
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the performance and degradation of Ru@Pt core–shell catalysts for fuel cells by advanced (scanning) transmission electron microscopy†
Alba
Garzón Manjón‡
 *a,
Miquel
Vega-Paredes‡
a,
Viktoriya
Berova
b,
Thomas
Gänsler
a,
Torsten
Schwarz
a,
Nicolas A.
Rivas Rivas
a,
Katharina
Hengge
b,
Tilman
Jurzinsky
b and
Christina
Scheu
*a
*a,
Miquel
Vega-Paredes‡
a,
Viktoriya
Berova
b,
Thomas
Gänsler
a,
Torsten
Schwarz
a,
Nicolas A.
Rivas Rivas
a,
Katharina
Hengge
b,
Tilman
Jurzinsky
b and
Christina
Scheu
*a
aMax-Planck-Institut für Eisenforschung GmbH, Max-Planck-Straße 1, 40237 Düsseldorf, Germany. E-mail: a.garzon@mpie.de; scheu@mpie.de
bFreudenberg Fuel Cell e-Power Systems GmbH, Bayerwaldstraße 3, 81737 München, Germany
First published on 4th November 2022
Abstract
Ru@Pt core–shell nanoparticles are currently being explored as carbon monoxide tolerant anode catalysts for proton exchange membrane fuel cells. However, little is known about their degradation under fuel cell conditions. In the present work, two types of Ru@Pt nanoparticles with nominal shell thicknesses of 1 (Ru@1Pt) and 2 (Ru@2Pt) Pt monolayers are studied as synthesized and after accelerated stress tests. These stress tests were designed to imitate the degradation occurring under fuel cell operating conditions. Our advanced (scanning) transmission electron microscopy characterization explains the superior initial electrochemical performance of Ru@1Pt. Moreover, the 3D reconstruction of the Pt shell by electron tomography reveals an incomplete shell for both samples, which results in a less stable Ru metal being exposed to an electrolyte. The degree of coverage of the Ru cores provides insights into the higher stability of Ru@2Pt during the accelerated stress tests. Our results explain how to maximize the initial performance of Ru@Pt-type catalysts, without compromising their stability under fuel cell conditions.
Introduction
Despite the pledge of the Paris agreement, in which 191 countries and the European Union agreed to substantially reduce global greenhouse emissions to limit the world's temperature rise,1 global carbon dioxide (CO2) emissions are still increasing.2 One key contributing factor is electricity production, since more than 60% of electricity is generated from fossil fuels (i.e., coal, natural gas and oil).3 Polymer electrolyte membrane fuel cells (PEMFCs) are electrochemical devices that can convert chemical energy stored in hydrogen (H2) or other fuels into electrical energy. As such, they are expected to play a major role in decarbonizing our electricity production systems.4 Furthermore, fuel cell vehicles could replace classical combustion engine vehicles. This would significantly reduce greenhouse gas emissions from the transportation sector, which nowadays account for over 15% of global CO2 emissions.5H2/oxygen (O2) PEMFCs are especially appealing, since the main product generated besides electricity is water.6 Pt is commonly used in these systems as a catalyst to enhance the cathodic and anodic reactions.7 However, Pt has some undesirable characteristics, such as its high price8 and low tolerance to impurities.9 The last factor can be critical at the anode of PEMFCs intended for heavy duty applications, such as ships or trucks. For those applications, H2 is typically generated on site, without the possibility of purifying it efficiently. Under these conditions, a mixture of H2 rich gas with carbon monoxide (CO) impurities (known as reformate) is fed to the anode.10 CO has strong affinity for Pt,11 and can adsorb strongly onto the surface of the catalyst, poisoning it and impacting the overall efficiency of the cell. It has been reported that concentrations of CO in fuel as low as 20 ppm can cause a significant drop in PEMFC performance.12 A typical strategy for enhancing the CO tolerance of the anode involves alloying Pt with another transition metal.13 Au, Co, Fe Cu, Ni and Ru have been explored as possible alloy candidates, with Ru showing the most promising results.13 There are two mechanisms thought to be responsible for the enhanced CO poisoning tolerance on Pt–Ru alloys, namely the bifunctional effect and the electronic effect.14 In the bifunctional effect, oxygen containing species adsorb on Ru surface sites, promoting the electrooxidation of CO adsorbed on Pt and liberating H2 adsorption sites.15 In the electronic effect, the changes in the electronic band structure of Pt atoms induced by Ru weaken the CO–Pt interaction, consequently reducing the CO coverage.15 Nonetheless, alloying Pt with a less noble metal such as Ru decreases the stability of a catalyst under electrocatalytic conditions,16 which has a negative impact on the long-term performance of the cell. Nanostructuring a catalyst into Ru@Pt core–shell nanoparticles (NPs) can help in mitigating its degradation by protecting the less noble Ru with a Pt shell, while still having enhanced CO tolerance.17 In perfect core–shell NPs, the bifunctional effect will not take place, since no Ru surface sites are available for the adsorption of oxygen containing species. However, the electronic effect will still take place, and studies of Pt thin films on Ru showed a significant decrease in the CO–Pt binding energy with respect to pure Pt.18 Furthermore, the surface strain caused by the lattice mismatch between Pt and Ru has an additional impact on the CO adsorption energy on Pt,18 which could reduce even further the CO poisoning of Ru@Pt catalysts. Therefore, Ru@Pt NPs are very promising as CO tolerant anode catalysts for PEMFCs. Although significant efforts have been made for synthesizing Ru@Pt NPs and characterizing their electrochemical properties17,19,20 and evaluating their structure at the highest resolutions,17,21,22 little is known about their degradation under fuel cell conditions.
Here, we report on the atomic scale characterization of two different state-of-the-art Ru@Pt PEMFC anode catalysts. The studied NPs possess a nominal shell thickness of 1 (Ru@1Pt) and 2 (Ru@2Pt) Pt monolayers. Aberration-corrected high resolution (scanning) electron microscopy (HR-(S)TEM) techniques are used to characterize the changes occurring in Ru@Pt NPs due to accelerated stress tests (ASTs; 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycles). A deep insight into these changes is key for understanding the underlying degradation mechanisms taking place during PEMFC operation, and how they are related to the PEMFC performance. Moreover, our studies help to explain the electrochemical performance of Ru@Pt core–shell particles described in a recent publication.23 Namely, the U–I curves indicate a superior initial performance against reformate (72% H2, 28% N2, and 10 ppm CO) with 1% air bleed for Ru@1Pt (0.65 V at 2 A mgPt−1) compared with Ru@2Pt (0.5 V at 2 A mgPt−1). However, after the ASTs the cell voltage for Ru@1Pt dropped to 0.5 V at 2 A mgPt−1, while for Ru@2Pt it increased to 0.55 V at the same mass current density. In addition, electrochemical surface area (ECSA) measurements from the CO stripping experiments revealed that the initially higher ECSA of Ru@1Pt decreased from 44 m2 mgPtRu−1 to 24 m2 mgPtRu−1, while for Ru@2Pt it decreased less strongly from 39 m2 mgPtRu−1 to 23 m2 mgPtRu−1.23
000 potential cycles). A deep insight into these changes is key for understanding the underlying degradation mechanisms taking place during PEMFC operation, and how they are related to the PEMFC performance. Moreover, our studies help to explain the electrochemical performance of Ru@Pt core–shell particles described in a recent publication.23 Namely, the U–I curves indicate a superior initial performance against reformate (72% H2, 28% N2, and 10 ppm CO) with 1% air bleed for Ru@1Pt (0.65 V at 2 A mgPt−1) compared with Ru@2Pt (0.5 V at 2 A mgPt−1). However, after the ASTs the cell voltage for Ru@1Pt dropped to 0.5 V at 2 A mgPt−1, while for Ru@2Pt it increased to 0.55 V at the same mass current density. In addition, electrochemical surface area (ECSA) measurements from the CO stripping experiments revealed that the initially higher ECSA of Ru@1Pt decreased from 44 m2 mgPtRu−1 to 24 m2 mgPtRu−1, while for Ru@2Pt it decreased less strongly from 39 m2 mgPtRu−1 to 23 m2 mgPtRu−1.23
HR-(S)TEM is used for determining the shell thickness via analysis of local displacements in the atomic lattice (strain), the size distribution, and the 3D morphology. The 3D reconstruction of the Pt shell via electron tomography is key for studying the encapsulation and protection of the Ru core, both in the as synthesized particles and after the ASTs. Furthermore, multivariate statistical analysis of energy dispersive X-ray spectroscopy (EDS) experiments was carried out to study the composition and elemental distribution of the catalyst particles.
