
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antifungal mono- and dimeric nitrogenous bisabolene derivatives from a sponge in the order Bubarida from Futuna Islands†
Maria
Miguel-Gordo
 a,
Maggie M.
Reddy
a,
Pilar
Sánchez
b,
Jordan J.
Buckley
a,
Thomas A.
Mackenzie
b,
Laurence K.
Jennings
a,
Fernando
Reyes
b,
Kevin
Calabro
a and
Olivier P.
Thomas
*a
a,
Maggie M.
Reddy
a,
Pilar
Sánchez
b,
Jordan J.
Buckley
a,
Thomas A.
Mackenzie
b,
Laurence K.
Jennings
a,
Fernando
Reyes
b,
Kevin
Calabro
a and
Olivier P.
Thomas
*a
aMarine Biodiscovery Laboratory, School of Chemistry and Ryan Institute, National University of Ireland Galway, University Road, Galway H91 TK33, Ireland. E-mail: olivier.thomas@nuigalway.ie
bFundación MEDINA, Centro de Excelencia en Investigación de Medicamentos Innovadores en Andalucía, Parque Tecnológico de Ciencias de la Salud, Avenida del Conocimiento 34, Armilla, Granada E-18016, Spain
First published on 4th January 2022
Abstract
An abundant sponge of the order Bubarida was selected for further chemical investigation following biological and chemical screening of sponges collected from Futuna Islands in the Indo-Pacific. Ten new nitrogenous bisabolene derivatives were isolated and identified: the monomeric theonellin formamide analogues named bubaridins A–F (1–6) with unusual oxidised linear chains, and the first isocyanide/formamide dimeric and cyclised bisabolenes 7–9. The structure elucidation of these nitrogenous bisabolenes involved HRESIMS, NMR, and ECD analyses, and the chiral compounds were found to be racemates. A biosynthetic hypothesis for the production of these metabolites is proposed and some chemotaxonomic considerations are discussed. Furthermore, the antimicrobial and antitumoral activity were evalutated and the trans-dimer theonellin isocyanide (7) was shown to exhibit potent and selective antifungal activity.
Introduction
Marine sponges and their associated microbiome are known as rich sources of chemical diversity. As such the phylum Porifera has been one of the most studied marine organism in the search for new bioactive metabolites.1 However, many questions pertaining to the pharmaceutical potential, ecological role and metabolic pathways of sponge natural products remain unanswered. Furthermore, the full extent of the diversity of sponges has still to be uncovered in many remote regions of the world.2 During the Tara Pacific expedition, the Futuna Islands located in the tropical Indo-Pacific were explored, and sponges and associated chemical diversity were inventoried for the first time in this region. Following a prioritisation process based on chemical and biological screenings (cytotoxicity and antimicrobial) a sponge belonging to the order Bubarida was selected for further chemical investigation. Although this sponge was represented by several morphotypes, they produced similar chemical profiles and were presumed to constitute a single species.A recent study highlighted the need for accurate taxonomic identification of sponges for natural product chemistry.3 However, identifying sponges, particularly from understudied regions of the world such as Futuna Island, remains a challenge because some taxa may have developed convergent morphological traits over their long evolutionary history and geographic isolation. This means that evolutionary distinct species may superficially resemble one another and will require DNA verification.4 An example is the order Bubarida which was described in 1894 by Topsent and later resurrected by Morrow and Cardenas in 2015.5 DNA from the generitypes of Acanthella, Dictyonella, Bubaris, Phakellia, Desmanthus and Cymbastella indicate that the order forms a well-supported clade. However, familial relationships are less clear as members of the Dictyonellidae are polyphyletic with respect to members of the Bubaridae. Nevertheless, the former family includes species of Acanthella, Dictyonella and Cymbastella that are known to produce nitrogenous terpenes with known pharmaceutical application.
The preliminary biological screening of fractions from this sponge showed potent antifungal activity. The infection by Candida spp. is the most common fungal infection with high mortality rates especially among immunocompromised patients. Furthermore, with the increasing occurrence of resistant Candida strains, the treatment of these infections is becoming challenging.6 The in-depth chemical study of this sponge led to the isolation and structure elucidation of 10 new nitrogenous bisabolenes (Fig. 1): seven monomeric formamide bisabolenes 1–6, including two epimers of 4, two isocyanide bisabolene dimers 7 and 8, and one formamide dimeric bisabolene 9, together with the known N,N‘-bis[(6R,7S)-7,8-dihydro-α-bisabolen-7-yl]urea,7 aureol,8 theonellin formamide,9 and 7-isothiocyanato-7,8-dihydro-α-bisabolene.7 The metabolites were screened against a range of microbial pathogens with a focus on pathogenic fungi, and the dimers were further tested against five tumoral cell lines.
 | ||
| Fig. 1 Structures of bisabolenes 1–9 isolated from a Bubarida sponge. | ||
Results and discussion
Compound 1 was isolated as a colourless oil with a molecular formula C16H25NO3 deduced from the protonated ion at m/z 280.1916 [M + H]+ in the (+)-HRESIMS spectrum. Inspection of the 1H NMR of 1 (Table 1) revealed three methyl singlets at δH 1.84 (br s, H3-15), δH 1.31 (s, H3-14) and δH 1.36 (s, H3-13) suggestive of the terpenoid family. The combination of 1H NMR, COSY and HSQC spectra revealed the presence of: an aldehyde functional group with characteristic signals at δH 9.42 (s, H-12) and δC 197.0 (CH, C-12), and a conjugated diene system with three deshielded signals at δH 7.05 (br d, J = 11.5 Hz, H-10), 6.81 (dd, J = 15.0, 11.5 Hz, H-9) and 6.41 (d, J = 15.0 Hz, H-8). Key HMBC correlations from the methyls allowed the structure elucidation of the linear chain (Fig. 2). First, the key H3-15/C-10, C-11 and C-12 correlations positioned the methyl C-15 on the non-protonated carbon C-11 of the diene conjugated to the aldehyde. The key H3-14/C-6, C-7 and C-8 HMBC correlations linked the diene system to an oxygenated and non-protonated carbon at δC 75.7 (C-7), and also to a key methine at δH 1.45 (tt, J = 12.3 Hz, 3.0 Hz 3, H-6) and δC 48.8 (CH, C-6). This methine at C-6 was found to be COSY-correlated to four non-equivalent methylene protons, H2-1/H2-5, which had further COSY correlations to the overlapping signals of H2-2/H2-4. Additional key H3-13/C-2, C4 and C-3 HMBC correlations revealed the presence of a cyclohexane ring, which was reminiscent of the bisabolene family of sesquiterpenoids. The chemical shift of the signal of the non-protonated carbon at δC 54.6 (C-3) suggested the presence of a nitrogen at this position. The deshielded singlet at δH 7.87 (H-16) was found to be HMBC correlated to the nitrogen substituted carbon C-3, therefore suggesting a formamide functional group. The skeleton of 1 was found to be identical to the one of theonellin formamide isolated from this specimen but also from other species of the genus Theonella.9 The apparent “doubling” of many signals in the 1H and 13C NMR spectra (Tables 1 and 2) was indicative of rotational isomers around the amide bond (cis and trans rotamers also called endo and exo in this case), a phenomenon already observed for formamide substituted terpenoids.10,11 Due to chemical exchange correlation, the negative nOe correlations between the formyl signals at δH 7.87 (s, H-16, endo) and 8.24 (s, H-16, exo) confirmed the presence of the two rotamers present in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in favour of the endo. The relative configuration around the cyclohexane ring was assigned through interpretation of coupling constant values and NOESY data. First, the multiplicity of H-6 as a triplet of triplet and coupling constant values of J = 12.5 and 3.0 Hz placed H-6 in an axial position of the chair conformation.12 Further H-6(ax)/H-1a(eq)/H-5a(eq) nOe correlations indicated that all three H-6, H-1a, and H-5a protons were located on the same side of the cycle. Thus, H-1a and H-5a were placed in equatorial position. Subsequently, the geminal H-1b(ax) and H-5b(ax) protons, were placed in axial positions on the ring. Then, the multiplicity and coupling constants of H-2b(ax)/H-4b(ax) as triplet of doublet with coupling constant values of J = 13.0 and 4.0 Hz positioned these protons in axial positions, leaving the H-2a(eq)/H-4a(eq) protons in equatorial positions. Interestingly, and despite a plane of symmetry of the 3,6-disubstituted cyclohexane ring, the pair of signals corresponding to the protons and carbons 1a/5a, 1b/5b, 2a/4a and 2b/4b were not equivalent but diastereotopic. This could be explained by the presence of a chiral center at C-7, in the alpha position of the cyclohexane. Although no conclusive nOe could be observed from H3-13 due to overlap with the signals corresponding to H-1b and H-5b, the nOe correlation from H-16 to H-2a/H-4a and H-2b/H-4b left no other option than placing the formamide group in an equatorial position. Therefore, the formamide group was in a trans relative configuration with the side chain also placed in equatorial position. The configuration of the C-8/C-9 double bond on the side chain was determined as E by the coupling constant of 15 Hz for 3JH8–H9. Then, the H3-15/H-9 nOe correlation was indicative of a E configuration for the second olefin at C-10/C-11.
