (CHPh), 129.7 (CHPh), 139.6 (CPh), 140.0 (CPh), 173.2 (COOi-Bu).
:10, 107 mg, 48%), as a colorless oil. Rf 0.27 (pentane/Et2O, 20:1). IR (ATR): ν 3027, 2957, 2929, 2858, 2800, 1747, 1726, 1603, 1496, 1453, 1361, 1254, 1128 cm−1. HRMS (ES+): calcd for C35H50NO3Si [M + H]+ 560.3554; found 560.3534. Spectroscopic data for the major anti (2S,3S)-diastereomer (13): 1H NMR (400 MHz, CDCl3), δ 0.10 (s, 3H, SiCH3), 0.14 (s, 3H, SiCH3), 0.74 (d, J = 6.8 Hz, 3H, OCH2CH(CH3)CH2CH3), 0.81 (t, J = 7.4 Hz, 3H, OCH2CH(CH3)CH2CH3), 0.97 (s, 9H, SiC(CH3)3), 1.18–1.28 (m, 2H, OCH2CH(CH3)CH2CH3), 1.46–1.56 (m, 1H, OCH2CH(CH3)CH2CH3), 2.86 (dd, J = 14.6, 4.8 Hz, 1H) and 3.01 (dd, J = 14.6, 9.6 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.35–3.42 (m, 1H, CHN), 3.57 (d, J = 14.0 Hz, 2H) and 3.90 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 3.69 (dd, J = 10.6, 6.6 Hz, 1H) and 3.78 (dd, J = 10.6, 6.2 Hz, 1H) (AB part of ABX syst., OCH2CH(CH3)CH2CH3), 4.69 (d, J = 2.4 Hz, 1H, CHOTBS), 7.00–7.04 (m, 2H, Ph), 7.06–7.11 (m, 4H, Ph), 7.13–7.24 (m, 9H, Ph). 13C NMR (100.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.5 (SiCH3), 11.1 (OCH2CH(CH3)CH2CH3), 16.2 (OCH2CH(CH3)CH2CH3), 18.2 (SiC(CH3)3), 25.8 (OCH2CH(CH3)CH2CH3), 25.9 (SiC(CH3)3), 32.4 (PhCH2CH), 33.9 (OCH2CH(CH3)CH2CH3), 54.4 (N(CH2Ph)2), 62.5 (CHN), 69.4 (OCH2CH(CH3)CH2CH3), 70.9 (CHOTBS), 125.8 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.6 (CHPh), 129.7 (CHPh), 139.6 (CPh), 140.0 (CPh), 173.3 (COOCH2).
:7, 21 mg, 11%), as a colorless oil. Rf 0.40 (pentane/Et2O, 20:1). IR (ATR): ν 3028, 2952, 2930, 2855, 2800, 1749, 1721, 1604, 1495, 1454, 1373, 1258, 1141, 1105 cm−1. HRMS (ES+): calcd for C33H46NO3Si [M + H]+ 532.3241; found 532.3217. Spectroscopic data for the major anti (2S,3S)-diastereomer (14): 1H NMR (360 MHz, CDCl3), δ 0.12 (s, 3H, SiCH3), 0.16 (s, 3H, SiCH3), 0.99 (s, 9H, SiC(CH3)3), 1.03 (d, J = 6.3 Hz, 3H, OCHCH3), 1.18 (d, J = 6.3 Hz, 3H, OCHCH3), 2.82 (dd, J = 14.8, 4.7 Hz, 1H) and 2.98 (dd, J = 14.8, 9.7 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.32 (ddd, J = 6.8, 4.7, 2.5 Hz, 1H, CHN), 3.52 (d, J = 14.0 Hz, 2H) and 3.93 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 4.68 (d, J = 2.5 Hz, 1H, CHOTBS), 4.86 (septuplet, J = 6.3 Hz, 1H, OCH(CH3)2), 6.94–7.02 (m, 2H, Ph), 7.05–7.12 (m, 4H, Ph), 7.15–7.23 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.4 (SiCH3), 18.2 (SiC(CH3)3), 21.5 (OCHCH3), 21.8 (OCHCH3), 25.9 (SiC(CH3)3), 32.4 (PhCH2CH), 54.3 (N(CH2Ph)2), 62.4 (CHN), 68.2 (OCH(CH3)2), 70.4 (CHOTBS), 125.7 (CHPh), 126.7 (CHPh), 127.8 (CHPh), 127.9 (CHPh), 128.7 (CHPh), 129.7 (CHPh), 139.6 (CPh), 140.1 (CPh), 172.9 (COOi-Pr).
:1) gave inseparable MAC reaction products 15/15′ (dr 90:10, 38 mg, 14%), as a viscous colorless oil. Rf 0.43 (pentane/Et2O, 10:1). IR (ATR): ν 3025, 2956, 2929, 2860, 2800, 1758, 1728, 1604, 1494, 1462, 1379, 1268, 1121 cm−1. HRMS (ES+): calcd for C35H48NO3Si [M + H]+ 558.3398; found 558.3371. Spectroscopic data for the major anti (2S,3S)-diastereomer (15): 1H NMR (300 MHz, CDCl3), δ 0.13 (s, 3H, SiCH3), 0.16 (s, 3H, SiCH3), 1.00 (s, 9H, SiC(CH3)3), 1.39–1.53 (m, 4H, OCH(CH2CH2)2), 1.56–1.71 (m, 4H, OCH(CH2CH2)2), 2.84 (dd, J = 14.7, 4.8 Hz, 1H) and 3.00 (dd, J = 14.7, 9.3 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.35 (ddd, J = 9.3, 4.8, 2.4 Hz, 1H, CHN), 3.55 (d, J = 14.0 Hz, 2H) and 3.93 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 4.68 (d, J = 2.4 Hz, 1H, CHOTBS), 4.96–5.03 (m, 1H, OCH(CH2CH2)2), 6.95–7.03 (m, 2H, Ph), 7.07–7.13 (m, 4H, Ph), 7.15–7.25 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.4 (SiCH3), 18.2 (SiC(CH3)3), 23.5 (OCHCH2CH2), 23.6 (OCHCH2CH2), 25.9 (SiC(CH3)3), 32.3 (PhCH2CH), 32.5 (OCH(CH2CH2)2), 54.4 (N(CH2Ph)2), 62.4 (CHN), 70.6 (CHOTBS), 77.6 (OCH(CH2CH2)2), 125.7 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.6 (CHPh), 129.7 (CHPh), 139.7 (CPh), 140.0 (CPh), 173.1 (COOCH).
