
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical synthesis of 4′-thio and 4′-sulfinyl pyrimidine nucleoside analogues†
Mieke
Guinan
ab,
Ningwu
Huang
c,
Chris S.
Hawes
 a,
Marcelo A.
Lima
bd,
Mark
Smith
c and
Gavin J.
Miller
*ad
a,
Marcelo A.
Lima
bd,
Mark
Smith
c and
Gavin J.
Miller
*ad
aLennard-Jones Laboratory, School of Chemical and Physical Sciences, Keele University, Keele, Staffordshire ST5 5BG, UK. E-mail: g.j.miller@keele.ac.uk
bSchool of Life Sciences, Keele University, Keele, Staffordshire ST5 5BG, UK
cRiboscience LLC, 428 Oakmead Pkwy, Sunnyvale, CA 94085, USA
dCentre for Glycoscience Research, Keele University, Keele, Staffordshire ST5 5BG, UK
First published on 11th November 2021
Abstract
Analogues of the canonical nucleosides required for nucleic acid synthesis have a longstanding presence and proven capability within antiviral and anticancer research. 4′-Thionucleosides, that incorporate bioisosteric replacement of furanose oxygen with sulfur, represent an important chemotype within this field. Established herein is synthetic capability towards a common 4-thioribose building block that enables access to thio-ribo and thio-arabino pyrimidine nucleosides, alongside their 4′-sulfinyl derivatives. In addition, this building block methodology is templated to deliver 4′-thio and 4′-sulfinyl analogues of the established anticancer drug gemcitabine. Cytotoxic capability of these new analogues is evaluated against human pancreatic cancer and human primary glioblastoma cell lines, with observed activities ranging from low μM to >200 μM; explanation for this reduced activity, compared to established nucleoside analogues, is yet unclear. Access to these chemotypes, with thiohemiaminal linkages, will enable a wider exploration of purine and triphosphate analogues and the application of such materials for potential resistance towards relevant hydrolytic enzymes within nucleic acid biochemistries.
1. Introduction
A significant proportion of current chemotherapeutic treatments for cancer involve the use of anti-metabolites, particularly modified nucleoside analogues that possess a capability to mimic native purine or pyrimidine nucleosides, which can disrupt metabolic and regulatory pathways.1 Notwithstanding this significant medicinal capability, therapeutic intervention using nucleoside analogues is often limited by poor cellular uptake, low conversion to the active triphosphate metabolite, rapid degradation or clearance and development of resistance profiles in certain cell types.2 Consequently, research activity in this field continues to develop syntheses for next generations of nucleoside analogues that can overcome these limitations and provide new therapeutic options.3–5Within this context, 4′-thionucleosides, where the furanose ring oxygen is substituted with sulfur, have received attention from several academic and industrial groups since a first disclosure of 4′-thioadenosine in the early 1960s;6,7Fig. 1 highlights some key achievements in this area. Bioisosteric replacement of furanose oxygen with larger sulfur seeks to explore the biological effect imparted through changes to furanose ring conformation and the hydrolytic stability of a thiohemiaminal glycosidic linkage.
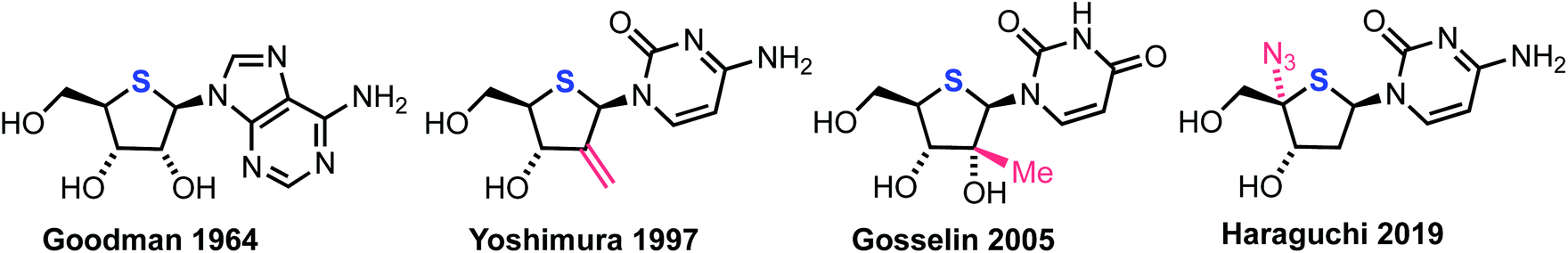 | ||
| Fig. 1 Historical and current examples of thionucleosides (blue sulphur) with additional ring substituent modifications (red). | ||
Insertion of a sulfur atom to access thiofuranose components generally start from available chiral pool materials and the use of thiourea to convert 4,5-epoxides to 4,5-thiiranes,8,9 alongside sodium sulfide-mediated formation of 2,5-bicyclic systems are notable examples of key intermediates developed to enable this.10–12 Open chain systems have also been used,13 for example, effecting double displacement of a 1,4-bismesylate with Na2S to access 1-deoxy thiofuranoses,14 and both C1 aldehyde and thioacetal oxidation levels have delivered access to anomeric thioglycosides.15,16 Two main approaches have been adopted to install appropriate nucleobases; firstly thioglycosylation of a silylated nucleobase,8,17 and, secondly a Pummerer-type reaction of the sulfoxide to condense with a corresponding nucleobase.16,18 Hypervalent iodine reagents have also been used to effect a similar transformation.19
As part of a program exploring chemical synthesis approaches to next generation nucleoside scaffolds, we were interested in accessing appropriate 4′-thionucleosides and templating this bioisosteric replacement onto established chemotypes, a tactic recently implemented by Liotta and colleagues for 2′-C-methyl-4′-thionucleosides.20 Herein we report our synthesis of 4′-thio and 4′-sulfinyl nucleosides; we targeted an open-chain C1-oxime to enable sulfur inclusion using displacement of an appropriate 4-position leaving group, with concomitant ring closure onto a C1 aldehyde. This was intended to provide a benchmark in then accessing related 2′-deoxy-2′,2′-gem-difluoro and D-arabino structures which, alongside recent examples 2′-deoxy-2′-fluoro and L-3′-deoxy-3′,3′-difluoro systems,13,21,22 provides capability to more broadly study 4′-thionucleoside chemotypes and their structure activity relationships. These aims are highlighted in Fig. 2.