:1 ratio in favour of the endo. The relative configuration around the cyclohexane ring was assigned through interpretation of coupling constant values and NOESY data. First, the multiplicity of H-6 as a triplet of triplet and coupling constant values of J = 12.5 and 3.0 Hz placed H-6 in an axial position of the chair conformation.12 Further H-6(ax)/H-1a(eq)/H-5a(eq) nOe correlations indicated that all three H-6, H-1a, and H-5a protons were located on the same side of the cycle. Thus, H-1a and H-5a were placed in equatorial position. Subsequently, the geminal H-1b(ax) and H-5b(ax) protons, were placed in axial positions on the ring. Then, the multiplicity and coupling constants of H-2b(ax)/H-4b(ax) as triplet of doublet with coupling constant values of J = 13.0 and 4.0 Hz positioned these protons in axial positions, leaving the H-2a(eq)/H-4a(eq) protons in equatorial positions. Interestingly, and despite a plane of symmetry of the 3,6-disubstituted cyclohexane ring, the pair of signals corresponding to the protons and carbons 1a/5a, 1b/5b, 2a/4a and 2b/4b were not equivalent but diastereotopic. This could be explained by the presence of a chiral center at C-7, in the alpha position of the cyclohexane. Although no conclusive nOe could be observed from H3-13 due to overlap with the signals corresponding to H-1b and H-5b, the nOe correlation from H-16 to H-2a/H-4a and H-2b/H-4b left no other option than placing the formamide group in an equatorial position. Therefore, the formamide group was in a trans relative configuration with the side chain also placed in equatorial position. The configuration of the C-8/C-9 double bond on the side chain was determined as E by the coupling constant of 15 Hz for 3JH8–H9. Then, the H3-15/H-9 nOe correlation was indicative of a E configuration for the second olefin at C-10/C-11.
 | ||
| Fig. 2 Key COSY, HMBC and NOESY NMR correlations of bubaridin A (1) in CD3OD. | ||
| No. | 1 | 2 | 3 | 4 and 11-epi-4d | 5 | 6 | |
|---|---|---|---|---|---|---|---|
|
Endo/exo (3:1) |
Endo/exo (3:1) |
Endo/exo (3:1) |
Endo/exo (3:1) |
11-Epimer |
Endo/exo (3:1) |
Endo/exo (3:1) |
|
| a Overlap (data from HSQC), b and c signals can be exchanged, d unable to distinguish between 4 and 11-epi-4. | |||||||
| 1ab | 1.79/1.75a, br d (13.0) | 1.75/1.80, br d (12.0) | 1.62/1.66a, dd (13.5, 3.0) | 1.70, m | — | 1.70/1.77, br d (14.0) | 1.67/1.71, br d (12.5) |
| 1bc | 1.29/1.31, br m | 1.23/1.31, m | 1.48/1.51, qd (13.5, 2.5) | 1.43, br q (14.0) | 1.46, m | 1.54/1.52, qd (12.5, 3.5) | |
| 2/4a | 2.02/1.84a, br dd (13.0, 2.5) | 2.00/1.81, br d (13.0) | 2.00/1.83, br d (12.5) | 2.02/1.83, dd (13.5, 2.5) | — | 2.02/1.84, br d (13.0) | 2.02/1.86, br d (13.0) |
| 2/4b | 1.67/1.58, br td (13.0, 4.0) | 1.63/1.55, td (13.0, 3.5) | 1.74/1.65a, td (12.5, 3.5) | 1.67/1.57, td (13.5, 3.5) | 1.71/1.60, td (12.5, 4.0) | 1.78/1.68, td (13.0, 3.5) | |
| 5ab | 1.69/1.73a, m | 1.70, br d (12.0) | 1.62/1.66a, dd (13.5, 3.0) | 1.62, br d (13.5) | — | 1.66, br d (13.5) | 1.67/1.71, br d (12.5) |
| 5bc | 1.34, br m | 1.27, m | 1.48/1.51, qd (13.5, 2.5) | 1.35, m | 1.35, m | 1.54/1.52, qd (12.5, 3.5) | |
| 6 | 1.45, tt (12.5, 3.0) | 1.38, tt (12.5, 3.5) | 1.97, tt (12.0, 3.0) | 1.23a | — | 1.31, br t (13.0) | 2.12, tt (12.5, 3.0) |
| 8 | 6.41, d (15.0) | 5.73, d (15.5) | 5.89/5.90, br d (10.5) | 3.32a | — | 3.47/3.48, br d (6.0) | 6.12, d (11.5) |
| 9 | 6.81/6.82, dd (15.0, 11.5) | 6.49, dd (15.5, 10.5) | 6.56, dd (15.5, 10.5) | 5.68, dd (15.5, 7.5) | 5.67, dd (15.5, 7.5) | 6.79/6.84, dd (16.0, 6.0) | 7.60, dd (15.5, 11.5) |
| 10 | 7.05, br d (11.5) | 6.07, br d (10.5) | 5.67/5.68, br d (15.5) | 6.02, br d (15.5) | — | 6.34/6.37, d (16.0) | 6.12, d (15.5) |
| 12 | 9.42, s | 3.99, s | 3.40, d (11.0) | 3.41, d (11.0) | 3.40, d (11.0) | 2.28, s | 2.29, s |
| 3.38, d (11.0) | 3.38, d (11.0) | 3.39, d (11.0) | |||||
| 13 | 1.36/1.32, s | 1.35/1.30, s | 1.41/1.36, s | 1.397/1.353, s | — | 1.40/1.36, s | 1.42/1.38, s |
| 14 | 1.31, s | 1.26, s | 1.77/1.78, s | 1.226/1.235, s | — | 1.23/1.24, s | 1.95/1.96, s |
| 15 | 1.84, br s | 1.79, s | 1.26, s | 1.251, s | 1.245, s | ||
| 16 | 7.87/8.24, s | 7.86/8.24, s | 7.88/8.26, s | 7.87/8.25, s | — | 7.88/8.26, s | 7.89/8.27, s |
| No. | 1 | 2 | 3 | 4 and 11-epi-4d | 5 | 6 | |
|---|---|---|---|---|---|---|---|
|
Endo/exo (3:1) |
Endo/exo (3:1) |
Endo/exo (3:1) |
Endo/exo (3:1) |
11-Epimer |
Endo/exo (3:1) |
Endo/exo (3:1) |
|
| a Data from HMBC, overlapped with solvent signal, b and c signals can be exchanged, d unable to distinguish between 4 and 11-epi-4. | |||||||
| 1b | 23.8/24.0, CH2 | 24.0/24.1, CH2 | 28.4/28.6, CH2 | 24.7/24.9, CH2 | — | 24.9/25.0, CH2 | 28.2/28.3, CH2 |
| 2c | 37.7/39.8, CH2 | 37.82/39.9, CH2 | 37.6/39.8, CH2 | 37.21/39.4, CH2 | — | 37.1/39.3, CH2 | 37.4/39.6, CH2 |
| 3 | 54.6/53.8, C | 54.6/53.8, C | 54.5/53.7, C | 54.5/53.8, C | — | 54.5/53.7, C | 54.4/53.6, C |
| 4c | 37.6/39.7, CH2 | 37.75/39.8, CH2 | 37.6/39.8, CH2 | 37.18/39.3, CH2 | — | 37.1/39.3, CH2 | 37.4/39.6, CH2 |
| 5b | 24.4/24.6, CH2 | 24.4/24.5, CH2 | 28.4/28.6, CH2 | 25.5/25.6, CH2 | — | 25.5/25.6, CH2 | 28.2/28.3, CH2 |
| 6 | 48.8a, CH | 49.6/49.8, CH | 47.9/47.8, CH | 46.9/46.7, CH | — | 46.9/46.6, CH | 48.4, CH |
| 7 | 75.7/75.6, C | 75.5/75.4, C | 143.1/142.8, C | 66.6/66.5, C | — | 68.0/67.8, C | 157.0/156.7, C |
| 8 | 152.2/152.1, CH | 140.2/140.1, CH | 124.4/124.6, CH | 64.1, CH | — | 62.2/62.1, CH | 123.5/123.6, CH |
| 9 | 124.2/124.3, CH | 124.9/125.0, CH | 126.33/126.26, CH | 125.2/125.1, CH | — | 143.9/143.8, CH | 142.2/142.1, CH |
| 10 | 150.6/150.5, CH | 125.6/125.5, CH | 137.0/137.1, CH | 142.24, CH | 142.20, CH | 134.3/134.4, CH | 129.6/129.7 CH |
| 11 | 138.2/138.3, C | 137.7/137.8, C | 74.2, C | 74.08, C | 74.07, C | 200.1a, C | 202.1, C |
| 12 | 197.0, CH | 68.50/68.45, CH2 | 70.9, CH2 | 70.6, CH2 | — | 27.3a, CH3 | 27.2, CH3 |
| 13 | 22.1/24.2, CH3 | 22.1/24.2, CH3 | 22.5/24.5, CH3 | 22.2/24.2, CH3 | — | 22.23/22.20, CH3 | 22.4/24.4, CH3 |
| 14 | 25.7/25.8, CH3 | 25.7/25.8, CH3 | 15.2/15.1, CH3 | 14.05, CH3 | 14.03, CH3 | 13.97/13.99, CH3 | 16.0, CH3 |
| 15 | 9.3, CH3 | 14.2, CH3 | 24.7, CH3 | 24.6, CH2 | 24.5, CH2 | ||
| 16 | 162.9/165.1, CH | 162.9/165.1, CH | 162.9/165.1, CH | 162.9/165.1, CH | — | 162.9/165.1, CH | 163.0/165.2, C |
Finally, the conjugated system was assigned as s-trans for both single bonds due to additional H-12/H-10/H-8 nOe correlations. For the absolute configuration of the unique chiral center at C-7, the ECD spectrum of 1 did not show any clear Cotton effect, despite the presence of a chromophore close to the chiral center and a UV band at 274 nm. This led to the conclusion that bubaridin A (1) is a racemic mixture of enantiomers at C-7. A very weak optical rotary power also corroborated this assumption.