:8; 322 mg, 0.62 mmol) in dry THF (15 mL) at 0 °C. After stirring for 2 h at 0 °C, the reaction mixture was quenched by addition of a saturated aqueous NH4Cl solution (5 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine (20 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash chromatography (pentane/CH2Cl2/Et2O, 10:1:1 then pentane/EtOAc, 5:1) to furnish the anti alcohol 17 (229 mg, 91%), as a colorless oil. Rf 0.34 (pentane/EtOAc, 5:1). [α]19D = +70.4 (c 1.0, CHCl3). IR (ATR): ν 3499, 3062, 3025, 2974, 2804, 1724, 1603, 1495, 1452, 1368, 1249, 1215, 1105 cm−1. HRMS (ES+): calcd for C26H30NO3 [M + H]+ 404.2220; found 404.2204. 1H NMR (360 MHz, CDCl3), δ 1.11 (t, J = 7.2 Hz, 3H, OCH2CH3), 2.78 (dd, J = 14.0, 7.2 Hz, 1H) and 3.00 (dd, J = 14.0, 7.2 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.12 (d, J = 6.1 Hz, 1H, CHOH), 3.44 (ddd, J = 7.2, 7.2, 2.1 Hz, 1H, CHN), 3.64 (d, J = 14.0 Hz, 2H) and 3.86 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 3.89–4.05 (m, 2H, OCH2CH3), 4.52 (dd, J = 6.1, 2.1 Hz, 1H, CHOH), 7.00–7.05 (m, 2H, Ph), 7.19–7.28 (m, 13H, Ph). 13C NMR (90.6 MHz, CDCl3), δ 13.8 (OCH2CH3), 31.9 (PhCH2CH), 54.5 (N(CH2Ph)2), 61.6 (OCH2CH3), 61.9 (CHN), 69.4 (CHOH), 126.0 (CHPh), 126.8 (CHPh), 128.0 (CHPh), 128.1 (CHPh), 128.8 (CHPh), 129.5 (CHPh), 139.0 (CPh), 139.6 (CPh), 174.6 (COOEt).
:8; 173 mg, 0.30 mmol) in dry THF (4 mL) at 0 °C under argon, was added dropwise tetrabutylammonium fluoride (1 M in THF, 450 μL, 0.45 mmol). After stirring for 50 minutes at 0 °C, the reaction mixture was quenched by addition of a saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine (20 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash chromatography (pentane/EtOAc, 10:1) to furnish the anti alcohol 19 (117 mg, 84%), as a colorless oil. Rf 0.32 (pentane/EtOAc, 5:1). [α]22D = +28.7 (c 1.0, CHCl3). IR (ATR): ν 3514, 3066, 3028, 2926, 2804, 1730, 1599, 1496, 1452, 1258, 1211, 1104 cm−1. HRMS (ES+): calcd for C31H32NO3 [M + H]+ 466.2376; found 466.2365. 1H NMR (300 MHz, CDCl3), δ 2.68 (dd, J = 14.1, 6.9 Hz, 1H) and 2.96 (dd, J = 14.1, 7.8 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.09 (d, J = 6.0 Hz, 1H, CHOH), 3.44 (ddd, J = 7.8, 6.9, 1.8 Hz, 1H, CHN), 3.61 (d, J = 14.0 Hz, 2H) and 3.86 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 4.60 (dd, J = 6.0, 1.8 Hz, 1H, CHOH), 4.88 (d, J = 12.3 Hz, 1H) and 4.92 (d, J = 12.3 Hz, 1H) (AB syst., OCH2Ph), 6.85–6.90 (m, 2H, Ph), 7.04–7.09 (m, 2H, Ph), 7.11–7.16 (m, 3H, Ph), 7.18–7.24 (m, 10H, Ph), 7.26–7.32 (m, 3H, Ph). 13C NMR (75.5 MHz, CDCl3), δ 31.9 (PhCH2CH), 54.5 (N(CH2Ph)2), 61.9 (CHN), 67.2 (OCH2Ph), 69.4 (CHOH), 126.0 (CHPh), 126.8 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.1 (CHPh), 128.3 (CHPh), 128.5 (CHPh), 128.7 (CHPh), 129.6 (CHPh), 134.8 (CPh), 138.9 (CPh), 139.6 (CPh), 174.5 (COOBn).
CH), 2.89–3.03 (m, 2H, PhCH2CH), 3.36–3.45 (m, 1H, CHN), 3.58 (d, J = 14.0 Hz, 2H) and 3.79 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 4.40 (dd, J = 15.6, 2.7 Hz, 1H) and 4.56 (dd, J = 15.6, 2.7 Hz, 1H) (AB syst., OCH2CCH), 4.63 (d, J = 3.9 Hz, 1H, CHOTBS), 7.05–7.13 (m, 6H, Ph), 7.16–7.25 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.8 (SiCH3), −4.7 (SiCH3), 18.1 (SiC(CH3)3), 25.8 (SiC(CH3)3), 32.6 (PhCH2CH), 51.9 (OCH2CCH), 54.4 (N(CH2Ph)2), 62.6 (CHN), 71.5 (CHOTBS), 75.0 (OCH2CCH), 77.3 (OCH2CCH), 125.8 (CHPh), 126.7 (CHPh), 128.0 (CHPh), 128.8 (CHPh), 129.6 (CHPh), 139.4 (CPh), 140.1 (CPh), 172.2 (COOCH2CCH).