Experimental section
Synthesis of Ru@Pt NPs
RuCl3·xH2O and H2PtCl6·6H2O were used as precursor metal salts for the synthesis of Ru@Pt NPs on carbon black via the polyol method.23 Two types of Ru@Pt NPs were synthesized with varying Pt shell thicknesses. The targeted shell thickness was 1 Pt layer for Ru@1Pt and 2 Pt layers for Ru@2Pt. In both cases, the Pt loading on carbon was kept constant and the Ru loading was chosen according to the intended shell thickness.Membrane electrode assembly fabrication
MEAs were composed of two electrodes (anode and cathode), and a 15 μm thick polymeric membrane in between.24 The anode catalyst layer contained the as synthesized Ru@Pt NPs (0.1 mgPt cmgeo−2), while on the cathode commercial Pt NPs (0.4 mgPt cmgeo−2) were deposited.Accelerated stress tests of the MEAs
ASTs were carried out in an automated test station equipped with a potentiostat.23 ASTs were performed at 1 bar, 353 K (80 °C), 95% relative humidity and N2/H2 flows of 150 nccm/500 nccm on the anode/cathode. The potential of the anode was cycled 10000 times between 0.06VRHE and 0.8VRHE (scan rate of 100 mV s−1).
The potential window used in this and our previous study23 differs from those of many ASTs found in the literature that have upper potential limits of 1.0VRHE16 or even as high as 1.5VRHE.24 These higher potential limits are used for studying degradation occurring during start-up or shut-down events, common in fuel cell vehicles. However, the Ru@Pt NPs investigated in this work are designed to be used as anode catalysts for reformate-operated fuel cells in stationary heavy duty applications, where the main degradation phenomenon of the catalyst originates from the dissolution of Ru.
TEM sample preparation
For TEM investigations, a drop of the as synthesized Ru@Pt dispersion was dropped on a holey carbon-coated Cu grid and was left drying overnight for adhesion. Moreover, cycled Ru@Pt NPs were removed by scratching the electrodes of the stressed MEAs, dispersed in deionized water (10 mM), and deposited on a holey carbon-coated Cu grid following the same procedure as before.Focused ion beam (FIB)
A FIB was used for preparing a thin lamella of MEAs for their characterization using TEM. (S)TEM lamella preparation was carried out with a dual-beam FIB-scanning electron microscope system (Thermo ScientificTM SciosTM 2 DualBeamTM). First, a protective Pt layer of 200 nm was deposited with the electron beam across the entire MEA section to be lifted out. Subsequently, 3 μm of Pt were deposited on top of the protective layer to prevent damage to the lamella during the lift-out and thinning process, and this was performed with a Ga beam. Bulk ion beam cuts were made with an acceleration voltage of 30 kV and a current of 3.0 nA. Thinning of the lamella was consecutively carried out with 30 kV and decreasing the current starting from 0.15 nA down to 80 pA. Finally, the last polishing step with a voltage of 5 kV and 15 pA current was performed, resulting in a lamella of ∼100 nm thickness.HR-TEM characterization
Structural analysis was performed using a Titan Themis microscope (Thermo Fisher Scientific) operated at 300 kV and equipped with an image corrector. The corrector cancels the positive spherical aberration (Cs) induced by an objective lens, and is set to introduce a small negative Cs. The negative Cs applied together with an overfocus results in HR-TEM micrographs with bright-atom contrast.25 For strain analysis, two dimensional Gaussians are fitted to the local maxima of intensity (i.e., the bright atoms), and their positions were compared to those of a reference lattice, set in the center of the NPs. The strain was calculated by taking the gradient from the local displacement in the atomic positions with respect to the reference lattice. All the TEM images were recorded using a CMOS (metal–oxide–semiconductor) camera with 4k × 4k pixels.STEM characterization
The Ru@Pt NPs and MEAs were further investigated using a Titan Themis microscope (Thermo Fisher Scientific) operated at 300 kV and equipped with a probe corrector. STEM images were recorded with a HAADF detector that covers the angular range of 73–352 mrad. A convergent angle of 23.8 mrad was chosen, which results in a probe size of ∼0.1 nm. EDS spectrum imaging was performed with a beam current of ∼150 pA. The particle size distribution (diameter) was determined by measuring 300–700 particles from each sample. The chemical composition was determined by quantifying the EDS data using the Cliff-Lorimer method26,27 (from around 50 particles). In all the samples, different regions were studied for the determination of the particle size distribution and composition. PCA of the EDS spectral maps of the cycled MEAs was performed.28 In particular, non-negative matrix factorization was used for decomposing the otherwise noisy data sets into their principal spectral components. Electron tomography was carried out to study the 3D morphology of Ru@Pt NPs and their surface to volume ratio. Tilt series were acquired using a single tilt tomography holder (FEI). Tilt angles were chosen in the range of ± 60° with increments of 10°. These angular increments were selected as a compromise between the number of images used in the reconstruction (resolution) and electron dose (stability of the carbon support material). A HAADF detector was used for acquiring the tilt series. This detector was selected to minimize the diffraction contrast on the images while allowing us to distinguish between Pt and Ru due to their difference in the atomic number. 3D volume reconstruction was carried out using a simultaneous interactive reconstruction methodology and refined by the discrete algebraic reconstruction technique. Visualization and S/V ratio calculations were performed using UCSF ChimeraX.29 Only relative values are given in these calculations to account for the error introduced by the missing wedge, as a consequence of the limited angular tilt range inside the microscope.Results and discussion
Characterization of the as synthesized NPs