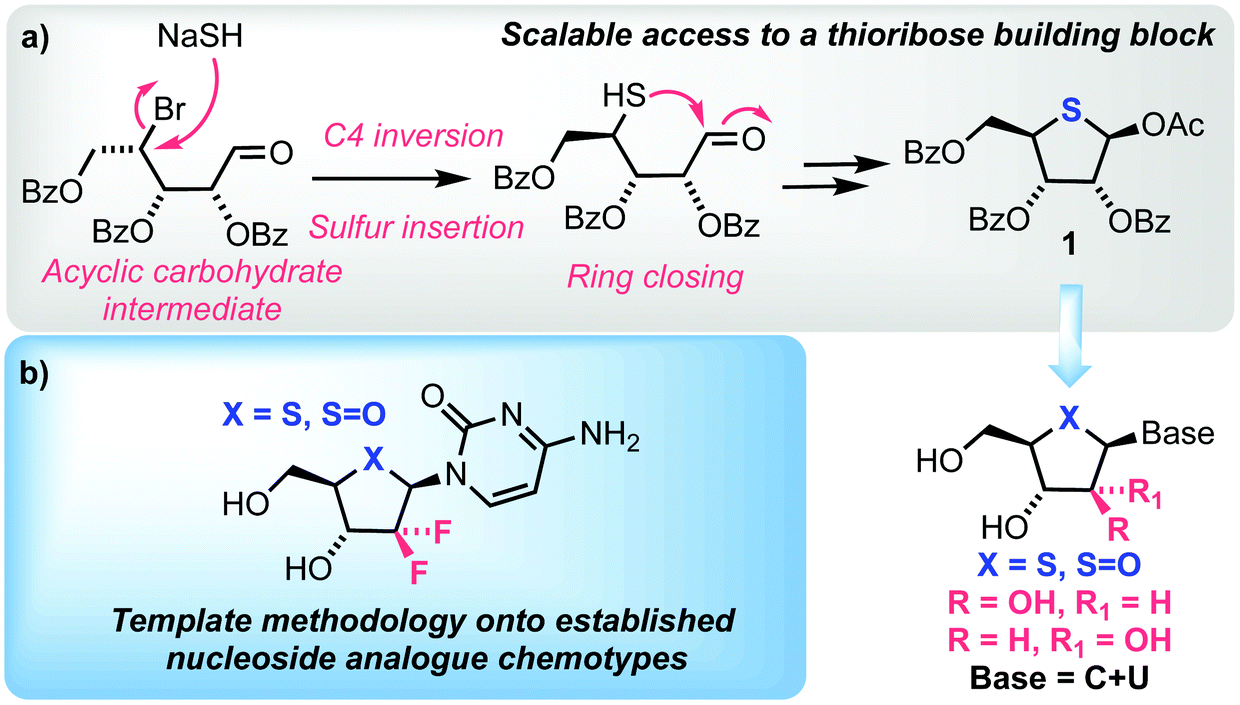 | ||
| Fig. 2 (a) Strategy to access 4-thioribose derivative 1 as a key intermediate for 4′-thionucleoside synthesis and (b) potential therefrom to template to established nucleoside analogue classes (blue box). | ||
2. Results and discussion
2.1. Synthesis of a 4-thioribose building block and extension to a 2-deoxy-2,2-difluoro-4-thioribose analogue
In order to access our intended series of thionucleoside derivatives, a scalable and reliable method for the synthesis of 4-thio-D-ribofuranose 1 was required (Scheme 1). Starting from commercially available 2, the anomeric acetate was hydrolysed using H2O/BF3·OEt2 in 91% yield on 100 g scale. The resultant hemi-acetal was used crude and converted to the corresponding oxime 3via treatment with O-methoxyhydroxylamine hydrochloride and Et3N. This material was isolated as a mixture of oxime isomers (3/1 ratio) in 90% yield, again on multigram scale. Manipulation of the C4 position in 3 to facilitate conversion to the final thiofuranose, with retention of the C4-ribo-configuration, was explored next. Direct halogenation and inversion of stereochemistry at C4 using CBr4 or Br2 with PPh3 proved unsuccessful, returning only starting material. Instead, the C4-OH in 3 was converted to an arylsulfonyl leaving group. Initially here we targeted a 4-nitrobenzenesulfonate, but persistent low yields (maximum obtained = 31%) and a requirement for chromatography led us to explore other options. An earlier report of a less common 2,3,5-tricholorobenzensulfonate proved fruitful,23 delivering 4 in 72% yield. Purification of 4 was achieved using trituration from Et2O, enabling easy isolation as a white solid. This material could be prepared in multigram amounts in 56% overall yield for three steps from 2 and required no column chromatography.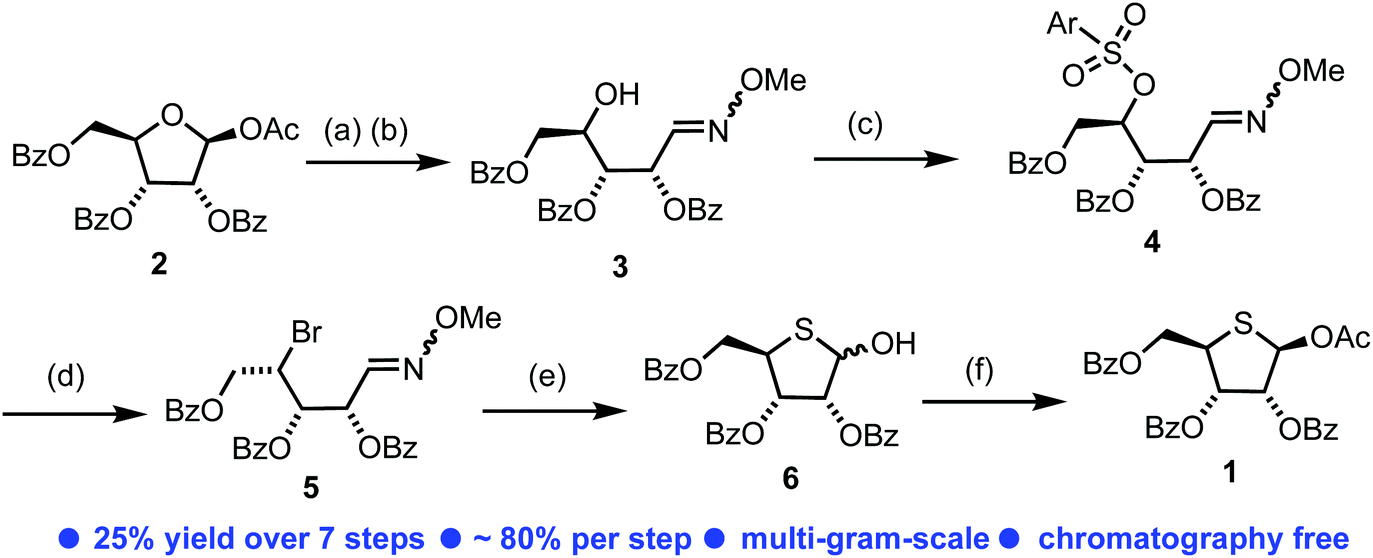 | ||