Compound 2 was obtained as colourless oil and exhibited a protonated ion at m/z 282.2077 [M + H]+ by (+)-HRESIMS, consistent with the chemical formula C16H27NO3. The 1H NMR, and 13C NMR spectra (Tables 1 and 2) were similar to those of compound 1, suggesting the same formamide bisabolene skeleton. Three methyl singlets (δH 1.35, 1.79 and 1.26), a conjugated diene system (δH 6.07, 6.49, and 5.73), the signals of the cyclohexane core and the same formamide peaks in a proportion 3:1 (endo/exo) were present for 2. The presence of diastereotopic methylenes on the cyclohexane was again evidenced in the 13C NMR spectrum of 2 and suggested a chiral center at C-7 in 2. However, when compared to 1, the shielding of the olefinic signals and the absence of the aldehyde signal in the 1H NMR spectrum of 2 suggested that the aldehyde was no longer present in 2. Instead, new signals at δH 3.99 (s, H-12) and δC 68.5 (C-12) were consistent with a primary alcohol at C-12 and this was further supported by the key H3-15/C-12 HMBC correlation. The H-12/H-10/H-8 nOe correlations confirmed the same relative configurations for the conjugated dienic system of the side-chain and the absence of Cotton effect in the ECD spectrum also indicated that bubaridin B (2) was also a racemic mixture.
Compound 3 was isolated as a colourless oil with the same molecular formula C16H27NO3 as 2 as evidenced by a protonated ion at m/z 282.2088 [M + H]+ by (+)-HRESIMS. The 1H NMR, and 13C NMR spectra (Tables 1 and 2) showed the characteristic peaks of the formamide bisabolene skeleton with three methyl singlets at δH 1.41, 1.77 and 1.26 and the same formamide signals in a ratio 3:1 (endo/exo). The absence of a chiral center at C-7 was proposed as the signals of the cyclohexane ring at C-1 and C-5 were equivalent for 3. A modification on the side-chain was further supported by the deshielding of the signal of H-6 at δH 1.97. Examination of HMBC correlations from the methyls C-14 and C-15 revealed differences in the linear chain when compared to compounds 1 and 2. The key H3-15/C-10, C-11 and C-12 HMBC correlations located the methyl C-15 on the non-protonated oxygenated carbon C-11 at δC 74.2 which was still linked to a primary alcohol at C-12 as in compound 2. The presence of the chiral tertiary alcohol at C-11 was responsible for the AB system of the signals corresponding to the methylene H2-12. In addition, key H3-14/C-6, C-7 and C-8 HMBC correlation linked the methyl C-14 to the unsaturated carbon C-7 at δC 143.1. Compounds 2 and 3 might then be formed by isomerisation of the conjugated system/tertiary alcohol, probably through an intermediate carbocation. While the value of the coupling constant 3JH9–H10 15.5 Hz revealed an E configuration for C-9/C-10, the H-6/H-8 and H3-14/H-9 nOe correlations were consistent with an E configuration for the second C-7/C-8 olefin, and the H-8/H-10 nOe confirmed the s-trans configuration of the conjugated system. Again, the ECD spectrum of compound 3 showed no Cotton effects suggesting that bubaridin C (3) is also a racemate. Interestingly, during the NMR analyses performed in CDCl3, 3 slowly evolved into the aldehyde 1 through isomerisation and subsequent oxidation. This transformation would have occurred in the slightly acidic conditions of the solvent CDCl3.
Compound 4 was obtained as a colourless oil, and the chemical formula C16H27NO4 was deduced from the molecular ion peak at m/z 298.2023 [M + H]+ by (+)-HRESIMS. The 1H NMR, and 13C NMR spectra of compound 4 revealed some similarities with those of compound 3 (Tables 1 and 2) with the vicinal diol evidenced by the AB system at δH 3.41 (d, J = 11.0 Hz, H-12a) and 3.38 (d, J = 11.0 Hz, H-12b). However, the signals were apparently “duplicated” in a 1:1 ratio with another AB system at δH 3.40 (d, J = 11.0 Hz, H-12′a) and 3.39 (d, J = 11.0 Hz, H-12′b). This observation suggested that 4 was an inseparable mixture of a pair of diasteroisomers formed by the presence of another chiral center on the side chain. In the case of 4, only two olefinic protons at δH 5.68 (dd, J = 15.5, 7.5 Hz, H-9) and 6.02 (br d, J = 15.5 Hz, H-10) were observed in the 1H NMR spectrum suggesting that the second olefin of 3 was functionalised in 4. A new signal at δH 3.32 (H-8) and δC 64.1 (C-8) was in accordance with an epoxide at position C-7/C-8.13 The key H3-15/C-10, C-11 and C-12 HMBC correlations linked the double bond to the vicinal diol at C-11/C-12 and the H-9/H-8 COSY correlation placed the epoxide on the other side of the double bond. The configuration of the double bond was assigned as E by a coupling constant value of 3JH9−H10 15 Hz and the H-9/H-14 nOe suggested a trans-epoxide. To the best of our knowledge, only two compounds of the bisabolene family with were reported with an epoxide at C-7/C-8.10 The similarity in the 1H and 13C NMR chemical shifts for C-14, C-7 and C-8 confirmed the relative configuration of the epoxides. We therefore propose that 4 is a mixture of two epimers at C-11 in a 1:1 ratio. The ECD spectrum of 4 did display a small Cotton effect that could be explained by an enantiomeric excess of one or the two epimers. However, at that stage it was not possible to separate the epimers and therefore confirm this assumption. Consequently, bubaridin D (4) is a mixture composed of two non-racemic epimers (7R*, 8R*, 11R*) and (7R*, 8R*, 11S*).