The high-angle annular dark field (HAADF)-STEM micrographs of the two as synthesized core–shell samples are shown in Fig. 1a and d, together with the corresponding EDS elemental distribution maps (Fig. 1b and e) and particle size distribution histograms (Fig. 1c and f). The STEM micrographs (Fig. 1a and d) show that both samples have two different kinds of NPs regarding the size and shape. One of the families is made of smaller, spherical shaped NPs (indicated in the micrographs by the orange arrows), while the other consists of bigger, somewhat elongated particles (red arrows). In the bigger particles, contrast differences between the outer (shell) and inner (core) parts can be observed. Since Pt is heavier than Ru, it scatters electrons more strongly and appears brighter in HAADF-STEM micrographs, which have Z2 contrast.30 Thus, the darker core of the particles surrounded by the brighter shell indicates that Ru is encapsulated with Pt. This is further confirmed by the EDS composition maps (Fig. 1b and e), since they show the Ru (in red) core totally or partially surrounded by the Pt (in yellow) shell. However, both the HAADF micrographs and the EDS maps show an incomplete encapsulation of the Ru cores, and Ru is detected in the outer part of the particles. This is further addressed when discussing electron tomography reconstruction. In contrast, the smaller spherical particles for both samples are found to be made purely out of Pt in the EDS composition maps, and therefore the samples do not have a core–shell like structure. This can be further corroborated by extracting and analyzing the EDS spectra of such particles, since no significant Ru signal can be detected over the background noise (ESI FS1†). A small peak is present at around 2.3–2.6 keV, which could be attributed to Ru Lα or to Cl Kα. Since the precursor salts used for the synthesis of NPs contain Cl as counterions, it explains the presence of such a peak. The signal to noise ratio of these spectra is relatively low even after long acquisition times (20 min for Ru@1Pt and 30 min for Ru@2Pt) due to the small size of the Pt particles, and it can not be further improved by increasing the acquisition time or the electron dose due to stability issues.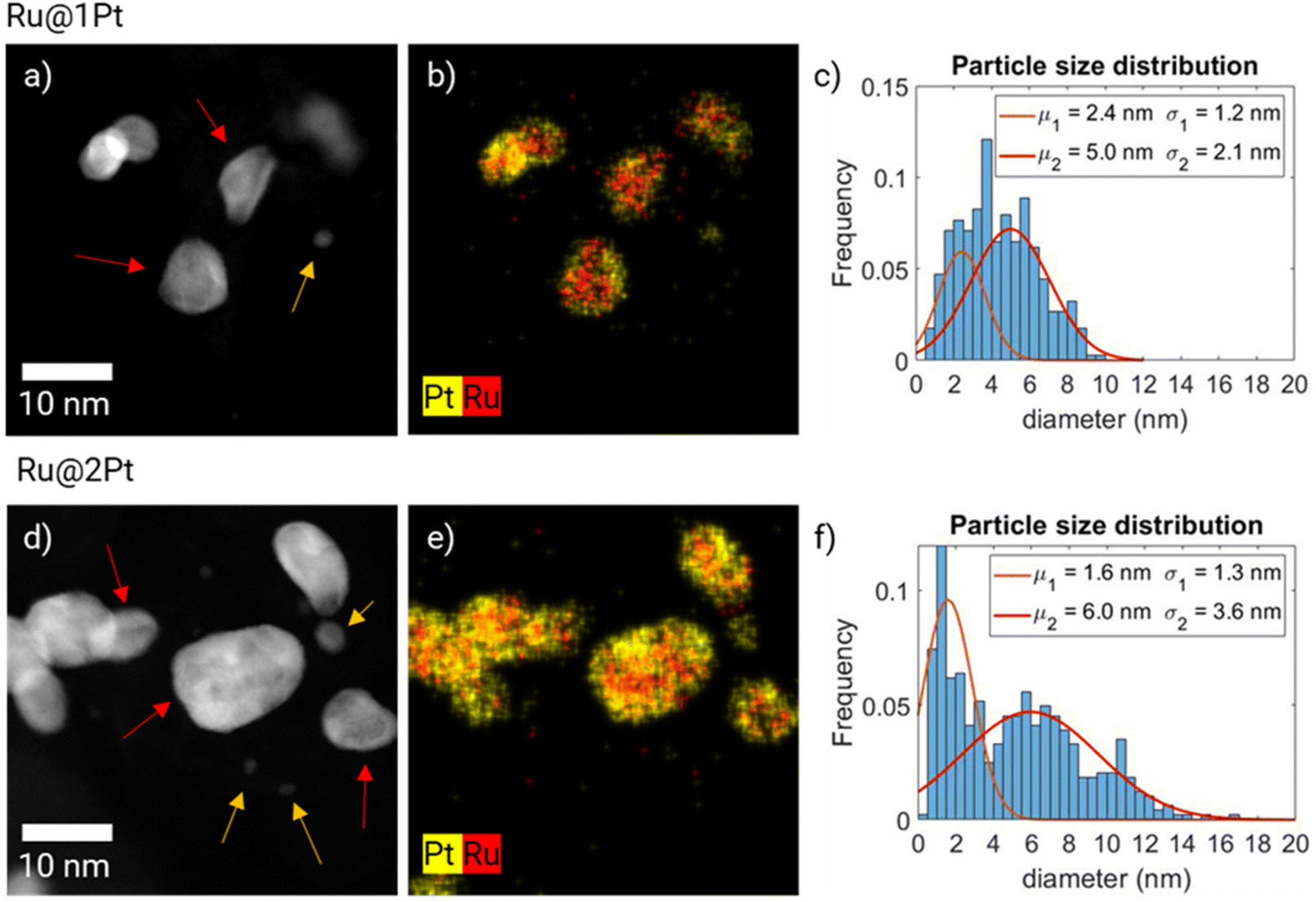 | ||
| Fig. 1 HAADF-STEM micrographs of Ru@1Pt (a) and Ru@2Pt (d), and the corresponding EDS elemental distribution maps (b and e) and particle size distribution histograms (c and f), where two Gaussian curves are fitted. Two types of particles can be observed in each sample, indicated by the orange and red arrows in (a) and (d). | ||
The dual nature of the particles is reflected in the particle size distribution histograms of Ru@1Pt and Ru@2Pt (Fig. 1c and f). This is especially clear for Ru@2Pt, where its particle size distribution histogram shows a bimodal distribution with average sizes of μ1 = 1.6 ± 1.3 nm and μ2 = 6.0 ± 3.6 nm. In the Ru@1Pt histogram two normal distributions, with μ1 = 2.4 ± 1.2 nm and μ2 = 5.0 ± 2.1 nm, can also be fitted. However, since the difference in the average size for the two types of particles is smaller, the bimodal nature is not clear, even if two distinct types of particles can be observed in the STEM micrographs and EDS composition maps. In addition, the particle size distribution histograms show that the frequency of the smaller pure Pt particles is higher for Ru@2Pt than for Ru@1Pt, as can also be seen in the HAADF-STEM micrographs (Fig. 1a and d). Chemical composition quantification of individual particles can be performed using EDS composition maps. The elemental average compositions of the core–shell like particles were 54.1 at% Pt and 45.9 at% Ru with a standard deviation of ± 12.0 at% for Ru@1Pt and 67.9 at% Pt and 31.9 at% Ru with a standard deviation of 8.7 at% for Ru@2Pt. These relatively big values for the standard deviation of the composition indicate significant differences in the chemical composition of the individual NPs, which is most likely due to the industrial-like nature of the catalyst synthesis process.
PCA analysis using the EDS hypermaps of the as synthesized NPs can be carried out for extracting the principal spectral components of the core–shell NPs and small Pt particles (ESI FS2 and FS3†).
Besides the size and elemental composition, it is also key to measure the shell thickness, since the catalytic properties of the NPs greatly depend on it.17,22,23 Since the acquisition of atomically resolved EDS maps is not possible due to the low stability of the carbon support material under the electron beam, another approach was adopted.
The atomic arrangement within a given core–shell particle can be addressed using HR-TEM micrographs (Fig. 2a and f), which show well-ordered and crystalline Ru cores for Ru@1Pt and Ru@2Pt. Since both the atomic radius (r_Ru = 130 pm and r_Pt = 135 pm)31 and crystalline structure (Ru = hexagonal close packed (hcp) and Pt = face centered cubic (fcc)) vary for the two elements present in the particles, differences in the atomic positions can be expected between the Ru core and the Pt shell. By calculating the number of monolayers with different atomic spacings from the displacement or strain maps (Fig. 2b, c, g and h), it is possible to estimate the thickness of the Pt shell. The strain maps for Ru@1Pt (Fig. 2b and c) reveal a negative (tensile) strain for the outermost atomic layer. Thus, the atomic positions for such atoms are expanded with respect to the reference lattice (set in the center of the particle, i.e., the Ru core). Furthermore, the interplanar distances between the two outermost atomic planes and the inner atomic planes are found to be 2.3 Å and 2.1 Å, respectively. These values are in agreement with the tabulated interplanar distances for {111} Pt (d = 2.26 Å)32 and {01−1−1} Ru (d = 2.08 Å),33 which indicates that the Pt atoms of the shell take the lowest energy plane {111}.34 Similarly, the strain maps for Ru@2Pt (Fig. 2g and h) show an equivalent tensile strain in the 2–3 outermost atomic layers, confirming the ticker shell of these particles.