:5, 210 mg, 80%), as a colorless oil. Rf 0.05 (pentane/Et2O, 20:1). IR (ATR): ν 3027, 2953, 2929, 2857, 2801, 1751, 1729, 1601, 1495, 1453, 1361, 1254, 1130 cm−1. HRMS (ES+): calcd for C38H48NO3Si [M + H]+ 594.3398; found 594.3372. Spectroscopic data for the major anti (2S,3S)-diastereomer (10): 1H NMR (300 MHz, CDCl3), δ 0.05 (s, 3H, SiCH3), 0.11 (s, 3H, SiCH3), 0.95 (s, 9H, SiC(CH3)3), 2.75 (t, J = 7.2 Hz, 2H, OCH2CH2Ph), 2.84 (dd, J = 14.4, 4.8 Hz, 1H) and 2.98 (dd, J = 14.4, 9.0 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.32–3.43 (m, 1H, CHN), 3.57 (d, J = 14.2 Hz, 2H) and 3.83 (d, J = 14.2 Hz, 2H) (AB syst., N(CH2Ph)2), 4.12 (t, J = 7.2 Hz, 2H, OCH2CH2Ph), 4.63 (d, J = 3.3 Hz, 1H, CHOTBS), 6.97–7.04 (m, 2H, Ph), 7.06–7.13 (m, 6H, Ph), 7.16–7.35 (m, 12H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.8 (SiCH3), −4.6 (SiCH3), 18.2 (SiC(CH3)3), 25.9 (SiC(CH3)3), 32.5 (PhCH2CH), 34.8 (OCH2CH2Ph), 54.4 (N(CH2Ph)2), 62.5 (CHN), 65.1 (OCH2CH2Ph), 71.3 (CHOTBS), 125.7 (CHPh), 126.5 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.4 (CHPh), 128.6 (CHPh), 128.7 (CHPh), 129.7 (CHPh), 137.4 (CPh), 139.6 (CPh), 140.0 (CPh), 173.0 (COOCH2CH2Ph).
:9, 154 mg, 64%), as a colorless oil. Rf 0.44 (pentane/Et2O, 20:1). IR (ATR): ν 3031, 2956, 2928, 2857, 2801, 1750, 1726, 1603, 1494, 1455, 1362, 1253, 1126 cm−1. HRMS (ES+): calcd for C35H50NO3Si [M + H]+ 560.3554; found 560.3531. Spectroscopic data for the major anti (2S,3S)-diastereomer (11): 1H NMR (400 MHz, CDCl3), δ 0.10 (s, 3H, SiCH3), 0.14 (s, 3H, SiCH3), 0.83 (d, J = 6.8 Hz, 6H, OCH2CH2CH(CH3)2), 0.97 (s, 9H, SiC(CH3)3), 1.34 (q, J = 6.8 Hz, 2H, OCH2CH2CH(CH3)2), 1.44–1.52 (m, 1H, OCH2CH2CH(CH3)2), 2.88 (dd, J = 14.8, 4.8 Hz, 1H) and 3.00 (dd, J = 14.8, 9.2 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.34–3.41 (m, 1H, CHN), 3.57 (d, J = 13.6 Hz, 2H) and 3.87 (d, J = 13.6 Hz, 2H) (AB syst., N(CH2Ph)2), 3.90–4.02 (m, 2H, OCH2CH2CH(CH3)2), 4.66 (d, J = 2.8 Hz, 1H, CHOTBS), 7.01–7.05 (m, 2H, Ph), 7.06–7.11 (m, 4H, Ph), 7.14–7.23 (m, 9H, Ph). 13C NMR (100.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.5 (SiCH3), 18.2 (SiC(CH3)3), 22.3 (OCH2CH2CHCH3), 22.4 (OCH2CH2CHCH3), 24.7 (OCH2CH2CH(CH3)2), 25.9 (SiC(CH3)3), 32.5 (PhCH2CH), 37.1 (OCH2CH2CH(CH3)2), 54.4 (N(CH2Ph)2), 62.6 (CHN), 63.2 (OCH2CH2CH(CH3)2), 71.1 (CHOTBS), 125.8 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.7 (CHPh), 129.7 (CHPh), 139.6 (CPh), 140.1 (CPh), 173.2 (COOCH2).
:9, 105 mg, 46%), as a colorless oil. Rf 0.39 (pentane/Et2O, 20:1). IR (ATR): ν 3026, 2957, 2931, 2857, 2800, 1750, 1730, 1603, 1495, 1453, 1363, 1251, 1129 cm−1. HRMS (ES+): calcd for C34H48NO3Si [M + H]+ 546.3398; found 546.3375. Spectroscopic data for the major anti (2S,3S)-diastereomer (12): 1H NMR (250 MHz, CDCl3), δ 0.10 (s, 3H, SiCH3), 0.15 (s, 3H, SiCH3), 0.76 (d, J = 6.7 Hz, 3H, OCH2CHCH3), 0.79 (d, J = 6.7 Hz, 3H, OCH2CHCH3), 0.98 (s, 9H, SiC(CH3)3), 1.73 (nonuplet, J = 6.7 Hz, 1H, OCH2CH(CH3)2), 2.87 (dd, J = 14.5, 5.0 Hz, 1H) and 3.01 (dd, J = 14.5, 9.0 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.34–3.44 (m, 1H, CHN), 3.57 (d, J = 14.0 Hz, 2H) and 3.89 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 3.65 (dd, J = 10.5, 6.7 Hz, 1H) and 3.70 (dd, J = 10.5, 6.7 Hz, 1H) (AB part of ABX syst., OCH2CH(CH3)2), 4.70 (d, J = 2.8 Hz, 1H, CHOTBS), 6.97–7.06 (m, 2H, Ph), 7.06–7.12 (m, 4H, Ph), 7.14–7.24 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.5 (SiCH3), 18.2 (SiC(CH3)3), 19.1 (OCH2CH(CH3)2), 25.9 (SiC(CH3)3), 27.5 (OCH2CH(CH3)2), 32.4 (PhCH2CH), 54.4 (N(CH2Ph)2), 62.5 (CHN), 70.9 (OCH2CH(CH3)2), 71.0 (CHOTBS), 125.8 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.6 (CHPh), 129.7 (CHPh), 139.6 (CPh), 140.0 (CPh), 173.2 (COOi-Bu).