The chemical formula C15H24NO3 of compound 5 was deduced from the protonated ion at m/z 266.1760 [M + H]+ in the (+)-HRESIMS spectrum. The 1H and 13C NMR spectra of 5 were similar to those of 4 (Tables 1 and 2) with characteristic rotamers due to the formamide in a ratio 3:1 (endo/exo), the epoxy group at C-7/C-8 and the double bond at C-9/C-10. A difference was noticed in the absence of the AB system of the primary alcohol. Instead, a methyl ketone was evidenced by a key HMBC correlation from the methyl H3-12 (δH 2.28, s) to C-10 and C-11 (δC 200.1). The large 3J-coupling constant value of 16 Hz between H-9 and H-10 confirmed the E configuration of the double bond. In this case, no Cotton effect was observed in the ECD spectrum of 5, suggesting bubaridin E (5) to be a racemic mixture.
The molecular formula of 6, was found to be C15H27NO2 by the proton adduct at m/z 250.1818 [M + H]+ in the (+)-HRESIMS spectrum. Compound 6 displayed the rotameric signals in a ratio 3:1 (endo/exo) characteristic of the formamide, and the same methoxy ketone C-11 (δC 202.1) and CH3-12 (δH 2.28 and δC 27.2) as compound 5 (Tables 1 and 2). The molecular formulae of compounds 5 and 6 differed by the number of oxygens, with one less oxygen for 6. A second and trisubstituted double bond was evidenced in 6 by the signals at δC 157.0 (C-7) and δH 6.12 (d, J = 11.5 Hz, H-8)/δC 123.5 (C-8) suggesting the replacement of the epoxide of 5 by a double bond in 6. The H-8/H-9/H-10 COSY correlations confirmed the conjugated dienic system and the H3-14/C-7, C-8 and C-9 HMBC correlations its location. The coupling constant value 3JH9−H10 15.5 Hz proved the E configuration for the double bond and the H-9/H3-12/H3-14 nOe correlations were indicative of a E configuration for the second double bond and s-trans configuration for the conjugated diene system with H3-12, H3-14 and H-9 on the same side. Bubaridin F (6) is therefore achiral.
Compound 7 was obtained as colourless oil with a chemical formula C32H46N2 deduced from the protonated molecular peak at m/z 459.3723 [M + H]+ in the (+)-HRESIMS spectrum. A first inspection of the 1H NMR (Table 3) and HSQC spectra revealed the presence of six methyl singlets, and five olefinic methines. The signals of two methylenes integrating for four hydrogens were assigned to two symmetrical cyclohexane rings. Taking also into consideration the molecular formula, these NMR data were consistent with a dimeric nature for compound 7. The lack of apparent “doubling” of the signals and the presence of equivalent methylenes for the cyclohexane rings were then indicative of an absence of formamides on the rings and chiral centers close to the rings. A closer inspection of the 13C NMR, COSY and HMBC spectra confirmed this assumption. The shielded methyl H3-15 at δH 0.89 exhibited key H3-15/C-10, C-11, C-12 and C-9′ HMBC correlations and allowed a connection of both bisabolene subunits through the C-9′/C-11 bond. For the first bisabolene subunit, a conjugated diene system formed by the double bonds at C-10/C-9 and C-8/C-7 were linked to the methine C-6 of the cyclohexane ring through the H-8/H-9/H-10 COSY-correlations and the key H3-14/C-6, C-7 and C-8 HMBC correlations. For the second bisabolene unit, the methine at δH 2.82 (br d, J = 10.0 Hz, H-9′) was connected to the double bond at C-8′/C-7′ and then with the other cyclohexane ring due to the H-8′/H-9′/H-10′ COSY correlations and the H3-14′/C-6′, C-7′ and C-8′ HMBC correlations. In addition, the key H3-15′/C-10′, C-11′ and C-12′ HMBC correlations linked the bis-allylic methine C-9′ to another olefinic bond at C-10′/C-11′ and then to the methylene H2-12′ (δH 1.94). Finally, H2-12′ was COSY correlated to H2-12 (δH 1.54) of the first bisabolene. Thus, the two monomers were connected through the C-12/C-12′ and C-11/C-9′ bonds, which formed the bis-bisabolene skeleton previously described in compounds isolated from the sponge Axinyssa variabilis.14 Finally, two isocyanides with a characteristic signals at δC 152.4 were located at C-3 and C-3′ of both bisabolenes based on the molecular formula and mass fragments at m/z 432 and 405 in the (+)-HRESIMS, indicating the loss of one or both units respectively. The configuration of the conjugated double bonds at C-9/C-10 and C-7/C-8 were established as E and the diene as s-trans by the 3JH8-H9 of 15.5 Hz, and the H3-14/H-9 and H-10/H-8 nOe correlations. The clear H3-15/H-8′ and H-10/H-9′ nOe correlations determined the trans orientation of the methyl H3-15 and the proton H-9′. Additional H-8′/H-6′ as well as H-9′/H-14′ nOe correlations led to the E configuration for the trisubstituted double bond of the second bisabolene. Based on chemical shifts and nOe correlations similar to those of the monomers, the same relative configurations were assumed for the cyclohexanes, with the isocyanide functional group in a trans relative configuration with the linear chain. With regard to the absolute configurations at the chiral centers C-11 and C-9′, no Cotton effects were displayed in the ECD spectrum and the weak optical rotation led to the conclusion that trans-dimer theonellin isocyanide (7) is a racemic mixture of the enantiomers 11R*, 9′R*.
| No. | 7 | 8 |
9
Endo/exo (1:1) |
|||
|---|---|---|---|---|---|---|
| δ H, mult. (J in Hz) | δ C | δ H, mult. (J in Hz) | δ C | δ H, mult. (J in Hz) | δ C | |
| a Data from HSQC, overlapped. | ||||||
| 1/5a | 1.70a, m | 26.7, CH2 | 1.68a, m | 26.7, | 1.62, m | 27.5, CH2 |
| 1/5b | 1.41a, m | 1.41a, m | CH2 | 1.43, m | ||
| 2/4a | 1.90, m | 38.3, CH2 | 1.93, br d (13.0) | 38.5, | 1.96/1.77, m | 37.0/39.6, CH2 |
| 2/4b | 1.83, m | 1.83, td (12.5, 3.5) | CH2 | 1.67a/1.54, m | ||
| 3 | — | 57.0, C | — | 56.8, C | — | 54.0/52.8, C |
| 6 | 1.96, m | 44.9, CH | 1.97, m | 45.1, CH | 1.89, tt (12.0, 3.5) | 46.4, CH |
| 7 | — | 138.6, C | — | 138.4, C | — | 139.2, C |
| 8 | 5.78, br d (10.5) | 124.6, CH | 5.79, br d (10.5) | 124.7, CH | 5.81, dd (10.5, 4.0) | 124.2, CH |
| 9 | 6.16, dd (15.5, 10.5) | 122.5, CH | 6.14, dd (15.5, 10.5) | 123.77, CH | 6.16, ddd (15.5, 10.5, 2.0) | 123.9, CH |
| 10 | 5.62, d (15.5) | 143.2, CH | 5.71, d (15.5) | 139.6, CH | 5.71, dd (15.5, 10.0) | 139.2, CH |
| 11 | — | 37.9, C | — | 38.1, C | — | 38.1, C |
| 12 | 1.54, m | 33.6, CH2 | 1.62, br d (13.0) | 34.1, CH2 | 1.63a, m | 34.1, CH2 |
| 1.56, td (13.0, 6.5) | 1.53, m | |||||
| 13 | 1.42, s | 25.2, CH3 | 1.44, s | 25.2, CH3 | 1.39/1.32, s | 22.6/25.0, CH3 |
| 14 | 1.70, s | 15.3, CH3 | 1.69, s | 15.3, CH3 | 1.72/1.71, s | 15.3, CH3 |
| 15 | 0.89, s | 21.0, CH3 | 1.01, s | 25.7, CH3 | 1.02, s | 25.7, CH3 |
16 ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) N- or H-NH N- or H-NH |
— | 152.4, C | — | 152.4, C | 8.05, d (1.0)/8.32, d (12.0) | 160.4/162.7, CH |
| –NH | 5.19, br d/5.64, br d (12.0) | — | ||||
| 1′/5′a | 1.70a, m | 26.7, CH2 | 1.65a, m | 26.4, CH2 | 1.62, m | 27.5, CH2 |
| 1′/5′b | 1.41a, m | 1.44a, m | 1.43, m | |||
| 2′/4′a | 1.90, m | 38.3, CH2 | 1.83, m | 37.7 CH2 | 2.02a/1.83, m | 37.0/39.6, CH2 |
| 2′/4′b | 1.83, m | 1.75, m | 1.72a/1.57a, m | |||
| 3′ | — | 57.0, C | — | 57.1, C | — | 54.0/52.8, C |
| 6′ | 1.96, m | 44.9, CH | 1.97, m | 44.1, CH | 1.89, tt (12.0, 3.5) | 46.4, CH |
| 7′ | — | 137.9, C | — | 137.2, C | — | 138.2, C |
| 8′ | 4.97, br d (10.0) | 125.1, CH | 4.91, d (10.0) | 126.2, CH | 4.93, t (9.0) | 126.0, CH |