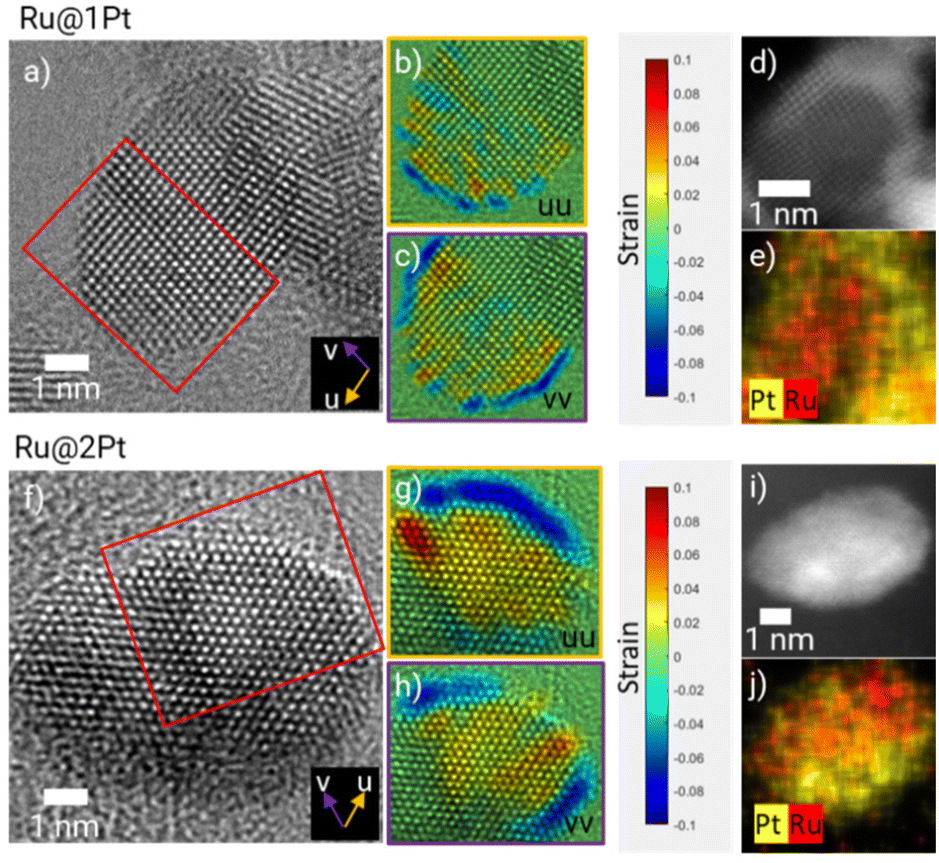 | ||
| Fig. 2 HR-TEM micrographs (a and f), strain maps along the u (b and c) and v (g and h) directions and the corresponding HAADF-STEM micrographs (d and i) and EDS maps (e and j) of Ru@1Pt and Ru@2Pt, respectively. | ||
These results are in concordance with the theoretical shell thickness calculated from the Pt to Ru ratio and the core size of 1.3 and 2.4 Pt monolayers for Ru@1Pt and Ru@2Pt, respectively.23 However, it is important to keep in mind that these theoretical shell thicknesses are calculated assuming complete and homogeneous core coverage, which is not the case for Ru@1Pt and Ru@2Pt, as seen in the HAADF micrographs (Fig. 1a and d). Therefore, it can be expected to find variations in the shell thickness throughout the particles.
Complementary HAADF-STEM micrographs and EDS elemental maps are shown in Fig. 2d, e, i and j, confirming the core–shell like nature of the studied particles. However, based on these figures, the shell appears to be significantly thicker. For instance, for Ru@1Pt, 4 bright atomic layers can be distinguished in the outer part of the particle. This apparent difference in the shell thickness could be explained by the curvature of the particles, which results in an overestimation of the quantity of Pt at the edges of the particle. Additionally, there could be some Pt diffusion that broadens the interface and thus the shell thickness was measured.
Besides measuring the interplanar distances between the outer and inner layers, it is also possible to determine the interatomic distances within the shell from the HR-TEM micrographs. In ESI FS4† the intensity line profiles along the shell's atomic columns of two selected NPs from Ru@1Pt and Ru@2Pt are shown, indicating that there are no differences in the atomic distances within the shell between the two samples (2.59 Å for Ru@1Pt and 2.58 Å for Ru@2Pt), which is consistent with DFT calculations found in the literature.17
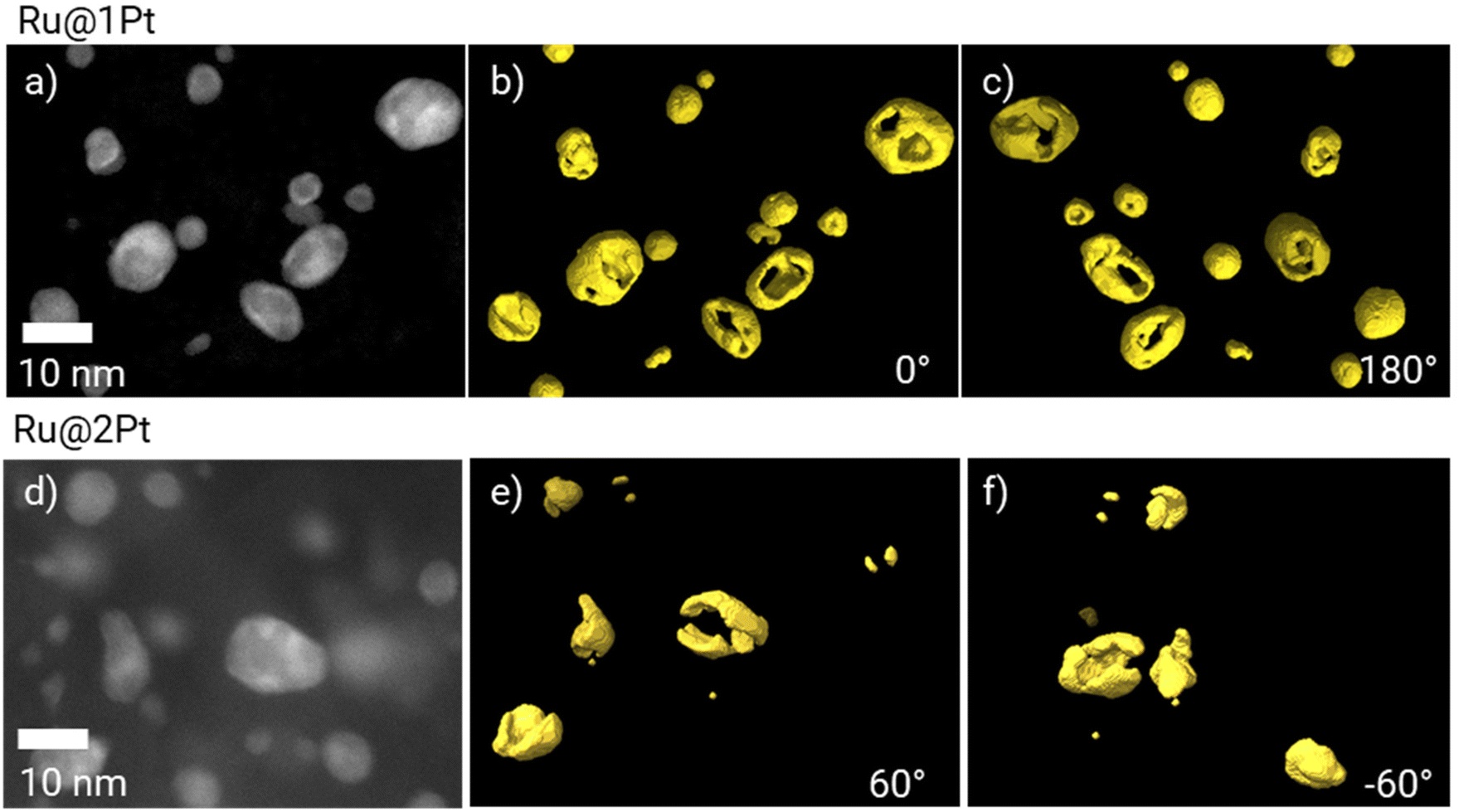
As previously mentioned, in the core–shell particles the Ru core is not fully encapsulated by the Pt shell. The coverage of the Ru cores in 3D can be further studied using electron tomography. The 3D reconstruction of Pt is shown in Fig. 3 at two different orientations (0° and 180°), together with the corresponding HAADF-STEM micrograph at 0°. The animated view of the reconstruction can be found in ESI Movie 1.†
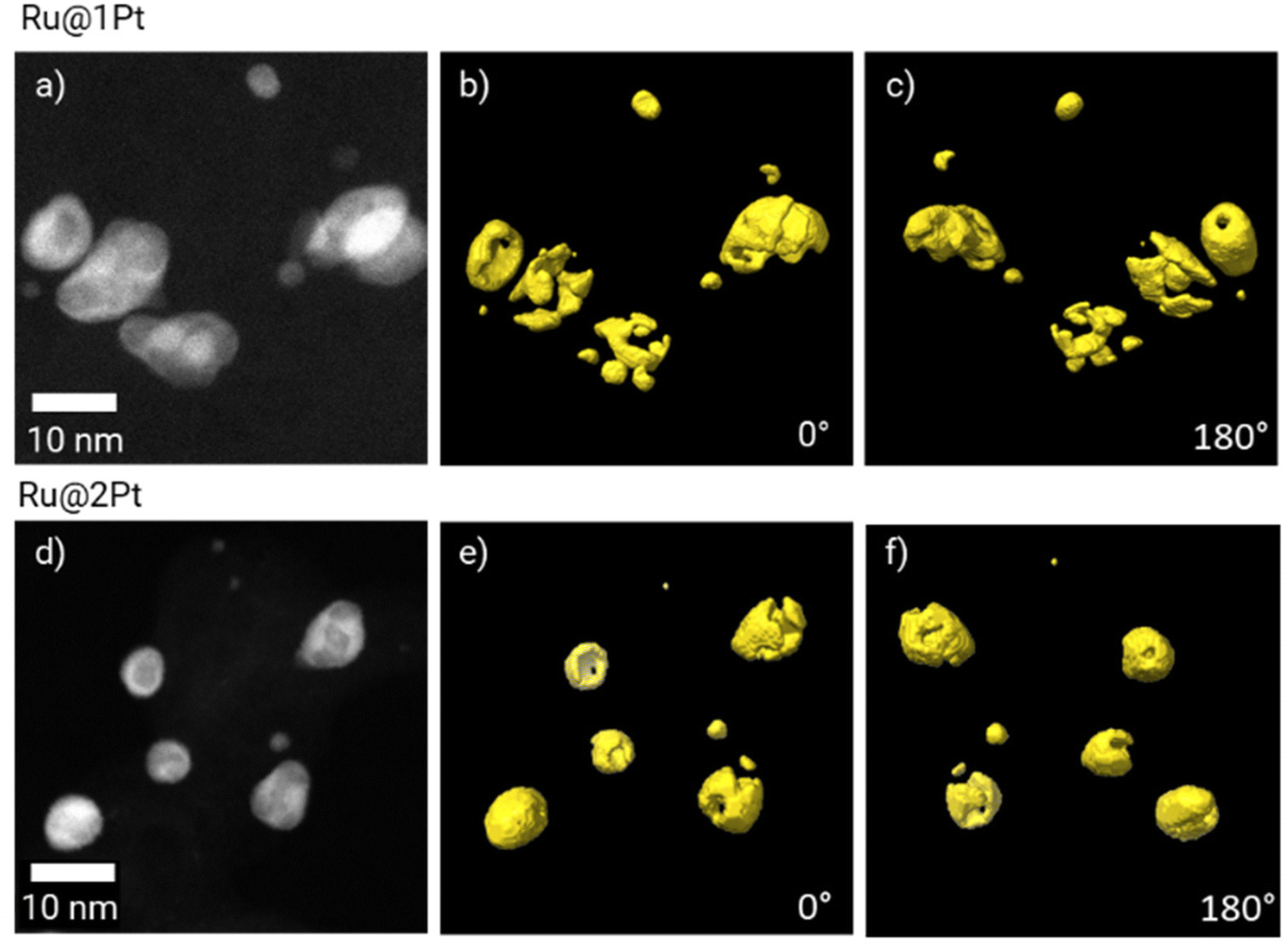 | ||
| Fig. 3 HAADF-STEM micrographs of Ru@1Pt (a) and Ru@2Pt (d), and the corresponding 3D electron tomography reconstruction showing the Pt distribution (in yellow) at 0° (b and e) and 180° (c and f). | ||