:10, 107 mg, 48%), as a colorless oil. Rf 0.27 (pentane/Et2O, 20:1). IR (ATR): ν 3027, 2957, 2929, 2858, 2800, 1747, 1726, 1603, 1496, 1453, 1361, 1254, 1128 cm−1. HRMS (ES+): calcd for C35H50NO3Si [M + H]+ 560.3554; found 560.3534. Spectroscopic data for the major anti (2S,3S)-diastereomer (13): 1H NMR (400 MHz, CDCl3), δ 0.10 (s, 3H, SiCH3), 0.14 (s, 3H, SiCH3), 0.74 (d, J = 6.8 Hz, 3H, OCH2CH(CH3)CH2CH3), 0.81 (t, J = 7.4 Hz, 3H, OCH2CH(CH3)CH2CH3), 0.97 (s, 9H, SiC(CH3)3), 1.18–1.28 (m, 2H, OCH2CH(CH3)CH2CH3), 1.46–1.56 (m, 1H, OCH2CH(CH3)CH2CH3), 2.86 (dd, J = 14.6, 4.8 Hz, 1H) and 3.01 (dd, J = 14.6, 9.6 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.35–3.42 (m, 1H, CHN), 3.57 (d, J = 14.0 Hz, 2H) and 3.90 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 3.69 (dd, J = 10.6, 6.6 Hz, 1H) and 3.78 (dd, J = 10.6, 6.2 Hz, 1H) (AB part of ABX syst., OCH2CH(CH3)CH2CH3), 4.69 (d, J = 2.4 Hz, 1H, CHOTBS), 7.00–7.04 (m, 2H, Ph), 7.06–7.11 (m, 4H, Ph), 7.13–7.24 (m, 9H, Ph). 13C NMR (100.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.5 (SiCH3), 11.1 (OCH2CH(CH3)CH2CH3), 16.2 (OCH2CH(CH3)CH2CH3), 18.2 (SiC(CH3)3), 25.8 (OCH2CH(CH3)CH2CH3), 25.9 (SiC(CH3)3), 32.4 (PhCH2CH), 33.9 (OCH2CH(CH3)CH2CH3), 54.4 (N(CH2Ph)2), 62.5 (CHN), 69.4 (OCH2CH(CH3)CH2CH3), 70.9 (CHOTBS), 125.8 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.6 (CHPh), 129.7 (CHPh), 139.6 (CPh), 140.0 (CPh), 173.3 (COOCH2).
:7, 21 mg, 11%), as a colorless oil. Rf 0.40 (pentane/Et2O, 20:1). IR (ATR): ν 3028, 2952, 2930, 2855, 2800, 1749, 1721, 1604, 1495, 1454, 1373, 1258, 1141, 1105 cm−1. HRMS (ES+): calcd for C33H46NO3Si [M + H]+ 532.3241; found 532.3217. Spectroscopic data for the major anti (2S,3S)-diastereomer (14): 1H NMR (360 MHz, CDCl3), δ 0.12 (s, 3H, SiCH3), 0.16 (s, 3H, SiCH3), 0.99 (s, 9H, SiC(CH3)3), 1.03 (d, J = 6.3 Hz, 3H, OCHCH3), 1.18 (d, J = 6.3 Hz, 3H, OCHCH3), 2.82 (dd, J = 14.8, 4.7 Hz, 1H) and 2.98 (dd, J = 14.8, 9.7 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.32 (ddd, J = 6.8, 4.7, 2.5 Hz, 1H, CHN), 3.52 (d, J = 14.0 Hz, 2H) and 3.93 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 4.68 (d, J = 2.5 Hz, 1H, CHOTBS), 4.86 (septuplet, J = 6.3 Hz, 1H, OCH(CH3)2), 6.94–7.02 (m, 2H, Ph), 7.05–7.12 (m, 4H, Ph), 7.15–7.23 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.4 (SiCH3), 18.2 (SiC(CH3)3), 21.5 (OCHCH3), 21.8 (OCHCH3), 25.9 (SiC(CH3)3), 32.4 (PhCH2CH), 54.3 (N(CH2Ph)2), 62.4 (CHN), 68.2 (OCH(CH3)2), 70.4 (CHOTBS), 125.7 (CHPh), 126.7 (CHPh), 127.8 (CHPh), 127.9 (CHPh), 128.7 (CHPh), 129.7 (CHPh), 139.6 (CPh), 140.1 (CPh), 172.9 (COOi-Pr).