| 9′ | 2.82, br d (10.0) | 43.3, CH | 2.75, d (10.0) | 44.8, CH | 2.74, d (9.0) | 44.7, CH |
| 10′ | 5.06, br s | 123.9, CH | 5.06, br s | 123.81, CH | 5.07, br s | 124.0, CH |
| 11′ | — | 133.1, C | — | 133.2, C | — | 133.0, C |
| 12′a | 1.94, m | 27.8, CH2 | 2.00, m | 28.0, CH2 | 2.01a, m | 28.0, CH2 |
| 12′b | 1.94, m | 1.94, m | ||||
| 13′ | 1.43, s | 25.6, CH3 | 1.36, s | 26.0, CH3 | 1.43, d (3.0)/1.36, s | 22.6/25.0, CH3 |
| 14′ | 1.56, s | 15.3, CH3 | 1.58, s | 15.2, CH3 | 1.59/1.58, s | 14.9, CH3 |
| 15′ | 1.65, br s | 23.5, CH3 | 1.66, br s | 23.5, CH3 | 1.66, br s | 23.5, CH3 |
| 16′ N- or H-NH |
— | 152.4, C | — | 152.4, C | 8.04, d (2.0)/8.30, d (12.0) | 160.4/162.7, CH |
| NH | 5.19, br d/5.64, br d (12.0) | — | ||||
Compound 8 was isolated as a colourless oil and exhibited the same chemical formula as 7, C32H46N2 as shown by HRESIMS analysis. The 1H and 13C NMR spectra showed very similar signals with slight differences in the chemical shifts especially for the signals of protons located around the central cyclohexene. The main differences were identified for the signals of the methylene H2-12 (δH 1.62 and 1.56) and H2-12′ (δH 2.02 and 1.94). Together with the H3-15/H-9′ and H-10/H-8′ nOe correlation, these changes were in accordance with a change in the relative configuration between the methyl H3-15 and the proton H-9′ which would be cis for compound 8. Based on biosynthetic considerations and similar nOe correlations, the rest of the molecule was considered to have the same configurations as 7. Furthermore, the cis-dimer theonellin isocyanide (8) was also found to be a racemic mixture of the enantiomers 11S*, 9′R* as the ECD and optical rotation analyses did show only very weak absorbance.
The molecular formula C32H50N2O2 for compound 9 was obtained from the proton adduct [M + H]+ at m/z 495.3967 in the (+)-HRESIMS spectrum. After inspection of its 1H and 13C NMR spectra, 9 was found to have the same skeleton as 8. However, the apparent “doubling” of most of the signals around the terminal cyclohexane rings and the presence of formyl peaks at δH 8.05 (d, J = 1.0 Hz, H-16, endo) and 8.32 (d, J = 12.0 Hz, H-16, exo), and also at δH 8.04 (d, J = 2.0 Hz, H-16′, endo) and 8.30 (d, J = 12.0 Hz, H-16′, exo) evidenced the presence of amide rotamers in a 1:1 ratio. Therefore, both isocyanides of 8 were replaced by two formamides in 9, a modification confirmed by the key H-16/C-3 and H-16′/C-2′/4′HMBC correlations. The H3-15/H-9′/H-8′, H-10/H-8′ nOe correlations, indicated also a cis relative configuration between the methyl C-15 and the proton H-9′. The absence of Cotton effects in the ECD spectra and the weak optical rotation suggested that cis-dimer theonellin formamide (9) was also a racemic mixture of the enantiomers 11S*, 9′R*.
In the marine environment, nitrogen-functionalised terpenes are almost exclusively found in sponges and nudibranchs that feed on them.15 These metabolites possess a terpene scaffold bearing a nitrogen-containing functional group, the most common being isocyanide (–NC), isocyanate (–NCO), isothiocyanate (–NCS) or formamide (–NH-CHO), but also dichloroimine, carbamate, urea or amine.16 This growing class of metabolites (over 300 to date) includes mainly sesquiterpenoids and diterpenoids that contain one or up to three nitrogenous functions around a variety of terpene skeletons. In the case of the Bubarida sponge studied here, all new compounds possess the same bisabolene skeleton characterised by a 3,6-methylalkyl disubstituted cyclohexane ring and a nitrogen always substituted at the position C-3 (mainly formamide).17 The intriguing biosynthesis of N-functionalized terpenes, and particularly the origin of these functional groups, has been questioned since the first report of a marine isocyanide, axisonitrile-1, in 1973.18 Several biosynthetic feeding experiments with sponges maintained in aquaria have confirmed the incorporation of cyanide ions into the terpene skeleton, but the source of the cyanide is still questionned.15,16 The main hypothesis remains that these terpenes are products of enzymatic processes by the host sponges, but microbial symbionts could be involved in the production or incorporation of the cyanide, as cyanogenesis has been well documented in bacteria.19 Moreover, the interconversion of the precursors isocyanide-thiocyanate has been demonstrated in Axinyssa sp.,20 but their transformation to formamide or amines has not been investigated in detail. It is also possible that the isocyanide is transformed into the formamide functional group through acid hydrolysis.14,21 In our case, the formamide derivatives 1–6 and 9 were also found in the extract as demonstrated by the UHPLC-HRMS profiles which suggests that these metabolites were naturally present in our specimens. From a biosynthetic point of view, the diversity of metabolites found in this sponge suggests the presence of a pathway linking all metabolites with theonellin isocyanide as a precursor (Scheme 1). A highly reactive conjugated triene would be a key intermediate towards the oxidised monomers but also the dimers through a Diels–Alder reaction. A racemic epoxidation on the terminal vinylic group of this triene will first lead to the unusual oxygenated side chain at C-12. The opening of the epoxide through hydrolysis will lead to the vicinal diol of 3 containing an unstable tertiary alcohol. Three different pathways could then be envisaged: (i) a second epoxidation would lead to the epimeric mixture of 4; (ii) the migration of a carbocation through the diene would lead to 2 and then 1 after oxidation into an aldehyde; (iii) an oxidative cleavage of the terminal diol would yield the unusual conjugated ketone of 5 and 6. The presence of racemic mixtures suggest that oxidation processes are not chiral or that a racemisation occurs at the tertiary oxygenated carbons C-7 and C-11. Interestingly, the skeleton of the dimers 7–9 has already been reported but without nitrogen substitution.14 The identification of the cis and trans dimers 7 and 8 suggest that the Diels–Alder reaction responsible for the linkage of both monomers is not fully stereoselective therefore questioning the enzymatic process. The lack of overall stereoselectivity in all these biosynthetic steps is intriguing and would require further biosynthetic studies. The presence of highly reactive tertiary oxygenated carbons on the side-chain of the bubaridins were prone to some interconversion during the extractive process. We therefore carried out a reactivity test using the substrate theonellin formamide which was isolated in large quantity and contained the reactive diene. Theonellin formamide was dissolved in DMSO-d6 slightly acidified with TFA and exposed to a flow of air during 7 days at room temperature. The results showed a slow transformation of theonellin formamide in other derivatives, but the 1H NMR spectrum of the resulting mixtures did not result in the formation of any of the isolated metabolites 1–9. As we were also able to annotate the isolated metabolites in the chromatogram from the UHPLC-MS/MS analysis of the methanolic fraction obtained before purification, a natural rather than artefactual origin of the bubaridins is demonstrated. Compound 3 is an exception as it was readily converted into 1 during the NMR analyses performed in CDCl3. Thus, 2 and 1 are likely produced after isomerisation and then oxidation of 3.
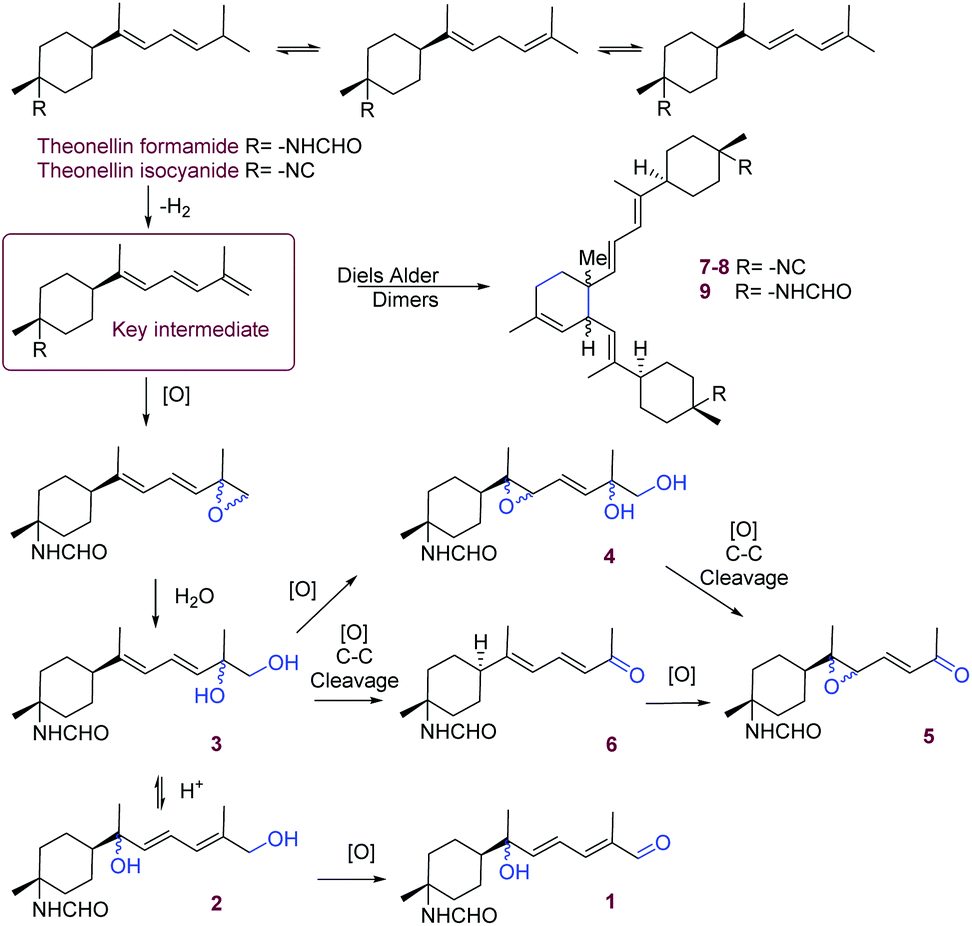 | ||
| Scheme 1 Biosynthetic pathway proposed for compounds 1–9. | ||
Our study also provided some chemotaxonomic insights into the classification of this sponge. Our DNA analyses firmly placed Bubarida sp. 1 MMR-2021 in the order Bubarida. All three specimens were resolved in a single well-supported clade (PP: 1; BP: 97) for the LSU (28S) gene. This clade was sister to the recently identified specimen collected from Hawaii, Bubarida sp. 2 JV-2020.22 These taxa combined were resolved within a larger clade shared with taxa currently assigned to the order Bubarida such as species of Acanthella, Dictyonella, Bubaris. The sister relationship between Bubarida sp. 1 MMR-2021 and Bubarida sp. 2 JV-2020 indicate that these specimens represent closely related but distinct taxa. The presence of nitrogenous bisabolenes in this species provided further meaningful insights into its classification. As mentioned earlier, there are only two reports of bisabolene dimers with the same cyclised skeleton named cis and trans dimers which have been previously isolated from Axinyssa variabilis (Order Suberitida) and Lipastrotethya ana (Order Bubarida, family Dictyonellidae).14,23 Interestingly, the authors pointed out that similar metabolites were not present in other species of Axinyssa. The family Dictyonellidae is currently represented by seven genera (Acanthella, Axinyssa, Cymbastela, Dictyonella, Lipastrotethya, Phakettia and Rhaphoxya), of which four (Acanthella, Axinyssa, Cymbastela, Dictyonella) have been sequenced for the 28S gene and one (Phakettia) for the COI gene. Our species did not cluster with any of the sequenced genera in the family Dictyonellidae but does produce nitrogenous bisabolene common in this family. However, as relationships within the order are likely to be revised as more sponge taxa are sequenced, especially from understudied regions of the world,24 ascribing taxa into families or genera at this stage would be premature. Nevertheless, based on our current understanding of sponge diversity and available sequences for comparison, our new species is resolved as a member of the Bubarida. Indeed, further taxonomic work will be required to clarify whether Bubarida sp. 1 MMR-2021 represents a new species, genus, or family or is merely the first barcode of a known taxon. We therefore encourage future studies on sponge natural products to provide DNA barcodes as a minimum in cases where names cannot be accurately assigned to taxa.
Bisabolene nitrogenous terpenes have been largely studied for their interesting biological activities such as cytotoxicity, antimicrobial and anti-malarial activity.25 The methanolic fraction of the sponge showed significant antifungal and cytotoxic activities against several tumoral cell lines. All the isolated compounds except the unstable bubaridin C (3) and 7-isothiocyanato-7,8-dihydro-α-bisabolene, were then evaluated against a fungal targeted panel of microbial pathogens, including the fungi Candida albicans, C. glabrata, C. krusei, C. parapsilosis, C. tropicalis, and Aspergillus fumigatus, the Gram-positive bacteria methicillin-sensitive Staphylococcus aureus (MSSA), and the Gram-negative bacteria Escherichia coli and Acinetobacter baumannii. In addition, the dimeric compounds 7–9 and N,N‘-bis[(6R,7S)-7-amino-7,8-dihydro-α-bisabolen-7-yl] were evaluated against five tumoral cell lines and none of the compounds tested displayed significant activity, indicating the selective antifungal bioactivity. Remarkably the dimer 7 exhibited potent antifungal activity against C. albicans, C. glabrata, C. parapsilosis and A. fumigatus with MIC values of 32, 1, 8 and 32 μg mL−1, respectively. Compounds 8 and N,N′-bis[(6R,7S)-7-amino-7,8-dihydro-α-bisabolen-7-yl] displayed moderate activity against C. glabrata with MIC values of 16–32 and 32 μg mL−1 respectively, while aureol presented a MIC of 16 μg mL−1 against MSSA. The other isolated metabolites did not display any significant bioactivity at the highest concentrations tested. Further analysis of the dose–response curves of compounds 7 and 8 against the five Candida species allowed a closer comparison of the antifungal activity. This revealed low micromolar AC50 values (Table 4) for both 7 and 8 against C. albicans, C. glabrata, and C. parapsilosis, and a lack of activity against C. krusei, and C. tropicalis. This also indicated that the trans configuration of the central ring present in 7, led to a greater potency of an order of magnitude. Furthermore, all compounds with a formamide functional group did not exhibit noticeable antifungal activity while 7 and 8 with an isocyanide were found to exhibit potent activity. Therefore, both the configuration and the nitrogen function of the bisabolene dimers have a strong influence on the antifungal activity. Previous structure–activity studies on nitrogenous bisabolene derivatives indicated a different mechanism of action when compared to the allylamine class of antifungal agents, although only moderate activity was reported for the former compounds.26 Compound 7 displayed a greater antifungal activity, with MIC values 10 fold lower than the other family of compounds, and a significant activity similar to the control amphotericin B. This potent and selective activity, along with a new possible mechanism of action, suggests that trans-theonellin isocyanide 7 is a promising lead antifungal candidate.
| Compound | MIC and AC50 (μM) | ||||
|---|---|---|---|---|---|
| C. albicans | C. glabrata | C. parapsilosis | C. tropicalis | C. krusei | |
| Note: First values are MIC and values in brackets are AC50 values calculated from dose–response curves. | |||||
| 7 | 70 [1.6] | 2.0 [1.6] | 17 [1.8] | 140 [107] | >140 [136] |
| 8 | >140 [12] | 70 [8.1] | 140 [18] | >140 | >140 |
| Amphotericin B | 2.0 [1.1] | 2.0 [0.85] | 4.0 [1.4] | 4.0 [0.93] | 2.0 [2.5] |
Conclusions
The chemical investigation of a common shallow-water sponge of the order Bubarida from Futuna Islands led to the identification of ten new nitrogenous bisabolene derivatives. Bubaridins A–F (1–6) are theonellin formamide analogues with an unusual oxidized linear chain mainly at the terminal position C-12, while the trans and cis-dimer theonellin isocyanide (7–8) and the cis-dimer theonellin formamide (9) are dimeric and cyclised bisabolenes bearing two isocyanide or formamide units. The metabolites were found to be mainly racemates and this interesting lack of stereoselectivity will require deeper biosynthetic studies. Although a few monomers could be artefacts of the isolation process, our data suggest a natural origin for most of the isolated metabolites. This study also represents the first report of nitrogenous and cyclised bisabolene dimers. The trans-dimer theonellin isocyanide 7 exhibited a potent and selective antifungal activity against several species of Candida, indicating this molecule as a good candidate for further pharmacological investigations. The remarkable antifungal activity of 7 confirmed the pharmaceutical potential of marine nitrogenous terpenoids and highlight the importance of the exploration of remote places as a source of new chemical diversity.Experimental
General experimental procedures
NMR experiments were recorded on an Agilent 600 MHz spectrometer (Agilent Technologies, Santa Clara, CA, USA) equipped with a 5 mm CryoProbe and on a Varian Innova 500 MHz spectrometer (Varian, Palo Alto, CA, USA) with a 5 mm OneNMR probe. Chemical shifts were referenced in ppm to the residual solvent signal (δH 7.26 ppm and δC 77.2 ppm for CDCl3, δH 3.31 ppm and δC 49.0 ppm for CD3OD). High-resolution mass spectrometry (HRESIMS) data were obtained from an Agilent QTOF 6540 coupled to a UPLC Agilent 1290 Infinity equipped with a diode array detector (Agilent Technologies, Santa Clara, CA, USA). Chemical profiling was performed on a Dionex Ultimate 3000 UPLC (Thermo Scientific, Waltham, MA, USA) equipped with a diode array detector coupled to an evaporative light scattering detector (Agilent 385-LC). HPLC separations and purifications were carried out on a Jasco HPLC with a LG-980-02 pump and a UV-1575 UV-Vis detector (JASCO, Tokyo, Japan). Optical rotations were recorded with an Autopol V polarimeter equipped with a 10 cm cell at 20 °C and at the Na D line, 589.3 nm (Rudolph Research Analytical, Hackettstown, NJ, USA). UV and ECD data were obtained from Chirascan™ V100 instrument (Applied Photophysics, Leatherhead, U.K.). Attenuated total reflectance-infrared spectroscopy (ATR-IR) spectra were recorded from 4000 to 650 cm−1 using a PerkinElmer Spectrum 400 (FT-IR/FT-NIR spectrometer) equipped with an UATR 1 bounce diamond/ZnSe Universal ATR sampling accessory (PerkinElmer, Waltham, MA, USA).Biological samples
The biological material was collected during the Tara Pacific expedition in 2016 around Futuna Islands. Five specimens of the same sponge species were collected by SCUBA from 10 to 18 m at the collections sites (GPS 14° 21′ 16′′ S, 178° 04′ 35′′ W) and (GPS 14° 21′ 47 S, 178° 02′ 55′′ W). Voucher samples are retained at the Marine Biodiscovery Laboratory (National University of Ireland, Galway) under the codes MBNUIG830/161213Fu05-01, MBNUIG851/161214Fu07-05 and MBNUIG852/161214Fu07-06. For taxonomic analyses the fragments were fixed in EtOH, while for chemical studies the biomass was freeze-dried and stored at −80 °C.Extraction and isolation
The freeze-dried biomass (50.3 g) was exhaustively extracted in CH3OH/CH2Cl2 (1:1, v/v) under sonication. The extract (6.6 g) was fractionated into 7 fractions using a sequence of solvents of decreasing polarity through C18 vacuum liquid chromatography: H2O, H2O/CH3OH (1:1, v/v), (1:3, v/v), CH3OH, CH3OH/CH2Cl2 (3:1, v/v), (1:1, v/v) and CH2Cl2. The fractions were analysed in the UHPLC-DAD-HRMS and UPLC-DAD-ELSD.
The CH3OH fraction revealed interesting metabolites on the chemical profiles, and was further separated into 14 subfractions via semipreparative RP-HPLC (Waters Xselect phenyl-hexyl column, 5 μm; 10 × 250 mm; flow rate 4.0 mL min−1, λ = 210 nm) using a gradient of solvents (A) H2O and (B) CH3CN (0–5 min 70% B, 20–30 min 90% B, 31–36 min 70% B) to give theonellin formamide (28.7 mg, 5.71 × 10−2% w/w), N,N′-bis[(6R,7S)-7-amino-7,8-dihydro-α-bisabolen-7-yl] (42.4 mg, 8.43 × 10−2% w/w) and compound 8 (7.0 mg, 1.39 × 10−2% w/w). The analytical separation of subfraction 3 (19.5 mg) by RP-HPLC (Waters Symmetry C18, 5 μm; 4.6 × 250 mm; flow rate: 1 mL min−1, λ = 210 nm) using a gradient of solvents (A) H2O and (B) CH3CN (0–5 min 15% B, 20–30 min 30% B, 31–36 min 15% B) afforded the compounds 4 (0.3 mg, 5.96 × 10−4% w/w), 2 (0.4 mg, 7.95 × 10−4% w/w), 3 (0.5 mg, 9.94 × 10−4% w/w), 1 (0.4 mg, 7.95 × 10−4% w/w), 5 (0.6 mg, 1.19 × 10−3% w/w), and 6 (0.6 mg, 1.19 × 10−3% w/w). The subfraction 7 was purified through semipreparative RP-HPLC (Phenomenex Synergi Fusion RP column, 4 μm; 10 × 250 mm; flow rate 4.0 mL min−1, λ = 210 nm) with an isocratic composition of solvents H2O/CH3CN (33:66 v/v, 36 min) to yield theonellin formamide (0.6 mg, 1.19 × 10−3% w/w) and 9 (5.0 mg, 9.94 × 10−4% w/w). The semipreparative purification of subfraction 8 using the same method as for fraction 7 gave the compound aureol (0.5 mg, 9.94 × 10−4% w/w). Subfraction 10 was separated with an isocratic composition of solvents H2O/CH3CN (32:68 v/v, 50 min) to afford the compound 7-Isothiocyanato-7,8-dihydro-α-bisabolene (0.2 mg, 3.97 × 10−4% w/w), and other minor and unstudied bisabolene derivatives. The subfraction 13 was purified through analytical RP-HPLC (Waters Symmetry C18, 5 μm; 4.6 × 250 mm; flow rate: 1 mL min−1, λ = 210 nm) with an isocratic composition of solvents H2O/CH3CN (16:84 v/v, 45 min) to separate the compounds 7 (5.4 mg, 1.07 × 10−2% w/w) and 8 (7.0 mg, 1.39 × 10−2% w/w).
ε): 276 (4.04) nm; (+)-HRESIMS m/z [M + H]+ 280.1916 (calcd. for C16H25NO3 280.1907); 1H NMR see Table 1 and 13C NMR see Table 2.
ε): 276 (2.89), 238 (3.91) nm; (+)-HRESIMS m/z [M + H]+ 282.2077 (calcd. 282.2064 for C16H27NO3); 1H NMR see Table 1 and 13C NMR see Table 2.
ε): 200 (+3.89), 277 (+2.94), nm; (+)-HRESIMS m/z 298.2023 [M + H]+ (calcd. 298.2013 for C16H27NO4); 1H NMR see Table 1 and 13C NMR see Table 2.
ε): 280 (2.74), 232 (3.40) nm; (+)-HRESIMS m/z 266.1760 [M + H]+ (calcd. 266.1751 for C15H23NO3); 1H NMR see Table 1 and 13C NMR see Table 2.
ε): 287 (3.69) nm; (+)-HRESIMS m/z 250.1818 [M + H]+ (calcd. 250.1802 for C15H23NO2); 1H NMR see Table 1 and 13C NMR see Table 2.
ε): 242 (4.16) nm; IR (neat) νmax: 2933, 2864, 2246, 2130, 1704, 1674, 1650, 1446, 1379, 1356, 1264, 1245, 1180, 1128, 969 cm−1; (+)-HRESIMS m/z 459.3726 [M + H]+ (calcd. 459.3724 for C32H46N2); 1H NMR and 13C NMR see Table 3.
ε): 281 (2.74), 224 (3.92) nm; IR (neat) νmax: 2931, 2861, 2130, 1728, 1463, 1273, 904, 766, 665 cm−1; (+)-HRESIMS m/z 459.3723 [M + H]+ (calcd. 459.3724 for C32H46N2); 1H NMR and 13C NMR see Table 3.
ε): 281 (2.74), 224 (3.92) nm; IR (neat) νmax: 2925, 2856, 1664, 1533, 1448, 1378, 970, 887, 735, 701 cm−1; (+)-HRESIMS m/z 495.3967 [M + H]+ (calcd. 495.3945 for C32H46N2); 1H NMR and 13C NMR see Table 3.
Biological activity assays
Antifungal activity against C. albicans, C. glabrata, C. krusei, C. parapsilosis, C. tropicalis, and Aspergillus fumigatus, and antibacterial activity against the Gram-positive bacteria MSSA, and the Gram-negative bacteria E. coli and A. baumannii were determined according to previously published procedures.27–29 MIC was defined as the concentration causing a growth inhibition of the parasite higher than 90%.The cell viability test against five different human cancer cell lines (A549 (lung carcinoma), A2058 (metastatic melanoma), MCF7 (breast adenocarcinoma), MIA PaCa-2 (pancreatic carcinoma), and HepG2 (hepatocyte carcinoma)) was based on the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay.30 Ten ½ serial dilutions starting at a concentration of 20 μg mL−1 in triplicate were tested for each compound. IC50 was determined as previously described.31
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are deeply grateful to the Tara Ocean Foundation teams and crew and the members of the Tara Pacific Consortium (see ESI†) for their support during the field trip to Futuna. Support and permission to undertake this study were provided by Atoloto Malau (Service de l'Environnement, Wallis and Futuna). We would like to express our gratitude to the administrative and customary authorities of the Territoire des iles Wallis et Futuna for their support in the sample collection. We are keen to thank the commitment of the people and the institutions mentioned in the ESI† for their financial and scientific support that made this singular expedition possible. Tara Pacific would not exist without the continuous support of the participating institutes. This work was mainly funded by a start-up research provided to O. P. T. by NUI Galway. M. M. G. acknowledges the James Hardiman Research Scholarship (NUI Galway) for supporting her PhD and Dr R. Doohan (NUI Galway) is acknowledged for her help in the NMR experiments.Notes and references
- A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2021, 38, 362–413 RSC.
- N. S. Webster and T. Thomas, mBio, 2016, 7, e00135–e00116 CrossRef PubMed.
- M. C. Leal, A. Hilario, M. H. G. Munro, J. W. Blunt and R. Calado, Nat. Prod. Rep., 2016, 33, 747–750 RSC.
- N. E. Redmond, C. C. Morrow, R. W. Thacker, M. C. Diaz, N. Boury-Esnault, P. Cardenas, E. Hajdu, G. Lobo-Hajdu, B. E. Picton, S. A. Pomponi, E. Kayal and A. G. Collins, Integr. Comp. Biol., 2013, 53, 388–415 CrossRef CAS PubMed.
- C. C. Morrow and P. Cárdenas, Front. Zool., 2015, 12, 7 CrossRef PubMed.
- W. H. Organization, Global Antimicrobial Resistance Surveillance System (GLASS): early implementation protocol for inclusion of Candida spp, World Health Organization, 2019 Search PubMed.
- B. W. Sullivan, D. J. Faulkner, K. T. Okamoto, M. H. M. Chen and J. Clardy, J. Org. Chem., 1986, 51, 5134–5136 CrossRef CAS.
- P. Djura, D. B. Stierle, B. Sullivan and D. J. Faulkner, J. Org. Chem., 1980, 45, 1435–1441 CrossRef CAS.
- H. Nakamura, J. I. Kobayashi, Y. Ohizumi, K. Mitsubishi and Y. Hirata, Tetrahedron Lett., 1984, 25, 5401–5404 CrossRef CAS.
- J.-Z. Sun, K.-S. Chen, H.-L. Liu, R. van Soest and Y.-W. Guo, Helv. Chim. Acta, 2010, 93, 517–521 CrossRef CAS.
- H.-L. Liu, D.-Q. Xue, S.-H. Chen, X.-W. Li and Y.-W. Guo, Helv. Chim. Acta, 2016, 99, 650–653 CrossRef CAS.
- D. E. N. Jacquot and T. Lindel, Curr. Org. Chem., 2005, 9, 1551–1565 CrossRef CAS.
- W. Cheng, D. Liu, F. Zhang, Q. Zhang, P. Pedpradab, P. Proksch, H. Liang and W. Lin, Tetrahedron, 2014, 70, 3576–3583 CrossRef CAS.
- S.-C. Mao, E. Manzo, Y.-W. Guo, M. Gavagnin, E. Mollo, M. L. Ciavatta, R. van Soest and G. Cimino, Tetrahedron, 2007, 63, 11108–11113 CrossRef CAS.
- M. J. Garson and J. S. Simpson, Nat. Prod. Rep., 2004, 21, 164–179 RSC.
- X. Zhang, L. Evanno and E. Poupon, Eur. J. Org. Chem., 2020, 1919–1929 CrossRef CAS.
- J. Emsermann, U. Kauhl and T. Opatz, Mar. Drugs, 2016, 14, 83 CrossRef.
- F. Cafieri, E. Fattorusso, S. Magno, C. Santacroce and D. Sica, Tetrahedron, 1973, 29, 4259–4262 CrossRef CAS.
- C. Blumer and D. Haas, Arch. Microbiol., 2000, 173, 170–177 CrossRef CAS PubMed.
- J. S. Simpson and M. J. Garson, Tetrahedron Lett., 2001, 42, 4267–4269 CrossRef CAS.
- J. S. Simpson and M. J. Garson, Org. Biomol. Chem., 2004, 2, 939–948 RSC.
- J. Vicente, M. K. Webb, G. Paulay, W. Rakchai, M. A. Timmers, C. P. Jury, K. Bahr and R. J. Toonen, Coral Reefs, 2021 DOI:10.1007/s00338-021-02109-7.
- S.-C. Mao, Y.-W. Guo, R. van Soest and G. Cimino, J. Asian Nat. Prod. Res., 2011, 13, 770–775 CrossRef CAS PubMed.
- M. M. Reddy, L. Jennings and O. P. Thomas, in Progress in the Chemistry of Organic Natural Products, ed. A. D. Kinghorn, H. Falk, S. Gibbons, Y. Asakawa, J. K. Liu and V. M. Dirsch, Springer, Cham, 2021, vol. 116, pp. 1–36 Search PubMed.
- M. J. Schnermann and R. A. Shenvi, Nat. Prod. Rep., 2015, 32, 543–577 RSC.
- M. T. Jamison, J. Macho and T. F. Molinski, Bioorg. Med. Chem. Lett., 2016, 26, 5244–5246 CrossRef CAS PubMed.
- C. Audoin, D. Bonhomme, J. Ivanisevic, M. D. L. Cruz, B. Cautain, M. C. Monteiro, F. Reyes, L. Rios, T. Perez and O. P. Thomas, Mar. Drugs, 2013, 11, 1477–1489 CrossRef CAS PubMed.
- G. Crespo, V. González-Menéndez, M. de la Cruz, J. Martín, B. Cautain, P. Sánchez, I. Pérez-Victoria, F. Vicente, O. Genilloud and F. Reyes, Planta Med., 2017, 234, 545–550 Search PubMed.
- M. C. Monteiro, M. de la Cruz, J. Cantizani, C. Moreno, J. R. Tormo, E. Mellado, J. R. De Lucas, F. Asensio, V. Valiante, A. A. Brakhage, J.-P. Latgé, O. Genilloud and F. Vicente, J. Biomol. Screening, 2012, 17, 542–549 CrossRef CAS PubMed.
- K. Präbst, H. Engelhardt, S. Ringgeler and H. Hübner, in Cell Viability Assays: Methods and Protocols, ed. D. F. Gilbert and O. Friedrich, Springer, New York, New York, NY, 2017, pp. 1–17 Search PubMed.
- R. R. Koagne, F. Annang, B. Cautain, J. Martin, G. Perez-Moreno, G. T. M. Bitchagno, D. Gonzalez-Pacanowska, F. Vicente, I. K. Simo, F. Reyes and P. Tane, BMC Complementary Med. Ther., 2020, 20, 8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob02297k |
| This journal is © The Royal Society of Chemistry 2022 |