The tomographic reconstruction (Fig. 3b, c, e and f) confirms that the shell in Ru@1Pt and Ru@2Pt is inhomogeneous and not fully encapsulating the Ru core, which explains why Ru is detected on the surface of some particles on the EDS measurements. However, the degree of inhomogeneity is not equal in both types of core–shell particles, since the shell is more incomplete for Ru@1Pt.
Characterization of the stressed membrane electrode assemblies
The as synthesized Ru@Pt NPs were used as anode catalysts of two distinct membrane electrode assemblies (MEAs). These MEAs underwent ASTs, in the course of 10000 potential cycles, as described in the Experimental section. The changes in the anode catalyst NPs with respect to the pristine core-shells were attributed to degradation (i.e., Ostwald ripening, particle aggregation and coalescence, and/or dissolution of the catalyst species).
The HAADF-STEM micrographs of the anode from the stressed MEAs (Fig. 4a and e) show that significant changes took place during the ASTs. For Ru@1Pt, a lot of atom clusters (<1 nm) can be observed (zoom-in images are shown in ESI FS5†). This is reflected in the particle size distribution histogram (Fig. 4d and h), where a narrow Gaussian with a mean of μ1 = 0.9 nm can be seen. In comparison, the smaller family of the as synthesized NPs had an average size of μ1 = 2.2 nm. The average size and standard deviation of the bigger core–shell-like particles increased from μ1 = 5.0 ± 2.1 nm for the pristine NPs to μ2 = 6.4 ± 3.6 nm after the cycles.
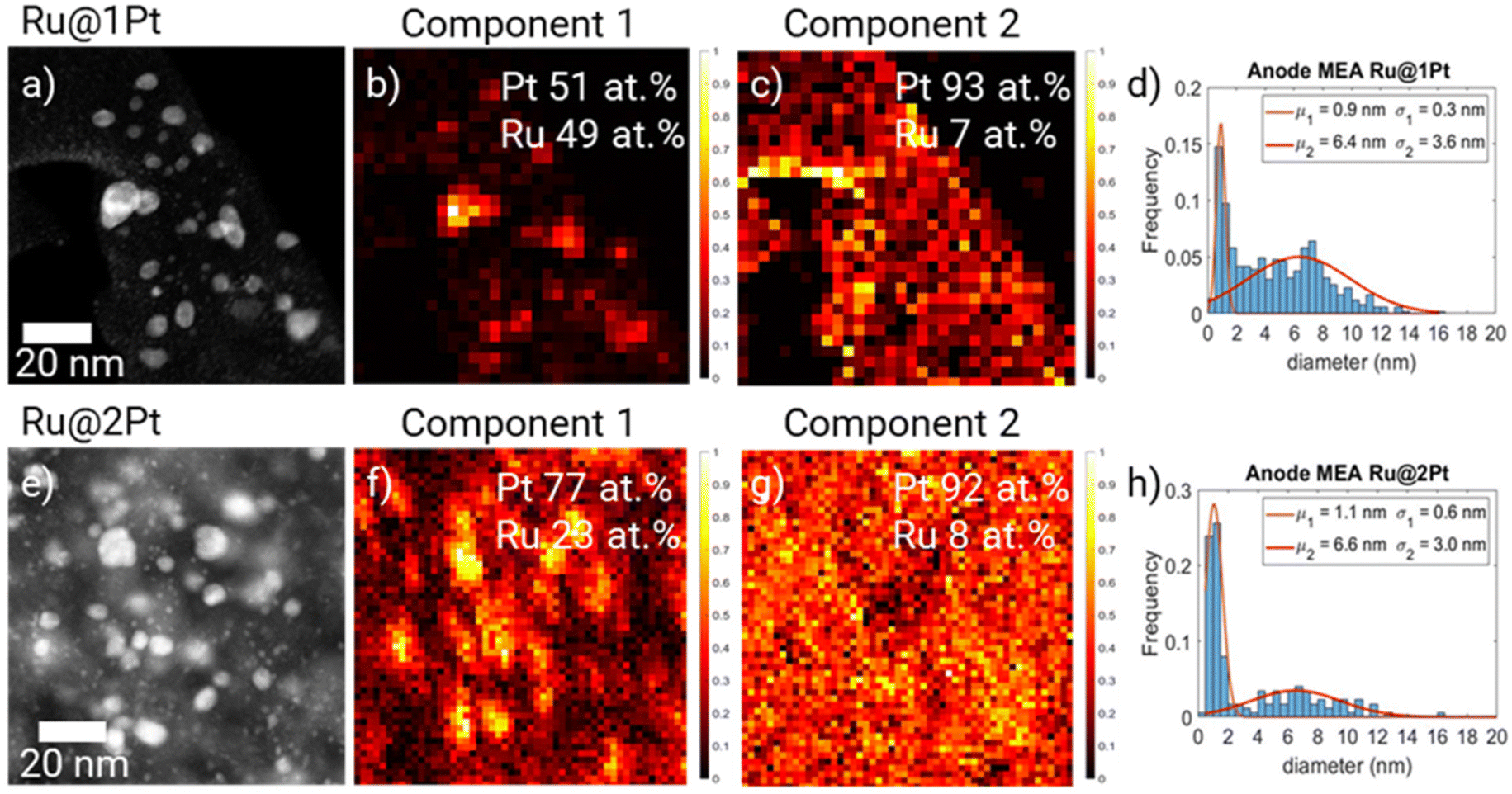 | ||
| Fig. 4 HAADF-STEM and particle size distribution of Ru@1Pt (a and d) and Ru@2Pt (e and h) NPs present in the MEA anode after ASTs. Two principal components obtained from the PCA of the EDS spectral maps of Ru@1Pt (b and c) and Ru@2Pt (f and g). In both cases C1 correlates with the bigger particles while C2 correlates with the smaller particles. | ||
Principal component analysis (PCA)28 of the EDS spectral maps (Fig. 4b and c) shows two main spectral components. The two spectral components are given in the ESI (FS6).† By comparing the spectral component maps (Fig. 4b and c) with the HAADF micrograph (Fig. 4a), it can be seen that the first component (C1) correlates with the core–shell-like particles, while the second (C2) corresponds to the <1 nm atom clusters. It is also possible to quantify the Pt and Ru contents present in each of the components using the Cliff-Lorimer method. In C1, a composition of 51 at% Pt and 49 at% Ru was found, while for C2 (small particles) it is 93 at% Pt and 7 at% Ru. In PCA the different spectra of the EDS hypermaps are separated into their main components by means of non-negative matrix factorization. Since the spectral components for pure Pt NPs, pure Ru NPs and mixed NPs would be different, different components would be assigned to each one of these types of NPs using PCA. Therefore, we can rule out the presence of a significant number of pure Pt NPs after cycling.