:1) gave inseparable MAC reaction products 15/15′ (dr 90:10, 38 mg, 14%), as a viscous colorless oil. Rf 0.43 (pentane/Et2O, 10:1). IR (ATR): ν 3025, 2956, 2929, 2860, 2800, 1758, 1728, 1604, 1494, 1462, 1379, 1268, 1121 cm−1. HRMS (ES+): calcd for C35H48NO3Si [M + H]+ 558.3398; found 558.3371. Spectroscopic data for the major anti (2S,3S)-diastereomer (15): 1H NMR (300 MHz, CDCl3), δ 0.13 (s, 3H, SiCH3), 0.16 (s, 3H, SiCH3), 1.00 (s, 9H, SiC(CH3)3), 1.39–1.53 (m, 4H, OCH(CH2CH2)2), 1.56–1.71 (m, 4H, OCH(CH2CH2)2), 2.84 (dd, J = 14.7, 4.8 Hz, 1H) and 3.00 (dd, J = 14.7, 9.3 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.35 (ddd, J = 9.3, 4.8, 2.4 Hz, 1H, CHN), 3.55 (d, J = 14.0 Hz, 2H) and 3.93 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 4.68 (d, J = 2.4 Hz, 1H, CHOTBS), 4.96–5.03 (m, 1H, OCH(CH2CH2)2), 6.95–7.03 (m, 2H, Ph), 7.07–7.13 (m, 4H, Ph), 7.15–7.25 (m, 9H, Ph). 13C NMR (90.6 MHz, CDCl3), δ −4.6 (SiCH3), −4.4 (SiCH3), 18.2 (SiC(CH3)3), 23.5 (OCHCH2CH2), 23.6 (OCHCH2CH2), 25.9 (SiC(CH3)3), 32.3 (PhCH2CH), 32.5 (OCH(CH2CH2)2), 54.4 (N(CH2Ph)2), 62.4 (CHN), 70.6 (CHOTBS), 77.6 (OCH(CH2CH2)2), 125.7 (CHPh), 126.7 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.6 (CHPh), 129.7 (CHPh), 139.7 (CPh), 140.0 (CPh), 173.1 (COOCH).
:8; 322 mg, 0.62 mmol) in dry THF (15 mL) at 0 °C. After stirring for 2 h at 0 °C, the reaction mixture was quenched by addition of a saturated aqueous NH4Cl solution (5 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine (20 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash chromatography (pentane/CH2Cl2/Et2O, 10:1:1 then pentane/EtOAc, 5:1) to furnish the anti alcohol 17 (229 mg, 91%), as a colorless oil. Rf 0.34 (pentane/EtOAc, 5:1). [α]19D = +70.4 (c 1.0, CHCl3). IR (ATR): ν 3499, 3062, 3025, 2974, 2804, 1724, 1603, 1495, 1452, 1368, 1249, 1215, 1105 cm−1. HRMS (ES+): calcd for C26H30NO3 [M + H]+ 404.2220; found 404.2204. 1H NMR (360 MHz, CDCl3), δ 1.11 (t, J = 7.2 Hz, 3H, OCH2CH3), 2.78 (dd, J = 14.0, 7.2 Hz, 1H) and 3.00 (dd, J = 14.0, 7.2 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.12 (d, J = 6.1 Hz, 1H, CHOH), 3.44 (ddd, J = 7.2, 7.2, 2.1 Hz, 1H, CHN), 3.64 (d, J = 14.0 Hz, 2H) and 3.86 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 3.89–4.05 (m, 2H, OCH2CH3), 4.52 (dd, J = 6.1, 2.1 Hz, 1H, CHOH), 7.00–7.05 (m, 2H, Ph), 7.19–7.28 (m, 13H, Ph). 13C NMR (90.6 MHz, CDCl3), δ 13.8 (OCH2CH3), 31.9 (PhCH2CH), 54.5 (N(CH2Ph)2), 61.6 (OCH2CH3), 61.9 (CHN), 69.4 (CHOH), 126.0 (CHPh), 126.8 (CHPh), 128.0 (CHPh), 128.1 (CHPh), 128.8 (CHPh), 129.5 (CHPh), 139.0 (CPh), 139.6 (CPh), 174.6 (COOEt).
:8; 173 mg, 0.30 mmol) in dry THF (4 mL) at 0 °C under argon, was added dropwise tetrabutylammonium fluoride (1 M in THF, 450 μL, 0.45 mmol). After stirring for 50 minutes at 0 °C, the reaction mixture was quenched by addition of a saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine (20 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash chromatography (pentane/EtOAc, 10:1) to furnish the anti alcohol 19 (117 mg, 84%), as a colorless oil. Rf 0.32 (pentane/EtOAc, 5:1). [α]22D = +28.7 (c 1.0, CHCl3). IR (ATR): ν 3514, 3066, 3028, 2926, 2804, 1730, 1599, 1496, 1452, 1258, 1211, 1104 cm−1. HRMS (ES+): calcd for C31H32NO3 [M + H]+ 466.2376; found 466.2365. 1H NMR (300 MHz, CDCl3), δ 2.68 (dd, J = 14.1, 6.9 Hz, 1H) and 2.96 (dd, J = 14.1, 7.8 Hz, 1H) (AB part of ABX syst., PhCH2CH), 3.09 (d, J = 6.0 Hz, 1H, CHOH), 3.44 (ddd, J = 7.8, 6.9, 1.8 Hz, 1H, CHN), 3.61 (d, J = 14.0 Hz, 2H) and 3.86 (d, J = 14.0 Hz, 2H) (AB syst., N(CH2Ph)2), 4.60 (dd, J = 6.0, 1.8 Hz, 1H, CHOH), 4.88 (d, J = 12.3 Hz, 1H) and 4.92 (d, J = 12.3 Hz, 1H) (AB syst., OCH2Ph), 6.85–6.90 (m, 2H, Ph), 7.04–7.09 (m, 2H, Ph), 7.11–7.16 (m, 3H, Ph), 7.18–7.24 (m, 10H, Ph), 7.26–7.32 (m, 3H, Ph). 13C NMR (75.5 MHz, CDCl3), δ 31.9 (PhCH2CH), 54.5 (N(CH2Ph)2), 61.9 (CHN), 67.2 (OCH2Ph), 69.4 (CHOH), 126.0 (CHPh), 126.8 (CHPh), 127.9 (CHPh), 128.0 (CHPh), 128.1 (CHPh), 128.3 (CHPh), 128.5 (CHPh), 128.7 (CHPh), 129.6 (CHPh), 134.8 (CPh), 138.9 (CPh), 139.6 (CPh), 174.5 (COOBn).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The award of a Guangzhou Elite Project Doctoral Research Scholarship (to X. H.) is acknowledged.Notes and references
- (a) D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239–258 CrossRef; (b) D. Seebach and D. Enders, Angew. Chem., Int. Ed. Engl., 1975, 14, 15–32 CrossRef.