| Scheme 1 (a) BF3·Et2O, H2O, MeCN, rt, 91%; (b) H2N-OMe·HCl, Et3N, MeOH, rt, 90%; (c) 2,4,5-Trichlorobenzenesulfonyl chloride, N-methylimidazole, MeCN, rt, 72%; (d) LiBr, butanone, 80 °C, 89%. (e) (i) Glyoxylic acid, MeCN, 70 °C, 92%; (ii) NaSH·H2O, H2O, DMF, 0 °C, 76%; (f) DMAP, Ac2O, rt, 72% (β-only); Ar = 2,4,5-trichlorophenyl. | ||
Nucleophilic displacement of the 4-sulfonyl group within 4 was explored using LiBr in a Finkelstein-type reaction. The use of DMI/THF as the solvent at 60 °C for 16 h resulted in a mixture of C4 diastereoisomers (3/1, S/R, see ESI Fig. S4†), presumably forming via a second nucleophilic displacement of bromide 5 to give the unwanted the C4-R epimer. Similar issues surrounding unwanted C4 epimerisation were encountered by Codée and co-workers during their synthesis of 4-thiofuranosides.14 Reducing the reaction time to 2 h in the same solvent system improved the diastereomeric outcome (10/1, S/R), however, this was accompanied by incomplete consumption of 4. Switching solvent to 2-butanone and heating at 80 °C for 18 h delivered diastereomerically pure C4-bromide 5 in 89% yield. 13C NMR analysis of 5 confirmed an upfield shift of C4 to δC 47.8 ppm, from δC 79.6 ppm. Treatment of bromide 5 with glyoxylic acid at 70 °C effected hydrolysis of the oxime to the corresponding aldehyde in 92% yield, which was used immediately for conversion to the desired 4-thiofuranose 6. This proceeded via reaction with NaSH, effecting SN2 displacement of the C4 bromide followed by ring closure onto the C1-aldehyde, obtaining 6 as a mixture of anomers in 76% yield. Crude 6 was treated with Ac2O in pyridine with catalytic DMAP to obtain the acetylated product 1 as a mixture of anomers (4/1, β/α). Simple trituration from MeOH isolated the β-anomer of 1 exclusively in a satisfactory 50% yield from 5, with analytical data confirming those reported previously for D-ribo thiofuranoses.9 The development of this synthetic route to 1 required no column chromatography and was completed starting from 100 g of 2, delivering 1 in an overall yield of 25% for the seven steps and in 25 g quantity.
The chemistry developed in Scheme 1 was next mapped onto a 2-deoxy-2,2-gem-difluoro ribose system to access 4′-thiogemcitabine and analogues thereof (Scheme 2). Commercially available lactone 7 was manipulated in six steps to deliver 2-deoxy-2,2-difluoro-1-(4-thio-D-ribofuranose) 10. Lactone 7 was first reduced using Li(OtBu)3AlH to the hemiacetal in 95% crude yield and then converted directly to oxime 8via reaction with O-methoxyhydroxylamine and Et3N in the presence of pyridinium p-toluenesulfonate. Interestingly, no reaction to convert the intermediate hemi-acetal was observed in the absence of the pyridinium salt. Oxime 8 then underwent successive derivatisation at C4, first to the sulfonate ester and then nucleophilic substitution to yield bromide 9. Finally, oxime hydrolysis was followed by incorporation of sulfur at C4 to restore the D-ribo configuration and subsequent cyclisation to thiohemiacetal 10. This enabled access to multigram quantities of 10 in 6 steps and 56% overall yield from the commercial starting material.
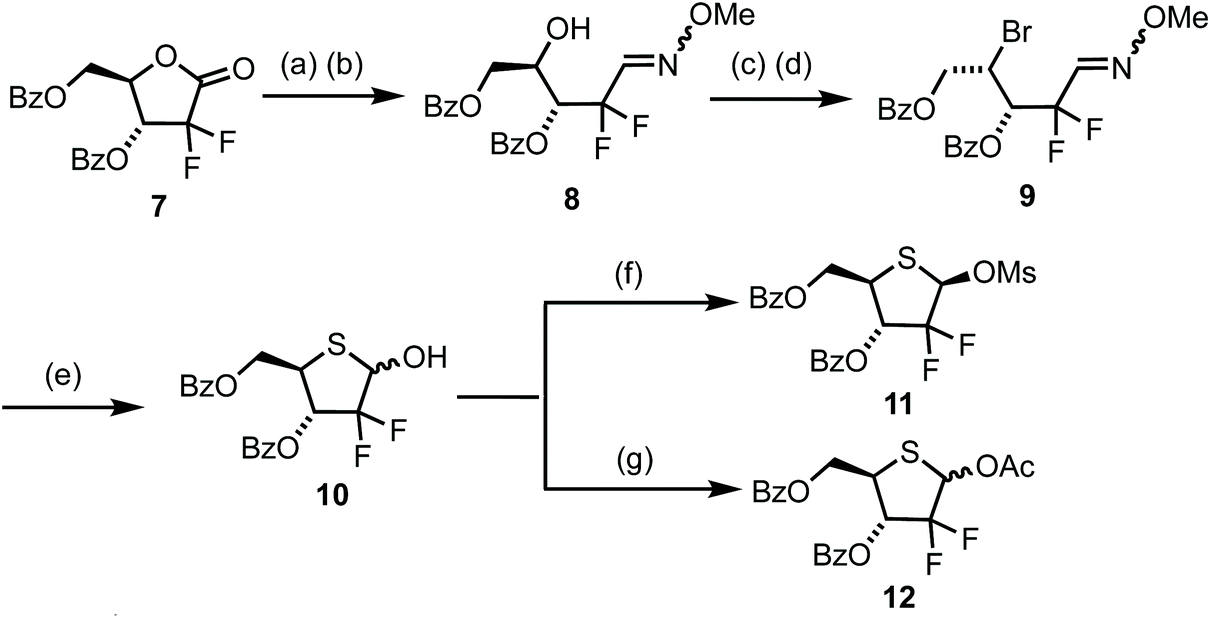 | ||
| Scheme 2 (a) Li(OtBu)3AlH, THF, 0 °C, 95%; (b) H2N-OMe·HCl, Et3N, MeCN, H2O, pyridinium p-toluenesulfonate, rt, 90% (c) 2,4,5-Trichlorobenzenesulfonyl chloride, N-methylimidazole, MeCN, rt 75% (d) LiBr, butanone, 80 °C. (e) (i) Glyoxylic acid, MeCN, 70 °C (ii) NaSH·H2O, H2O, DMF, 0 °C, 97% over 3 steps (f) MsCl, Et3N, CH2Cl2, rt, 79% (53% for β-anomer only) (g) Ac2O, Et3N, CH2Cl2, rt, 88%. | ||
The anomeric hydroxyl group in 10 was next converted to either mesylate 11 or acetate 12 as appropriate leaving groups to explore glycosylation of nucleobases. Treatment of 10 with MsCl and Et3N (Scheme 2) afforded 11 in 79% yield as a 1/4, α/β mixture. The β-anomer was isolated by crystallisation from hot Et2O and the resultant solid recrystallised by diffusion using CH2Cl2/hexanes to afford pure β-11. A sample of β-11 was analysed by X-ray crystallography and confirmed the double stereochemical inversion at C4, which retained a D-ribo configuration (Fig. 3). The anomeric mesylate and C5 were observed above the plane of the thiopentose ring, consistent with 1-β-D-ribo stereochemistry, with the ring adopting a C3-endo conformation in the solid state. Anomeric acetate 12 was accessed in 88% yield as a 3/2 mixture of anomers that, in our hands, proved inseparable by crystallisation or column chromatography.
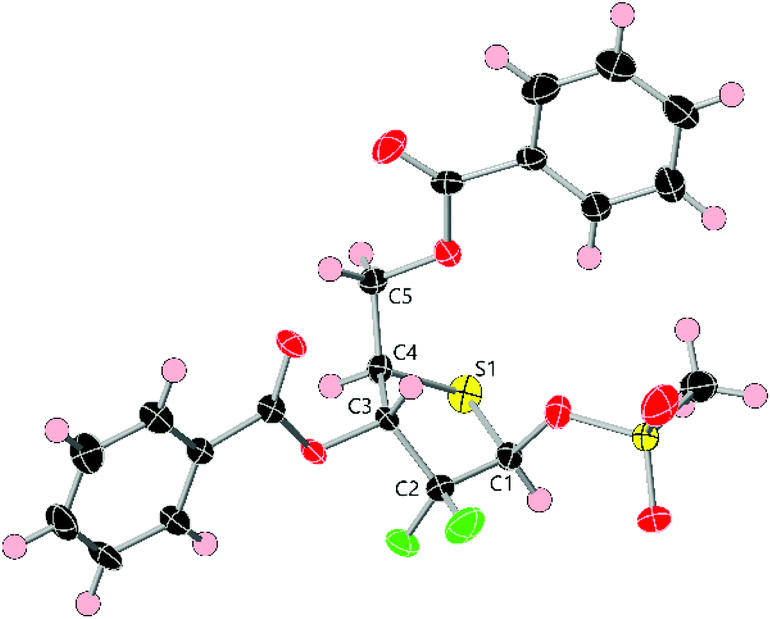 | ||
| Fig. 3 X-ray crystal structure of β-11 with numbering scheme for core atoms. ADPs are rendered at 50% probability level and ring disorder on the C5 phenyl ester substituent is omitted for clarity. Atom colouring scheme: black = carbon, red = oxygen, green = fluorine, yellow = sulfur, pink = hydrogen. | ||
2.2. Synthesis of 4′-thioribo- and 4′-thioarabino nucleosides
With access to multigram scale quantities of 1, a series of 4′-thioribo and 4′-thioarabino pyrimidines were targeted. A pathway to access these was proposed from 4′-thiouridine 14 (Scheme 3). Comparative literature for the synthesis of 2′,3′,5′-tri-O-benzoyl protected uridine involved refluxing the reactants for 3 h in acetonitrile, following silyation of the nucleobase.24 However, reaction under these conditions for 1 produced low yields of 13 (20–30%). By conducting the reaction just below the boiling point of MeCN, at 75 °C, the yield improved significantly, to 74%. The β-anomer of 13 formed preferentially (1H NMR of crude 13 indicated a 10/1, β/α ratio, [3JH1–H2 = 6.8 Hz for β-13]) and the reaction was scalable to 10 g.25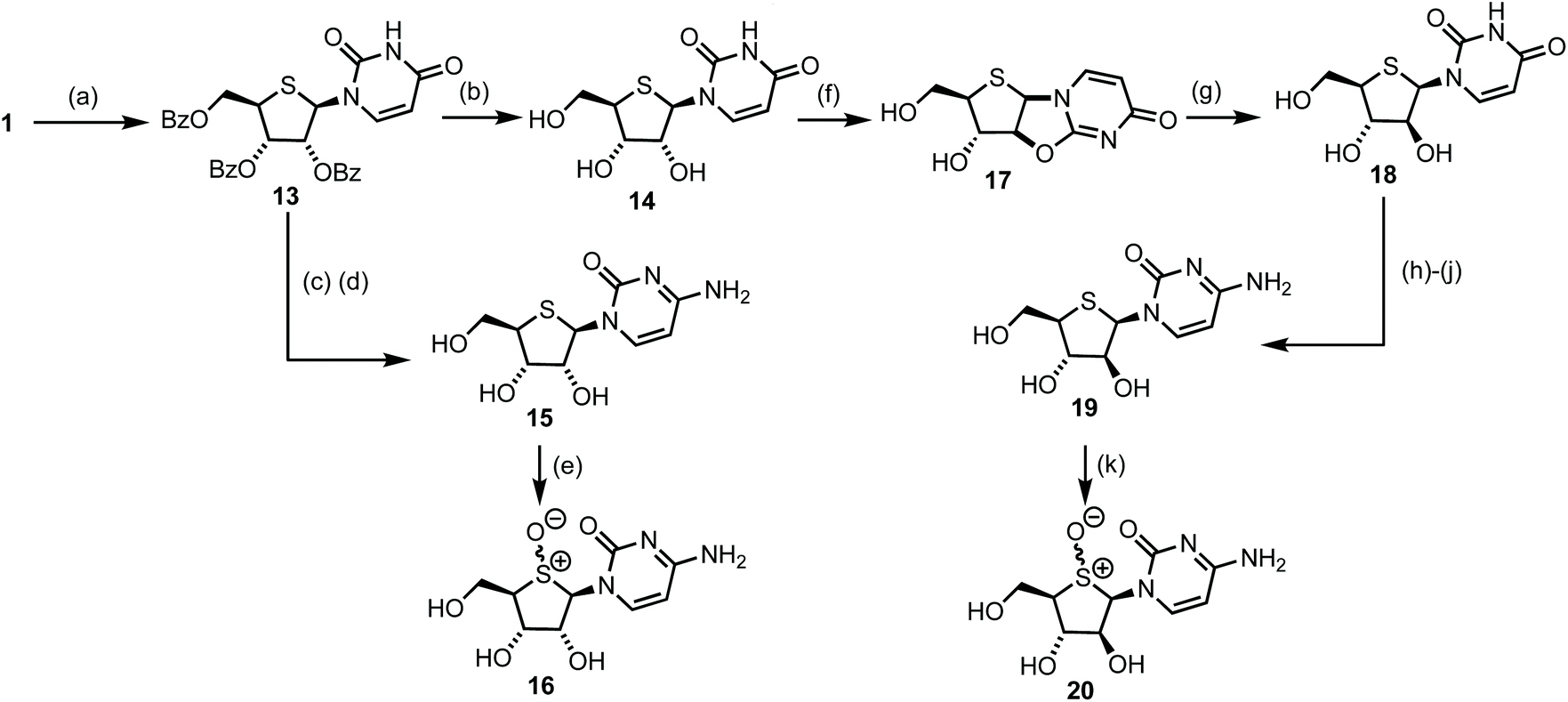 | ||
| Scheme 3 (a) (i) Uracil, HMDS, reflux (ii) TMSOTf, MeCN, 75 °C, 74% (b) 7 M NH3 in MeOH, MeOH, 40 °C, 96% (c) POCl3, 1,2,4-triazole, Et3N, MeCN, rt (d) NH4OH, 1,4-dioxane, rt, then 7 M NH3 in MeOH, MeOH, 40 °C, 77%, 2 steps from 13 (e) m-CPBA, H2O, MeCN, 0 °C, 90%, 1/1 d.r. at S (f) (PhO)2CO, NaHCO3, DMF, 100 °C, 98% (g) KOH, EtOH, H2O, rt, 90% (h) Ac2O, DMAP, pyridine, rt, 82% (i) POCl3, 1,2,4-triazole, Et3N, MeCN, rt (j) 7 M NH3 in MeOH, MeOH, 120 °C, 70%, 2 steps from 18 (k) m-CPBA, H2O, MeCN, 0 °C, 78%, 2.5/1 d.r. at S. | ||
Global deprotection of 13 was completed using 7 M NH3 in MeOH to obtain 4′-thiouridine 14 in 96% yield. This material was next converted to its cytosine form, first by treatment of 14 with POCl3 and then 1,2,4-triazole to furnish the heterocyclic intermediate, before a final treatment with NH3 in MeOH concomitantly substituted the triazole and removed the benzoyl protecting groups to give 4′-thiocytidine 15 in 77% yield over two steps.
4′-thiouridine 14 was also converted to its C2′ epimer 18via an intramolecular cyclisation of the C4-carbonyl, displacing a C2′-carbonate leaving group. This inverted the C2′ stereochemistry to give β-1′,2′-syn-bicycle 17 in 92% yield, with 13C NMR confirming a chemical shift change of δC 152.3 ppm to δC 175.4 ppm for C2′ in 17. Subsequent ring opening of 17 was achieved by stirring with KOH in EtOH/H2O, to give C2′-arabino configured 18 in almost quantitative yield. The observed 1H NMR coupling constant between H2′ and H3′ for 18 (3JH2′–H3′ = 9.1 Hz) was significantly different than that observed for 14 (3JH2′–H3′ = 3.7 Hz), supporting inversion of stereochemistry at C2′. 4′-thioarabinouridine 18 was also converted to its cytosine form using the method previously established for 15, delivering 4′-thioarabinocytidine 19 in 52% yield over three steps from 18. Earlier efforts to directly convert 15 to 19via a β-1′,2′-syn-intermediate bi-cycle were unsuccessful; multiple reaction products were observed and this conversion was abandoned. Finally, the corresponding sulfoxides of 15 and 19 were prepared via treatment with 1.5 equivalents of m-CPBA, to afford novel 16 and 20 in 90% and 78% yields respectively. 13C NMR analysis confirmed a significant downfield shift of C1′ and C4′ from δC 65.1 and 51.7 ppm in 16 to δC 73.1 and 65.7 ppm in 20 respectively. Attempted separation of the S-diastereoisomers by crystallisation and reverse phase preparative HPLC were unsuccessful.
2.3. Synthesis of 4′-thio and 4′-sulfinylgemcitabine
In order to access 2-deoxy-2,2-gem-difluorothionucleosides, we returned to anomeric acetate 12 and mesylate 11 to investigate nucleobase attachment. Prior reports suggested use of an anomeric mixture of 11 delivered the corresponding nucleosides as a 1/1, α/β mixture, regardless of the anomeric ratio of the starting mesyl glycoside.10,26 In order to target 4′-thiogemcitabine, silylated N4-benozyl cytosine was first glycosylated with donor 11, using TMSOTf as activator. These conditions were unsuccessful (no product was isolated) and SnCl4 was selected instead. Under these conditions the desired nucleoside was isolated, but in <10% yield. Accordingly, we switched glycosyl donor to anomeric acetate 12 and were pleased to observe an improved yield for glycosylation of silylated N4-benozyl cytosine, delivering 21 in 28% yield, with 25% of 12 recovered. Expectedly, no diastereocontrol was observed for this glycosylation; 21 was isolated as a 1.1/1 anomeric mixture, requiring subsequent separation either using fractional precipitation or preparative HPLC. Following protecting group removal, 22-α was confirmed through selective 1D NOESY, where though-space dipolar interactions between H1 and H3 were observed (Scheme 4 and ESI Fig. S5†). Anomer 22-β was then oxidised at sulfur to afford the 4′-sulfinyl derivative of 4′-thiogemcitabine. Oxidation was confirmed through a significant downfield shift of C1′ and C4′ from δC 59.7 and 46.3 ppm respectively in 22-β to δC 84.6 and 72.8 ppm respectively in 23.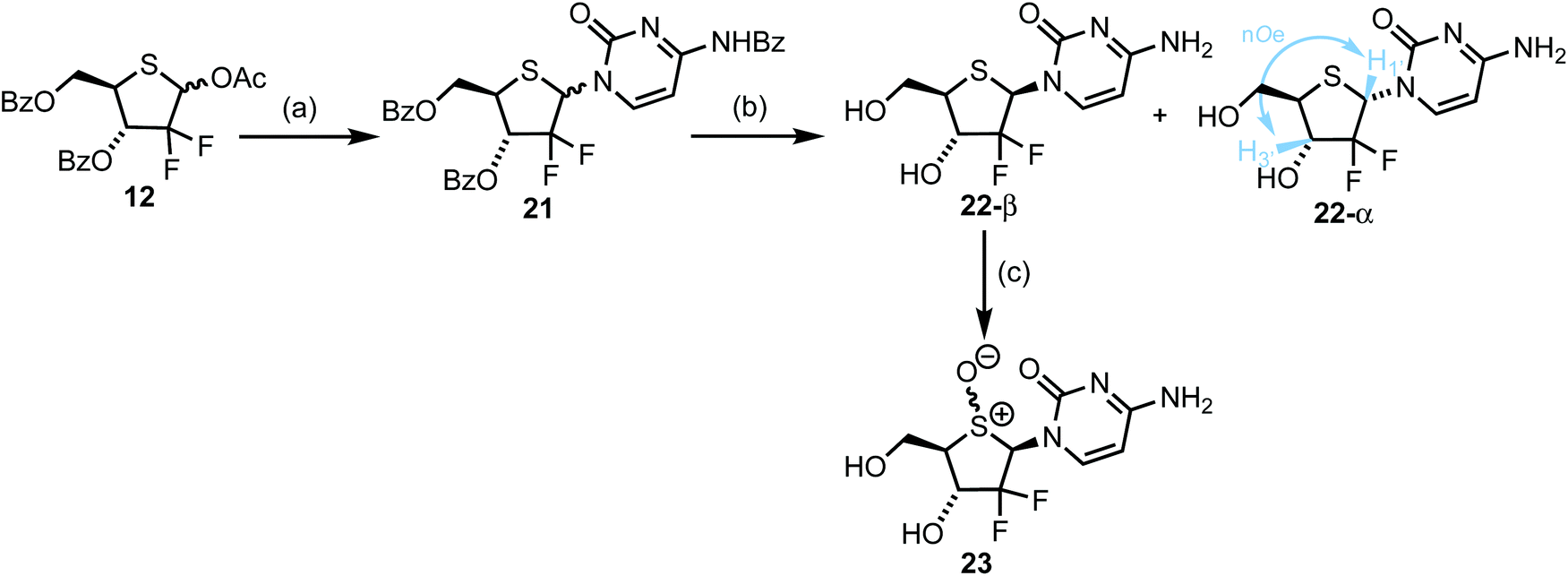 | ||
| Scheme 4 (a) N4-Benzoyl cytosine, HMDS, pyridine, SnCl4, DCE, reflux, 28% (25% returned 12), 1/1, α/β, then fractional precipitation, 10% for 21-α, 6% for 21-β. (b) From 21-α/β, NH3, MeOH, 40 °C, 96% crude, then preparative HPLC, 44% for 22-α, 34% for 22-β; From 21-β, NH3, MeOH, 63% (c) m-CPBA, H2O, MeCN, 15%, 4/1 d.r. at S. | ||
2.4. Biological Evaluation
Nucleoside analogues 15, 16, 19, 20, 22-β and 23 were evaluated in cytotoxicity assays against human pancreatic cancer (PANC-1) and human primary glioblastoma (U87-MG) cells and their CC50 values compared to commercial arabinocytidine (Ara-C) and gemcitabine (Table 1). Only analogue 19 displayed measurable cytotoxicity, with a CC50 value of 0.59 μM in U87-MG cells and 12 μM in PANC-1 cells (entry 3, Table 1); all other analogues had CC50 values >200 μM. The data for 19 were not immediately comparable to Ara-C or gemcitabine, as both displayed superior CC50 values in the cell lines evaluated (entries 1 and 2, Table 1). These results suggest that a thio arabino chemotype may impart some degree of cytotoxicity, but that replacement of furanose oxygen with sufinyl is not tolerated.| Entry | Compound | CC50 (μM) | CC50 (μM) |
|---|---|---|---|
| U87-MG | PANC-1 | ||
| 1 | Ara-C | 0.19 | 0.43 |
| 2 | Gemcitabine | 0.01 | 0.21 |
| 3 | 19 | 0.59 | 12.0 |
3. Conclusions
We have developed a scalable and column chromatography free synthesis of a protected 4-thioribose intermediate. This synthetic methodology has also been adapted for a multigram preparation of 2-deoxy-2-gem-difluoro 4-thioribose analogues. These building blocks enable entry to a range of pyrimidine-based 4′-thionucleoside analogues, including first examples of 4′-sulfinyl derivatives of ribocytidine, Ara-C and gemcitabine.From the compounds synthesised, 4′-thioarabinocytidine was shown to have moderate cytotoxicity towards U87-MG cells, but generally all the thionucleosides evaluated showed little cytotoxicity. This is most likely due to the compounds being poor substrates for nucleotide-forming kinases,27 essential for converting to the active triphosphate. Alternatively, the thionucleoside triphosphates might not be substrates for human polymerases. Further work to access monophosphate prodrug forms of these analogues is currently underway and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Keele University and Riboscience LLC are thanked for PhD studentship funding to M. G. UK Research and Innovation (UKRI, Future Leaders Fellowship, MR/T019522/1) are thanked for project grant funding to G. J. M.References
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS PubMed.
- C. Galmarini, J. Mackey and C. Dumontet, Leukemia, 2001, 15, 875–890 CrossRef CAS PubMed.
- M. Guinan, C. Benckendorff, M. Smith and G. J. Miller, Molecules, 2020, 25, 2050 CrossRef CAS.
- M. Meanwell, S. M. Silverman, J. Lehmann, B. Adluri, Y. Wang, R. Cohen, L.-C. Campeau and R. Britton, Science, 2020, 369, 725–730 CrossRef CAS PubMed.
- G. J. Miller, Science, 2020, 369, 623 CrossRef PubMed.
- E. J. Reist, D. E. Gueffroy and L. Goodman, J. Am. Chem. Soc., 1964, 86, 5658–5663 CrossRef CAS.
- M. Betson, N. Allanson and P. Wainwright, Org. Biomol. Chem., 2014, 12, 9291–9306 RSC.
- D. Dukhan, E. Bosc, J. Peyronnet, R. Storer and G. Gosselin, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 577–580 CrossRef CAS.
- Z.-H. Sun and B. Wang, J. Org. Chem., 2008, 73, 2462–2465 CrossRef CAS PubMed.
- Y. Yoshimura, K. Kitano, K. Yamada, H. Satoh, M. Watanabe, S. Miura, S. Sakata, T. Sasaki and A. Matsuda, J. Org. Chem., 1997, 62, 3140–3152 CrossRef CAS PubMed.
- Y. Yoshimura, K. Kitano, H. Satoh, M. Watanabe, S. Miura, S. Sakata, T. Sasaki and A. Matsuda, J. Org. Chem., 1996, 61, 822–823 CrossRef CAS.
- Y. Yoshimura, Y. Saito, Y. Natori and H. Wakamatsu, Chem. Pharm. Bull., 2018, 66, 139–146 CrossRef CAS PubMed.
- S. Dostie, M. Prévost, P. Mochirian, K. Tanveer, N. Andrella, A. Rostami, G. Tambutet and Y. Guindon, J. Org. Chem., 2016, 81, 10769–10790 CrossRef CAS.
- J. M. Madern, T. Hansen, E. R. van Rijssel, H. A. V. Kistemaker, S. van der Vorm, H. S. Overkleeft, G. A. van der Marel, D. V. Filippov and J. D. C. Codée, J. Org. Chem., 2019, 84, 1218–1227 CrossRef CAS.
- I. Birtwistle, P. Maddocks, J. M. O'Callaghan and J. Warren, Synth. Commun., 2001, 31, 3829–3838 CrossRef CAS.
- T. Naka, N. Minakawa, H. Abe, D. Kaga and A. Matsuda, J. Am. Chem. Soc., 2000, 122, 7233–7243 CrossRef CAS.
- K. Haraguchi, H. Kumamoto, K. Konno, H. Yagi, Y. Tatano, Y. Odanaka, S. S. Matsubayashi, R. Snoeck and G. Andrei, Tetrahedron, 2019, 75, 4542–4555 CrossRef CAS.
- I. O'Neil and K. Hamilton, Synlett, 1992, 791–792 Search PubMed.
- N. Nishizono, R. Baba, C. Nakamura, K. Oda and M. Machida, Org. Biomol. Chem., 2003, 1, 3692–3697 RSC.
- Z. W. Dentmon, T. M. Kaiser and D. C. Liotta, Molecules, 2020, 25, 5165 CrossRef CAS.
- F. Zheng, X.-H. Zhang, X.-L. Qiu, X. Zhang and F.-L. Qing, Org. Lett., 2006, 8, 6083–6086 CrossRef CAS.
- M. Takahashi, S. Daidouji, M. Shiro, N. Minakawa and A. Matsuda, Tetrahedron, 2008, 64, 4313–4324 CrossRef CAS.
- K. Nakamura, S. Shimamura, J. Imoto, M. Takahashi, K. Watanabe, K. Wada, Y. Fujino, T. Matsumoto, M. Takahashi and H. Okada, et al., Intermediate for Synthesis of 1-(2-Deoxy-2-Fluoro-4-Thio-Beta-D-Arabinofuranosyl) Cytosine, Intermediate for Synthesis of Thionucleoside, and Methods for Producing These Intermediates, EP 2,883,866A1, 2013 Search PubMed.
- H. Shirouzu, H. Morita and M. Tsukamoto, Tetrahedron, 2014, 70, 3635–3639 CrossRef CAS.
- M. Guinan, D. Lynch, M. Smith and G. J. Miller, in Carbohydrate Chemistry: Proven Synthetic Methods, CRC Press, 2020, vol. 5, pp. 227–232 Search PubMed.
- K. Brown, M. Dixey, A. Weymouth-Wilson and B. Linclau, Carbohydr. Res., 2014, 387, 59–73 CrossRef CAS.
- N. L. Golitsina, F. T. Danehy, R. Fellows, E. Cretton-Scott and D. N. Standring, Antiviral Res., 2010, 85, 470–481 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2115322. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob02097h |
| This journal is © The Royal Society of Chemistry 2022 |