For the MEA with Ru@2Pt, many atom clusters can also be seen in the HAADF-STEM micrographs (Fig. 4e). Nonetheless, the average particle size remained quite constant during the ASTs. For the core–shell-like family, the mean particle size μ2 changed from 6.0 nm in the pristine NPs to 6.6 nm, while for the smaller particles, μ1 decreased from 1.5 nm to 1.1 nm, mainly due to the presence of small atom clusters. Similarly to Ru@1Pt, PCA revealed the presence of two spectral components (FS6†), with C1 (Fig. 4f) being strongly correlated with the bigger core–shell like particles and with a composition of 77 at% Pt and 23 at% Ru, while C2 (Fig. 4g) is related to the atom clusters and possesses 92 at% Pt and 8 at% Ru.
During the ASTs, Pt and Ru species from the core–shell particles + Pt species from the small pure Pt particles are dissolved. These dissolved species can either redeposit on pre-existing core–shell particles or precipitate forming new small atom clusters, as seen in the STEM micrographs (Fig. 4). If more species are redepositing on the core–shell particles than those stemming out of them, the average particle size can increase. Besides this type of Ostwald ripening, other effects such as particle aggregation can also take place, which could also result in an increase in the average particle size of the core–shell particles.
STEM tomography experiments were also performed on the catalyst NPs of the anode after the ATSs (Fig. 5) to study how the shell thickness affects the change in the coverage of the Ru cores during the cycles. The animated view of the reconstruction can be found in ESI Movie 2.† It is confirmed from Fig. 5 that after the ASTs, part of the catalyst particles is still core–shell like. To analyse how the Pt shell changes during the cycles, the average Pt surface to volume (S/V) ratio of the core–shell particles can be calculated.29 For focusing on studying the degradation of the core–shell particles, the smaller non-core–shell-like particles are excluded from the calculations. The obtained results show that the Pt S/V ratio from Ru@1Pt decreases to 63% during the ASTs, while for Ru@2Pt no significant differences are observed in the Pt S/V ratio before and after cycles.
 | ||
| Fig. 5 HAADF-STEM micrographs of Ru@1Pt (a) and Ru@2Pt (b) after 10000 cycles, and the corresponding 3D electron tomography reconstruction showing the Pt distribution (in yellow) at 0° (b and e) and 180° (c and f). | ||
Understanding the performance of Ru@Pt catalysts
The overview STEM micrographs of Ru@1Pt and Ru@2Pt (Fig. 1a and d) show that both samples contain two types, or families, of particles with different average sizes. No significant Ru can be detected on the smaller particles (μ1 = 2.4 nm for Ru@1Pt and μ1 = 1.6 nm for Ru@2Pt), indicating that they are formed purely from Pt. These Pt particles could be one important factor in explaining the differences in the performance between Ru@1Pt and Ru@2Pt. In our earlier work23 we attributed the poorer performance of Ru@2Pt in reformate with respect to Ru@1Pt to the thinner shell of the latter, which would result in a stronger electronic effect and hence, enhanced CO poisoning tolerance. However, the presence of pure Pt NPs uncovered by the STEM investigations indicates an additional aspect that might contribute to such differences in performance. Since they do not possess detectable amounts of Ru (FS1)†, they will be prone to be poisoned by CO when reformate is fed to the anode, resulting in poor performance. Since the frequency of these small pure Pt particles is higher for Ru@2Pt, they will have a greater impact on the cell performance, which is in good agreement with the electrochemical results.Atomic position displacement analysis from the HR-TEM images (Fig. 2) can be used for estimating the thickness of the Pt shell. These analyses indicate that the targeted shell thickness was achieved for both the samples (Ru@1Pt ∼ 1 Pt monolayer and Ru@2Pt ∼ 2–3 Pt monolayers). However, the shell is neither complete nor homogeneous, and thickness variations can be expected. This has an important effect on the degradation of the Ru cores during operation and on the activity of the catalyst regarding CO oxidation, and will be addressed when discussing the electron tomography results. A high degree of control in the shell thickness during the synthesis of core–shell particles is key, since their electrochemical performance greatly depends on it. Schlapka et al.18 reported that for Pt thin films deposited on Ru(0001), the electronic influence of the substrate on the CO adsorption energy has largely vanished for more than three Pt layers. Since for Ru@1Pt and Ru@2Pt the average shell thickness is lower than this, the Ru core will impact significantly the electronic properties of the shell, decreasing Pt affinity towards CO (electronic effect). Furthermore, Schlapka et al.18 also reported that the effect of surface strain on CO adsorption remains intact for less than 5 Pt monolayers. The strain maps of Ru@1Pt (Fig. 2b and c) and Ru@2Pt (Fig. 2g and h) confirm the presence of such strain in both the samples. Thus, it can be expected that it leads to a further reduction in CO poisoning. Moreover, the HR-TEM micrographs show well ordered crystalline Ru cores for both Ru@1Pt and Ru@2Pt, which affects positively their catalytic properties. Zou et al.22 studied the impact of the atomic ordering of the metal core on Ru@Pt NPs. They found that ordered cores result in a higher apparent lattice strain on the shell, which induces a reduction in the CO adsorption energy and enhances the catalytic activities of the NPs.