- T. A. Hase, Umpoled Synthons, John Wiley, New York, 1987 Search PubMed.
- (a) D. Seebach, Synthesis, 1969, 17–36 CAS; (b) M. Yus, C. Najera and F. Foubelo, Tetrahedron, 2003, 59, 6147–6212 CrossRef CAS; (c) A. B. Smith III and C. M. Adams, Acc. Chem. Res., 2004, 37, 365–377 CrossRef PubMed; (d) B.-T. Gröbel and D. Seebach, Synthesis, 1977, 357–402 CrossRef; (e) A. B. Smith III, S. M. Condon and J. A. McCauley, Acc. Chem. Res., 1998, 31, 35–46 CrossRef.
- (a) R. Brehme, D. Enders, R. Fernandez and J. M. Lassaletta, Eur. J. Org. Chem., 2007, 5629–5660 CrossRef CAS; (b) D. Enders, L. Wortmann and R. Peters, Acc. Chem. Res., 2000, 33, 157–169 CrossRef CAS PubMed.
- (a) G. Stork and L. Maldonado, J. Am. Chem. Soc., 1971, 93, 5286–5287 CrossRef CAS; (b) J. D. Albright, Tetrahedron, 1983, 39, 3207–3233 CrossRef CAS.
- (a) G. Stork, A. A. Ozirio and A. Y. W. Leong, Tetrahedron Lett., 1978, 5175–5178 CrossRef CAS; (b) D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359–373 RSC; (c) T. Opatz, Synthesis, 2009, 1941–1959 CrossRef CAS; (d) V. Beaufort-Droal, E. Pereira, F. Vergne and D. J. Aitken, Synlett, 2010, 2913–2917 CAS.
- (a) H. Nemoto, Y. Kubota and Y. Yamamoto, J. Org. Chem., 1990, 55, 4515–4516 CrossRef CAS; (b) Y. Kubota, H. Nemoto and Y. Yamamoto, J. Org. Chem., 1991, 56, 7195–7196 CrossRef CAS; (c) H. Nemoto, Y. Kubota and Y. Yamamoto, J. Chem. Soc., Chem. Commun., 1994, 1665–1666 RSC; (d) Y. Yamamoto, Y. Kubota, Y. Honda, H. Fukui, N. Asao and H. Nemoto, J. Am. Chem. Soc., 1994, 116, 3161–3162 CrossRef CAS; (e) I. Kadota, A. Shibuya, Y. S. Gyoung and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 10262–10263 CrossRef CAS; (f) H. Nemoto, R. Ma, T. Ibaragi, I. Suzuki and M. Shibuya, Tetrahedron, 2000, 56, 1463–1468 CrossRef CAS; (g) N. T. Patil, I. Kadota, A. Shibuya, Y. S. Gyoung and Y. Yamamoto, Adv. Synth. Catal., 2004, 346, 800–804 CrossRef CAS; (h) H. Nemoto, T. Kawamura and N. Miyoshi, J. Am. Chem. Soc., 2005, 127, 14546–14547 CrossRef CAS PubMed; (i) H. Nemoto, T. Kawamura, K. Kitasaki, K. Yatsuzuka, M. Kamiya and Y. Yoshioka, Synthesis, 2009, 1694–1702 CrossRef CAS; (j) H. Nemoto, T. Ibaragi, M. Bando, M. Kido and M. Shibuya, Tetrahedron Lett., 1999, 40, 1319–1322 CrossRef CAS; (k) H. Nemoto, R. Ma, I. Suzuki and M. Shibuya, Org. Lett., 2000, 2, 4245–4247 CrossRef CAS PubMed; (l) H. Nemoto, R. Ma, H. Moriguchi, I. Suzuki and M. Shibuya, J. Organomet. Chem., 2000, 611, 445–448 CrossRef CAS; (m) H. Nemoto, R. Ma, X. Li, I. Suzuki and M. Shibuya, Tetrahedron Lett., 2001, 42, 2145–2147 CrossRef CAS; (n) H. Nemoto, R. Ma, T. Kawamura, M. Kamiya and M. Shibuya, J. Org. Chem., 2006, 71, 6038–6043 CrossRef CAS PubMed; (o) H. Nemoto, H. Moriguchi, R. Ma, T. Kawamura, M. Kamiya and M. Shibuya, Tetrahedron: Asymmetry, 2007, 18, 383–389 CrossRef CAS; (p) H. Nemoto, R. Ma, H. Moriguchi, T. Kawamura, M. Kamiya and M. Shibuya, J. Org. Chem., 2007, 72, 9850–9853 CrossRef CAS PubMed; (q) H. Nemoto, R. Ma, T. Kawamura, K. Yatsuzuka, M. Kamiya and M. Shibuya, Synthesis, 2008, 3819–3827 CrossRef CAS; (r) T. Kawamura, N. Matsuo, D. Yamauchi, Y. Tanabe and H. Nemoto, Tetrahedron, 2013, 69, 5331–5341 CrossRef CAS.
- (a) K. Yamatsugu, L. Yin, S. Kamijo, Y. Kimura, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2009, 48, 1070–1076 CrossRef CAS PubMed; (b) K. Yamatsugu, M. Kanai and M. Shibasaki, Tetrahedron, 2009, 65, 6017–6024 CrossRef CAS; (c) S. P. Roche, S. Faure and D. J. Aitken, Angew. Chem., Int. Ed., 2008, 47, 6840–6842 CrossRef CAS PubMed; (d) S. Mahapatra and R. G. Carter, J. Am. Chem. Soc., 2013, 135, 10792–10803 CrossRef CAS PubMed; (e) S. S. Kher, M. Penzo, S. Fulle, P. W. Finn, M. J. Blackman and A. Jirgensons, Bioorg. Med. Chem. Lett., 2014, 24, 4486–4489 CrossRef CAS PubMed; (f) Y. Nomura, F. Thuaud, D. Sekine, A. Ito, S. Maeda, H. Koshino, D. Hashizume, A. Muranaka, T. Cruchter, M. Uchiyama, S. Ichikawa, A. Matsuda, M. Yoshida, G. Hirai and M. Sodeoka, Chem. – Eur. J., 2019, 25, 8387–8392 CrossRef CAS PubMed; (g) C. He, H. Chu, T. P. Stratton, D. Kossler, K. J. Eberle, D. T. Flood and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 13683–13688 CrossRef CAS PubMed.
- (a) K. S. Yang, A. E. Nibbs, Y. E. Türkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 16050–16053 CrossRef CAS PubMed; (b) K. S. Yang and V. H. Rawal, J. Am. Chem. Soc., 2014, 136, 16148–16151 CrossRef CAS PubMed; (c) M. Esgulian, V. Belot, R. Guillot, S. Deloisy and D. J. Aitken, Org. Biomol. Chem., 2017, 15, 1453–1462 RSC; (d) K. Zhao, Y. Zhi, A. Wang and D. Enders, Synthesis, 2018, 50, 872–880 CrossRef CAS; (e) N. G. Jentsch, J. D. Hume, E. B. Crull, S. M. Beauti, A. H. Pham, J. A. Pigza, J. J. Kessl and M. G. Donahue, Beilstein J. Org. Chem., 2018, 14, 2529–2536 CrossRef CAS PubMed; (f) N. Kagawa, A. E. Nibbs and V. H. Rawal, Org. Lett., 2016, 18, 2363–2366 CrossRef CAS PubMed; (g) J. C. Hethcox, S. E. Shockley and B. M. Stoltz, Org. Lett., 2017, 19, 1527–1529 CrossRef CAS PubMed; (h) S. E. Shockley, J. C. Hethcox and B. M. Stoltz, Angew. Chem., Int. Ed., 2017, 56, 11545–11548 CrossRef CAS PubMed; (i) T. W. Butcher and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 13125–13129 CrossRef CAS PubMed; (j) J. B. Grimm, A. N. Tkachuk, L. Xie, H. Choi, B. Mohar, N. Falco, K. Schaefer, R. Patel, Q. Zheng, Z. Liu, J. Lippincott-Schwartz, T. A. Brown and L. D. Lavis, Nat. Methods, 2020, 17, 815–821 CrossRef CAS PubMed.
- (a) J. Chen, L. V. Kuznetsova, I. M. Ungreanu and I. Ojima, in Enantioselective Synthesis of β-Amino Acids, 2nd edn, ed. E. Juaristi and V. Soloshonok, Wiley, Hoboken, NJ, 2005, pp. 447–476 Search PubMed; (b) P. Spiteller and F. von Nussbaum, in Enantioselective Synthesis of β-Amino Acids, 2nd edn, ed. E. Juaristi and V. Soloshonok, Wiley, Hoboken, NJ, 2005, pp. 19–92 Search PubMed.
- Amino aldehyde oxyhomologations with dr (4:1) are reported in ref. 7k, 7m and 9c; similar reactions are reported without a dr in ref. 8c and 8e.
- M. Esgulian, M. Buchotte, R. Guillot, S. Deloisy and D. J. Aitken, Org. Lett., 2019, 21, 2378–2382 CrossRef CAS PubMed.
- Review: M. T. Reetz, Chem. Rev., 1999, 99, 1121–1162 CrossRef CAS PubMed.
- Illustrative examples: (a) F. P. Meirelis, B. G. N. Vieira and V. L. P. Pereira, Synthesis, 2020, 52, 3650–3656 CrossRef; (b) J. M. Concellón, H. Rodríguez-Solla and C. Concellón, J. Org. Chem., 2006, 71, 7919–7922 CrossRef PubMed; (c) Q. Pan, B. Zou, Y. Wang and D. Ma, Org. Lett., 2004, 6, 1009–1012 CrossRef CAS PubMed; (d) G. Cuny, R. Gámez-Montaño and J. Zhu, Tetrahedron, 2004, 60, 4879–4885 CrossRef CAS; (e) M. Brünjes, C. Kujat, H. Monenschein and A. Kirshning, Eur. J. Org. Chem., 2004, 1149–1160 CrossRef; (f) D. Ma, Q. Pan and F. Han, Tetrahedron Lett., 2002, 43, 9401–9403 CrossRef CAS; (g) J. M. Andrés, M. A. Martínez, R. Pedrosa and A. Pérez-Encabo, Tetrahedron: Asymmetry, 2001, 12, 347–353 CrossRef; (h) J. M. Concellón, P. L. Bernad and J. A. Pérez-Andrés, J. Org. Chem., 1997, 62, 8902–8906 CrossRef; (i) M. T. Reetz, M. W. Drewes and A. Schmitz, Angew. Chem., Int. Ed. Engl., 1987, 26, 1141–1143 CrossRef.
- Previous syntheses of (2S,3S)-allophenylnorstatin or protected derivatives thereof: (a) D. Hidasová, M. Janák, E. Jahn, I. Císařová, P. G. Jones and U. Jahn, Eur. J. Org. Chem., 2018, 5222–5230 CrossRef; (b) M. Shimizu, Y. Hayashi, R. Hamanaka and I. Hachiya, Heterocycles, 2007, 73, 191–195 CrossRef CAS; (c) D. Y. Jung, S. Kang, S. Chang and Y. H. Kim, Synthesis, 2006, 86–90 CAS; (d) L. Li, X. He and D. Bai, Synth. Commun., 2005, 35, 1535–1540 CrossRef; (e) T. Suzuki, Y. Honda, K. Izawa and R. M. Williams, J. Org. Chem., 2005, 70, 7317–7323 CrossRef CAS PubMed; (f) F. Fringuelli, F. Pizzo, M. Rucci and L. Vaccaro, J. Org. Chem., 2003, 68, 7041–7045 CrossRef CAS PubMed; (g) J. H. Lee, B. W. Lee, K. C. Jang, I.-Y. Jeong, M. S. Yang, S. G. Lee and K. H. Park, Synthesis, 2003, 829–836 CAS; (h) J.-M. Lee, H.-S. Lim, K.-C. Seo and S.-K. Chung, Tetrahedron: Asymmetry, 2003, 14, 3639–3641 CrossRef CAS; (i) R. Caputo, G. Cecere, A. Guaragna, G. Palumbo and S. Pedatella, Eur. J. Org. Chem., 2002, 3050–3054 CrossRef CAS; (j) J. M. Andrés, M. A. Martínez, R. Pedrosa and A. Pérez-Encabo, Tetrahedron: Asymmetry, 2001, 12, 347–353 CrossRef; (k) H.-J. Ha, Y.-G. Ahn and G. S. Lee, Tetrahedron: Asymmetry, 1999, 10, 2327–2336 CrossRef CAS; (l) B. C. H. May and A. D. Abell, Synth. Commun., 1999, 29, 2515–2525 CrossRef CAS; (m) M. Seki and K. Matsumoto, Synthesis, 1999, 924–926 CrossRef CAS; (n) N. Shibata, E. Itoh and S. Terashima, Chem. Pharm. Bull., 1998, 46, 733–735 CrossRef CAS PubMed; (o) L. Pégorier, M. Haddad and M. Larchevêque, Synlett, 1996, 585–586 CrossRef; (p) M. E. Bunnage, S. G. Davies, C. J. Goodwin and O. Ichihara, Tetrahedron, 1994, 50, 3975–3986 CrossRef CAS; (q) S. Hormuth, H.-U. Reissig and D. Dorsch, Liebigs Ann. Chem., 1994, 121–127 CrossRef CAS; (r) R. Herranz, J. Castro-Pichel and T. García-López, Synthesis, 1989, 703–706 CrossRef CAS; (s) R. Herranz, J. Castro-Pichel, S. Vinuesa and T. García-López, J. Chem. Soc., Chem. Commun., 1989, 938–939 RSC; (t) R. Nishizawa, T. Saino, T. Takita, H. Suda, T. Aoyagi and H. Umezawa, J. Med. Chem., 1977, 20, 510–515 CrossRef CAS PubMed.
- H. Nemoto, X. Li, R. Ma, I. Suzuki and M. Shibuya, Tetrahedron Lett., 2003, 44, 73–75 CrossRef CAS.
- I. Hori and H. Midorikawa, Sci. Pap. Inst. Phys. Chem. Res. (Jpn.), 1962, 56, 216–217 CAS.
- Details of the X-ray diffraction study of 4c are given in the ESI document.† CCDC file 2088378† contains the crystallographic data for this compound.
- W. Yuan, B. Munoz, C.-H. Wong, J. Z. Haeggström, A. Wetterholm and B. Samuelsson, J. Med. Chem., 1993, 36, 211–220 CrossRef CAS PubMed.
- K. Suzuki, Y. Hamada, J.-T. Nguyen and Y. Kiso, Bioorg. Med. Chem., 2013, 21, 6665–6673 CrossRef CAS PubMed.
- A. J. Harvey and A. D. Abell, Tetrahedron, 2000, 56, 9763–9771 CrossRef CAS.
- A. D. Abell and A. J. Harvey, ARKIVOC, 2006, iii, 72–76 Search PubMed.
- Use of methyl ester 16 in the patent literature: (a) glycogen phosphorylase inhibitors: S. E. Bradley, T. M. Krulle, P. J. Murray, M. J. Procter, R. J. Rowley, C. P. Sambrook Smith and G. H. Thomas, WO Pat, 2004104001A2, 2004 Search PubMed; (b) protein thyrosin phosphatase inhibitors: Y. Amanomiya and M. Taniuchi, Jpn. Pat, 2004256435A, 2004 Search PubMed; (c) HIV protease inhibitors: T. Kamijo, T. Yamaguchi, T. Yanagi, I. Tsuchiya and H. Takeuchi, Jpn. Pat, 09124629A, 1997 Search PubMed.
- Use of ethyl ester 18 in the patent literature: (a) HIV protease inhibitors: D. J. Kucera and R. W. Scott, US Pat, US 20050124673A1, 2005 Search PubMed; (b) cysteine protease inhibitors: M. Sato, H. Mukoyama, J. Kobayashi, S. Tsuyuki, K. Tokutake and S. Akaba, Jpn. Pat, 2001055366A, 2001 Search PubMed; (c) HIV protease inhibitors: A. Krantz, T. F. Tam, A. L. Castelhano and J. J. Nestor Jr., WO Pat, 9313066A1, 1993 Search PubMed.
- C.-P. Xu, Z.-H. Xiao, B.-Q. Zhuo, Y.-H. Wang and P.-Q. Huang, Chem. Commun., 2010, 46, 7834–7836 RSC.
- Of all the strategies considered to date (ref. 15), the oxyhomologation of N,N-dibenzyl-L-phenylalaninal appears to be one of the most direct. However, it appears to have been performed, in three steps via a cyanohydrin, on only two occasions: in 54% yield (ref. 15n) and in 49% yield (ref. 15j). In a related approach using a phosphane oxide derived reagent, the anti ester derivative 5 was obtained in 53% yield for two steps (ref. 14e).
- E. Richmond, K. B. Ling, N. Duguet, L. B. Manton, N. Çelebi-Ölçüm, Y.-H. Lam, S. Alsancak, A. M. Z. Slawin, K. N. Houk and A. D. Smith, Org. Biomol. Chem., 2015, 13, 1807–1817 RSC.
- Z. Neouchy, D. Gomez Pardo and J. Cossy, Org. Lett., 2018, 20, 6017–6021 CrossRef CAS PubMed.
- P. Dimopoulos, J. George, D. A. Tocher, S. Manaviazar and K. J. Hale, Org. Lett., 2005, 7, 5377–5380 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra and X-ray diffraction data. CCDC 2088378. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob02411f |
| This journal is © The Royal Society of Chemistry 2022 |