Understanding the degradation of Ru@Pt catalysts
The characterization of the cycled MEAs (Fig. 4) revealed, for both samples, the presence of many small particles (<1 nm) that were attributed to atom clusters (FS5†). Rasouli et al.37 also reported the presence of many atom clusters on the surface of the carbon support on Pt-based cathode catalysts of MEAs. The authors concluded that the presence of these single atoms and clusters plays an important role in the loss of the ECSA during ASTs, through redeposition or bridging of NPs. Another factor that greatly affects the ECSA of PEMFC electrodes is an increase in the average particle size of the catalyst. For Ru@1Pt, the average core–shell particle size increased by 27%, while for Ru@2Pt the average particle size remained quite constant, and only increased by 10%. Consequently, a higher loss of the ECSA can be expected for the anode with Ru@1Pt than for the one with Ru@2Pt, which is in good agreement with the ECSA calculations from the CO stripping experiments.23 There are four mechanisms proposed to be responsible for the loss of the ECSA from Pt NPs in PEMFCs, namely, Ostwald ripening, particle migration and coalescence, particle detachment and particle dissolution and re-precipitation.38 In identical location TEM experiments used for studying the degradation of Pt–Ru alloyed NPs, Hengge et al.16 reported that dissolution and dealloying were the main degradation mechanisms. However, agglomeration and Ostwald ripening also played an important role, especially for the first cycles. In the case of our core–shell particles, a small tail towards bigger particle sizes can be seen in the particle size distribution of the MEA containing Ru@2Pt (Fig. 4h), suggesting that particle agglomeration takes place in these systems. A tail towards smaller particle sizes would be a hint for Ostwald ripening.16 However, due to the presence of small clusters, the presence of such a tail would be hidden in our case.Besides the loss of efficiency caused by an increase in the ECSA, the changes in the shell thickness can also have an effect on the PEMFC performance. In Ru@1Pt, the average diameter of the core–shell particles increases from 5.0 nm to 6.4 nm (1.4 nm), while for Ru@2Pt the increase is smaller, from 6.0 to 6.6 nm (0.6 nm) (Fig. 1 and 4). Considering that the {111} planes for Pt have an interplanar distance of 0.226 nm, each additional layer of Pt on the shell would increase the diameter of the particle by ∼0.45 nm (since atoms would be added in each side of the particle). Therefore, for Ru@1Pt the shell is expected to have grown on average by ∼3 additional layers, and for Ru@2Ptit was ∼ 1 layer. However, it should be noted that the shell before ASTs in the as synthesized NPs is quite incomplete (Fig. 3). Thus, these additional layers most likely will also nucleate and grow in an inhomogeneous way. This is consistent with the reconstructions obtained from electron tomography (Fig. 5), which reveals a still incomplete shell after the cycles. Even after ASTs, the average shell thickness for both samples would be below 5 monolayers, and therefore the strain effects of the core on the Pt shell (and the consequently enhanced CO poisoning tolerance) are expected to remain intact. Moreover, Ru diffusion or dissolution from the core and redeposition on the core–shell particles would result in oxophilic Ru atoms being on the surface, which would allow for the bifunctional effect to take place. However, the electronic effects of the Ru core are expected to change. For Ru@2Pt, since the average shell thickness after ASTs is ∼3 Pt monolayers, the electronic effect of Ru on the CO adsorption energy would still be relevant,18 while for Ru@1Pt the thicker ∼4 Pt monolayer shell can result in a weaker electronic effect. This could be a factor explaining the loss of performance of Ru@1Pt in reformate after the ASTs compared to Ru@2Pt.23
PCA from the EDS spectral maps (Fig. 4c and g) reveals that these small clusters consist mainly of Pt atoms, although 7 and 8 at% Ru can be detected for Ru@1Pt and Ru@2Pt, respectively. Since in the as synthesized samples Ru was only detected in the core–shell like particles (Fig. 1 and FS1†), during the ASTs, dissolution and re-precipitation of Pt and Ru take place. Ru dissolution can occur since the shell is incomplete.
Electron tomography experiments can be used for studying the 3D encapsulation of the Ru cores (Fig. 3). The 3D reconstruction of Pt shows that the shell is incomplete for Ru@1Pt and Ru@2Pt, which would explain why Ru is detected on the surface of some particles on the EDS maps (Fig. 2b and e). The fact that the Pt shell is not fully protecting the Ru core has important consequences on the electrochemical performance and durability of Ru@1Pt and Ru@2Pt. The primary reason behind nanostructuring the catalyst into core–shell NPs is to protect the less stable Ru, since it is well known that surface Ru can dissolve under operating conditions.35 In fact, previous studies have shown that when solid solutions36,39 or bimetallic13 Pt–Ru NPs are used as anode catalysts, Ru crosses over from the anode towards the cathode through the membrane. On the other hand, Hsieh et al.17 demonstrated superior stability of well-ordered Ru@Pt NPs during accelerated stress tests. Since the Ru cores in the samples investigated in the present study are not fully encapsulated, Ru can also leach out during operation. The shell is especially incomplete for Ru@1Pt (Fig. 3), which can be explained by the lower Pt to Ru ratio added in the synthesis of these particles. Therefore, the degree of Ru protection under fuel cell conditions in Ru@2Pt is expected to be higher than in Ru@1Pt.
This is in good agreement with EDS measurements performed on the cathode, which shows that the amount of Ru detected in that electrode is higher for MEA with Ru@1Pt (10 at%) than for MEA with Ru@2Pt (3 at%).23
In Ru@1Pt, the Pt S/V ratio calculated from the tomographic reconstruction of the pristine core–shell particles (Fig. 3) is 2.7 times larger than that of the core–shell particles present in the corresponding cycled MEA (Fig. 5). These results suggest that important changes in the Pt shell take place during operation. On the other hand, in Ru@2Pt no significant differences in the Pt S/V ratio before and after ASTs can be found, suggesting a more stable shell. These results are in good agreement with the electrochemical measurements that indicate a higher degree of stability for Ru@2Pt.23
In summary, the advanced (S)TEM characterization presented in this work explains why in our earlier studies23 higher initial performance is seen when Ru@1Pt NPs are used as anode catalysts. Moreover, the higher stability under fuel cell conditions of Ru@2Pt is also addressed. The obtained insights show that there is no intrinsic trade-off between high initial performance and long-term stability. By tuning the synthesis conditions, it could be possible to maximize the initial performance in reformate (e.g., by avoiding the synthesis of the smaller pure Pt NPs) while augmenting the long-term stability (e.g., by ensuring a homogeneous Pt shell that fully covers and protects the Ru core).
Conclusions
In this work we investigated two PEMFC anode catalysts: Ru–Pt core–shell NPs with different shell thicknesses (Ru@1Pt and Ru@2Pt). The catalyst NPs were studied both as synthesized and after 10000 stress tests under fuel cell conditions. The changes that occurred in these NPs during the stress tests were attributed to operational degradation, and related to the electrochemical performance of the cells reported in our earlier study.23
Two families of particles were found in each sample: smaller pure Pt particles (μ1 = 2.4 ± 1.2 nm for Ru@1Pt and μ1 = 1.6 ± 1.3 nm for Ru@2Pt) and bigger core–shell like particles (μ2 = 5.0 ± 2.1 nm for Ru@1Pt and μ1 = 6.0 ± 3.6 nm for Ru@2P). Pure Pt particles were more numerous in Ru@2Pt, which contributes to the poorer performance of Ru@2Pt when reformate is fed to the anode. The shell thickness of the core–shell like particles was estimated by computing the strain maps from the local differences in atomic arrangement observed in the HR-TEM micrographs. An average shell thickness of 1 Pt monolayer was found for Ru@1Pt, and of 2–3 Pt monolayers was found for Ru@2Pt. However, the 3D morphological studies of the as synthesized particles via electron tomography showed an incomplete shell. Thus, it was not fully encapsulating the Ru cores, especially for Ru@1Pt, which explains the higher Ru dissolution of this sample.
Furthermore, the core–shell particles of Ru@1Pt grew on average of 0.7 nm in radius (∼4 Pt layers), while for Ru@2Pt a smaller radius increase of 0.3 nm (∼1 Pt monolayer) was found, which explains why the ECSA of Ru@1Pt decreased more than that of Ru@2Pt. This increase in the shell thickness is expected to decrease the CO poisoning tolerance of the catalyst particles, especially for Ru@1Pt, which is in good agreement with the results of our previous work.23
Our results indicate a direction for maximizing the performance in reformate without compromising the long-term stability of the anode catalyst NPs under fuel cell conditions.
Author contributions
AGM and MVP performed the (S)TEM characterization and the corresponding data analysis and wrote the manuscript. VB performed the synthesis of the samples and ASTs. TG assisted in the tomographic reconstruction. TS and NRR performed the FIB sample preparation. KH provided scientific support. TJ and CS initiated the study, coordinated the research and provided scientific support. All authors contributed to the manuscript and interpretation of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research project (FKZ 03ETB018) was supported by the Federal Ministry for Economic Affairs and Climate Action (BMWi) based on a decision taken by the German Bundestag. The authors would like to thank B. Distl for his corrections of the manuscript, A. Garzón Romero for his contribution to manuscript formatting and C. Liebscher for his help with the strain analysis. Open Access funding provided by the Max Planck Society.References
- The Paris Agreement, https://www.un.org/en/climatechange/paris-agreement.
- H. Ritchie and M. Roser, CO2 and Greenhouse Gas Emissions, https://ourworldindata.org/co2-and-other-greenhouse-gas-emissions.
- H. Ritchie and M. Roser, Electricity Mix, https://ourworldindata.org/energy.
- R. Jinnouchi, K. Kudo, K. Kodama, N. Kitano, T. Suzuki, S. Minami, K. Shinozaki, N. Hasegawa and A. Shinohara, Nat. Commun., 2021, 12, 4956, DOI:10.1038/s41467-021-25301-3.
- H. Ritchie and M. Roser, Emissions by sector, https://ourworldindata.org/emissions-by-sector.
- M. Boaventura, I. Alves, P. Ribeirinha and A. Mendes, Int. J. Hydrogen Energy, 2016, 41, 19771–19780, DOI:10.1016/j.ijhydene.2016.06.201.
- Y. Sun, S. Polani, F. Luo, S. Ott, P. Strasser and F. Dionigi, Nat. Commun., 2021, 12, 1–14, DOI:10.1038/s41467-021-25911-x.
- Y. J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467, DOI:10.1021/cr500519c.
- X. Ren, Q. Lv, L. Liu, B. Liu, Y. Wang, A. Liu and G. Wu, Sustainable Energy Fuels, 2019, 4, 15–30, 10.1039/C9SE00460B.
- G. Franchi, M. Capocelli, M. De Falco, V. Piemonte and D. Barba, Membranes, 2020, 10, 10, DOI:10.3390/membranes10010010.
- P. Pei, M. Wang, D. Chen, P. Ren and L. Zhang, Prog. Nat. Sci.: Mater. Int., 2020, 30, 751–763, DOI:10.1016/j.pnsc.2020.08.015.
- C. Molochas and P. Tsiakaras, Catalysts, 2021, 11, 1127, DOI:10.3390/catal11091127.
- K. Hengge, C. Heinzl, M. Perchthaler, D. Varley, T. Lochner and C. Scheu, J. Power Sources, 2017, 364, 437–448, DOI:10.1016/j.jpowsour.2017.08.042.
- A. C. Garcia, V. A. Paganin and E. A. Ticianelli, Electrochim. Acta, 2008, 53, 4309–4315, DOI:10.1016/j.electacta.2008.01.006.
- P. P. Lopes and E. A. Ticianelli, J. Electroanal. Chem., 2010, 644, 110–116, DOI:10.1016/j.jelechem.2009.06.011.
- K. Hengge, T. Gänsler, E. Pizzutilo, C. Heinzl, M. Beetz, K. J. J. Mayrhofer and C. Scheu, Int. J. Hydrogen Energy, 2017, 42, 25359–25371, DOI:10.1016/j.ijhydene.2017.08.108.
- Y. C. Hsieh, Y. Zhang, D. Su, V. Volkov, R. Si, L. Wu, Y. Zhu, W. An, P. Liu, P. He, S. Ye, R. R. Adzic and J. X. Wang, Nat. Commun., 2013, 4, 2466, DOI:10.1038/ncomms3466.
- A. Schlapka, M. Lischka, A. Groß, U. Käsberger and P. Jakob, Phys. Rev. Lett., 2003, 91, 016101, DOI:10.1103/PhysRevLett.91.016101.
- Y. F. Xing, Y. Zhou, Y. B. Sun, C. Chi, Y. Shi, F. B Wang and X. H. Xia, J. Electroanal. Chem., 2020, 872, 114348, DOI:10.1016/j.jelechem.2020.114348.
- A. Jackson, A. Strickler, D. Higgins and T. F. Jaramillo, Nanomaterials, 2018, 8, 38, DOI:10.3390/nano8010038.
- S. Alayoglu, P. Zavalij, B. Eichhorn, Q. Wang, A. I. Frenkel and P. Chupas, ACS Nano, 2009, 3, 3127–3137, DOI:10.1021/nn900242v.
- J. Zou, M. Wu, S. Ning, L. Huang, X. Kang and S. Chen, ACS Sustainable Chem. Eng., 2019, 7, 9007–9016, DOI:10.1021/acssuschemeng.9b01270.
- V. Berova, A. Garzón Manjón, M. Vega Paredes, T. Schwarz, N. A. Rivas, K. Hengge, T. Jurzinsky and C. Scheu, J. Power Sources, 2023, 554, 232327, DOI:10.1016/j.jpowsour.2022.232327.
- S. Henning, R. Shimizu, J. Herranz, L. Kühn, A. Eychmüller, M. Uchida, K. Kakinuma and T. J. Schmidt, J. Electrochem. Soc., 2018, 165, F3001–F3006, DOI:10.1149/2.0531802jes.
- C. L. Jia, M. Lentzen and K. Urban, Microsc. Microanal., 2004, 10, 174–184, DOI:10.1017/S1431927604040425.
- G. Cliff and G. W. Lorimer, J. Microsc., 1975, 103, 203–207, DOI:10.1111/j.1365-2818.1975.tb03895.x.
- A. G. Manjón, T. Löffler, M. Meischein, H. Meyer, J. Lim, V. Strotkötter, W. Schuhmann, A. Ludwig and C. Scheu, Nanoscale, 2020, 12, 23570–23577, 10.1039/D0NR07632E.
- S. Zhang and C. Scheu, Microscopy, 2018, 67, i133–i141, DOI:10.1093/jmicro/dfx091.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 70–82, DOI:10.1002/pro.3943.
- Scanning Transmission Electron Microscopy, ed. S. J. Pennycook and P. D. Nellist, SpringerNew York, NY, 1st edn, 2011, DOI:10.1007/978-1-4419-7200-2.
- J. C. Slater, J. Chem. Phys., 1964, 41, 3199–3204, DOI:10.1063/1.1725697.
- Pt fcc (Pt) Crystal Structure: Datasheet from “PAULING FILE Multinaries Edition – 2012”, ed. P. Villars and K. Cenzual, in SpringerMaterials, 2012 (https://materials.springer.com/isp/crystallographic/docs/sd_0250899) Search PubMed.
- Ruhcp (Ru) Crystal Structure: Datasheet from “PAULING FILE Multinaries Edition – 2012”, ed. P. Villars and K. Cenzual, in SpringerMaterials, 2012 (https://materials.springer.com/isp/crystallographic/docs/sd_1244358) Search PubMed.
- J. M. Zhang, F. Ma and K. W. Xu, Chin. Phys., 2004, 13, 1082–1090, DOI:10.1088/1009-1963/13/7/020.
- P. Ochal, J. L. Gomez De La Fuente, M. Tsypkin, F. Seland, S. Sunde, N. Muthuswamy, M. Rønning, D. Chen, S. Garcia, S. Alayoglu and B. Eichhorn, J. Electroanal. Chem., 2011, 655, 140–146, DOI:10.1016/j.jelechem.2011.02.027.
- E. Antolini, J. Solid State Electrochem., 2011, 15, 455–472 CrossRef CAS.
- S. S. Rasouli, Degradation Mechanisms of Pt and Pt Alloy Nanocatalysts in Proton Exchange Membrane Fuel Cells, The University of Texas, Austin, 2017 Search PubMed.
- P. J. Ferreira, G. J. La O’, Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256, DOI:10.1149/1.2050347.
- M. Vega Paredes, A. Garzón Manjón, B. Hill, T. Schwarz, N. A. Rivas, T. Jurzinsky, K. Hengge, F. Mack and C. Scheu, Nanoscale, 2022, 14, 11543–11551, 10.1039/D2NR02892A.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr04869h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |