
Ultrafast insights into full-colour light-emitting C-Dots†
Nandan
Ghorai‡
 a,
Soumyadip
Bhunia‡
b,
Subham
Burai
d,
Hirendra N.
Ghosh
ac,
Pradipta
Purkayastha
b and
Somen
Mondal
*d
a,
Soumyadip
Bhunia‡
b,
Subham
Burai
d,
Hirendra N.
Ghosh
ac,
Pradipta
Purkayastha
b and
Somen
Mondal
*d
aInstitute of Nano Science and Technology, Mohali, Punjab 140306, India
bIndian Institute of Science Education and Research Kolkata, Mohanpur 741246, WB, India
cBhabha Atomic Research Centre, Mumbai 400085, India
dInstitute of Chemical Technology, Mumbai, Marathwada Campus, Jalna, Maharashtra 431203, India. E-mail: s.mondal@marj.ictmumbai.edu.in
First published on 6th October 2022
Abstract
Designing carbon dots (C-Dots) in a controlled way requires a profound understanding of their photophysical properties, such as the origin of their fluorescence and excitation wavelength-dependent emission properties, which has been a perennial problem in the last few decades. Herein, we synthesized three different C-Dots (blue, green, and red-emitting C-Dots) from the same starting materials via a hydrothermal method and separated them by silica column chromatography. All the purified C-Dots exhibited three different emission maxima after a certain range of different excitations, showing a high optical uniformity in their emission properties. It was also observed that the average distributions of the particle size in all the C-Dots were the same with a typical size of 4 nm and the same interplanar d spacing of ∼0.21 nm. Here, we tried to establish a well-defined conclusive answer to the puzzling optical properties of C-Dots via successfully investigating the carrier dynamics of their core and surface state with a myriad use of steady-state, time-resolved photoluminescence, and ultrafast transient absorbance spectroscopy techniques. The ultrafast charge-carrier dynamics of the core and surface state clearly indicated that the graphitic nitrogen in the core state and the oxygen-containing functional group in the surface state predominately contribute to controlling their wide range of emission properties. We believe that these findings will give the C-Dots their own designation in the fluorophore world and create a new avenue for designing and developing C-Dot-based new architectures.
Introduction
Carbon Dots (C-Dots) have been considered mysterious nanoparticles since their inception, although numerous research groups in physical chemistry, materials chemistry, and nanotechnology have performed research on them and investigated them for potential applications, making research on C-Dot among the most fascinating and emerging research fields.1,2 C-Dots are a highly promising material for developing light-emitting diodes, photocatalytic H2 production, and biomedical and optoelectronic applications due to their high quantum yield, water solubility, high biocompatibility, and low cytotoxicity.3–5 The most mysterious and unexplored properties of C-Dots are the origins of their fluorescence and their excitation wavelength-dependent emission, even though these have potential interest in various fields. The understanding of the fundamental photophysics and carrier dynamics of different-colour-emitting C-Dots is still blurred, even after the attempts of various research groups, due to their complex photophysical behaviour.6–8 Despite this, most reported works only focus on the synthesis and broad potential applications of C-Dots, especially in bio-imaging, bio-sensing, and photocatalysis, rather than on deciphering their unique properties.9–14 In most cases, a strong blue emission from C-Dots is observed that decreases gradually with a shift of the emission maximum, which can mislead the search for the origin of the fluorescence.15,16 This puzzle is often complicated due to the lack of proper purification of the C-Dots. Thus, it appears to be important to isolate the products from a mixture of the C-Dots using proper purification techniques. Most of the reported works used dialysis to purify the C-Dots. However, in utilizing this method, only the ions and the small capping ligands can be removed from the mixture, but separating the differently emitting C-Dots, such as the blue-emitting C-Dots (B-C-Dots), green-emitting C-Dots (G-C-Dots), and red-emitting C-Dots (R-C-Dots), is not possible. There are a few reports where high-separation-efficiency techniques were employed, such as high-performance liquid chromatography (HPLC), column chromatography, or gel electrophoresis (PAGE).17,18Therefore, exploring the optical properties of different-colour-emitting pure C-Dots along with their excited-state carrier dynamics is of the utmost importance for technology development. There are several reports on C-Dots that can help to understand the origin of photoluminescence (PL) and its excitation-dependent behaviour, where the C-Dots were synthesized by different methods.19,20 A careful literature survey reveals two different opinions on the origin of the C-Dot PL: (a) the presence of two different emissive species within C-Dots: one being the core state, i.e. composed of an sp2-hybridized aromatic network surrounded by an amorphous sp3-hybridized domain, and the other being the surface states, i.e. the functional groups present at the edge of the C-Dot core, and (b) the formation of molecular fluorophores in the C-Dot core or an aggregated structure formation during the time of the C-Dot synthesis.19,21–23 Wang et al. investigated the common origin of the green luminescence of C-Dots and graphene quantum dots using transient absorption (TA) spectroscopy. They revealed that the graphitic core acts as a temporary reservoir for the photogenerated electrons and holes, which are rapidly transferred to the different trap states through a non-radiative pathway.19 A comprehensive study showed that the short wavelength emission originates from the sp2-carbon network of the C-Dot core and that a longer wavelength is responsible for another deactivation pathway.24 In this regard, we previously attempted to understand the photophysical as well as photosensitizing properties of C-Dots using ultrafast spectroscopy. The existence of the core and the surface states were observed in the N–C-Dots, and upon doping with phosphate or boron, the N–C-Dots significantly changed their excited-state dynamics.25,26 Some recent studies have shown that the emission of C-Dots from blue to red can be controlled by using solvents of varying polarity, thus amplifying the number of electron-withdrawing groups on the surface of the C-Dots, which then increases the degree of oxidation and the negative surface charge. Moreover, different degrees of dehydration and carbonization play important roles in controlling the size of the sp2-conjugated domains within the prepared C-Dots.27–30 Some recent reviews have suggested that the oxygen-containing functional groups act as emissive surface states as well as trap states of C-Dots. Carbonyl groups of carboxylic acid, esters, and aldehyde groups play an important role regarding the tuning of band gap, whereby it has been suggested that the band gap becomes narrower with the increase in –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in the surface state. Moreover, different methods have been proposed to control the narrow-bandwidth emission in C-Dots.31–33 To the best of our knowledge, most of the reported works have focused on exploring the carrier dynamics of either blue-emitting or green-emitting C-Dots.24,25 Here, for the first time, we explored the carrier dynamics and different relaxation processes of pure blue-, green-, and red-emitting C-Dots in detail, which have remained unexplored previously. Our results are expected to shed considerable light on the actual origin of the PL in C-Dots.
O groups in the surface state. Moreover, different methods have been proposed to control the narrow-bandwidth emission in C-Dots.31–33 To the best of our knowledge, most of the reported works have focused on exploring the carrier dynamics of either blue-emitting or green-emitting C-Dots.24,25 Here, for the first time, we explored the carrier dynamics and different relaxation processes of pure blue-, green-, and red-emitting C-Dots in detail, which have remained unexplored previously. Our results are expected to shed considerable light on the actual origin of the PL in C-Dots.
In the present work, we aimed to identify the underlying carrier dynamics of blue (B-C-Dots), green (G-C-Dots), and red (R-C-Dots)-emitting C-Dots with the aid of femtosecond transient absorption spectroscopy. To explore the dynamics of the different-colour-emitting C-Dots, we synthesized B-C-Dots, G-C-Dots, and R-C-Dots from common starting materials (citric acid and urea), which is also essential for understanding the origin of the PL of the differently emitting C-Dots. Foremost, the three C-Dot-variants were separated from the mixture of the products by column chromatography. Here, we used steady-state and time-resolved PL along with ultrafast TA spectroscopy to resolve these mysteries of the carrier dynamics and the existence of the perennial problems. Herein, we highlight the different relaxation processes of the differently emitting C-Dots (B-C-Dots, G-C-Dots, and R-C-Dots) through selective excitation of the core and the surface states. Moreover, we tried to answer two unique questions: first: is blue emission an inherent property of all emitting C-Dots?; second: how does the emission centre differ with changing the surface polarity of C-Dots? These two fundamental questions are never discussed in the literature in creating a proper environment. We believe that a deep understanding of the carrier dynamics in the core state as well as the surface state in all the emitting pure C-Dots and our new findings would create new and novel outcomes for C-Dots to help build better photosensitizers, and for applications in catalysis and optoelectronic devices.
Results and discussion
We initially synthesized a mixture of full-colour-emitting C-Dots by dissolving citric acid and urea in dimethylformamide (DMF) using a solvothermal method.19 The targeted products were separated from the crude mixture by column chromatography, as shown in Fig. 1a. The B-C-Dots were eluted from the column using a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :97 (v:v) methanol (MeOH) and dichloromethane (DCM) mixture. The G-C-Dots and R-C-Dots were eluted using 6:94 and 25:75 mixtures of MeOH and DCM, respectively. The structural morphology of the synthesized and separated C-Dots was examined by transmission electron microscopy (TEM). The wide area and HRTEM images are shown in Fig. S1a–c† and Fig. 1b–d. All the C-Dots showed spherical shapes with an average particle size of ∼4 nm (left bottom: Fig. 1b–d). Here, the wide area TEM images are shown in Fig. S1† where the shape of these nanocrystals remained spherical and also identical in size. The HRTEM images and line profiles of the C-Dots (right bottom: Fig. 1b–d) showed their crystalline nature with an interplanar d spacing of ∼0.21 nm, resembling the lattice plane (100) of graphite.34 In Fig. S2,† the X-ray diffraction patterns (XRD) showed peaks at around 15° and 26°, corresponding to the graphitic structure with (100) and (002) lattice planes, respectively. This result well supports the bulk graphite and implies a good crystalline nature. The fluorescence quantum yields (QY) of the B-C-Dots, G-C-Dots, and R-C-Dots were measured using Coumarin 102 as a reference after the purification and the calculated relative QYs were 41.1%, 18.6% and 16.2%.
:97 (v:v) methanol (MeOH) and dichloromethane (DCM) mixture. The G-C-Dots and R-C-Dots were eluted using 6:94 and 25:75 mixtures of MeOH and DCM, respectively. The structural morphology of the synthesized and separated C-Dots was examined by transmission electron microscopy (TEM). The wide area and HRTEM images are shown in Fig. S1a–c† and Fig. 1b–d. All the C-Dots showed spherical shapes with an average particle size of ∼4 nm (left bottom: Fig. 1b–d). Here, the wide area TEM images are shown in Fig. S1† where the shape of these nanocrystals remained spherical and also identical in size. The HRTEM images and line profiles of the C-Dots (right bottom: Fig. 1b–d) showed their crystalline nature with an interplanar d spacing of ∼0.21 nm, resembling the lattice plane (100) of graphite.34 In Fig. S2,† the X-ray diffraction patterns (XRD) showed peaks at around 15° and 26°, corresponding to the graphitic structure with (100) and (002) lattice planes, respectively. This result well supports the bulk graphite and implies a good crystalline nature. The fluorescence quantum yields (QY) of the B-C-Dots, G-C-Dots, and R-C-Dots were measured using Coumarin 102 as a reference after the purification and the calculated relative QYs were 41.1%, 18.6% and 16.2%.
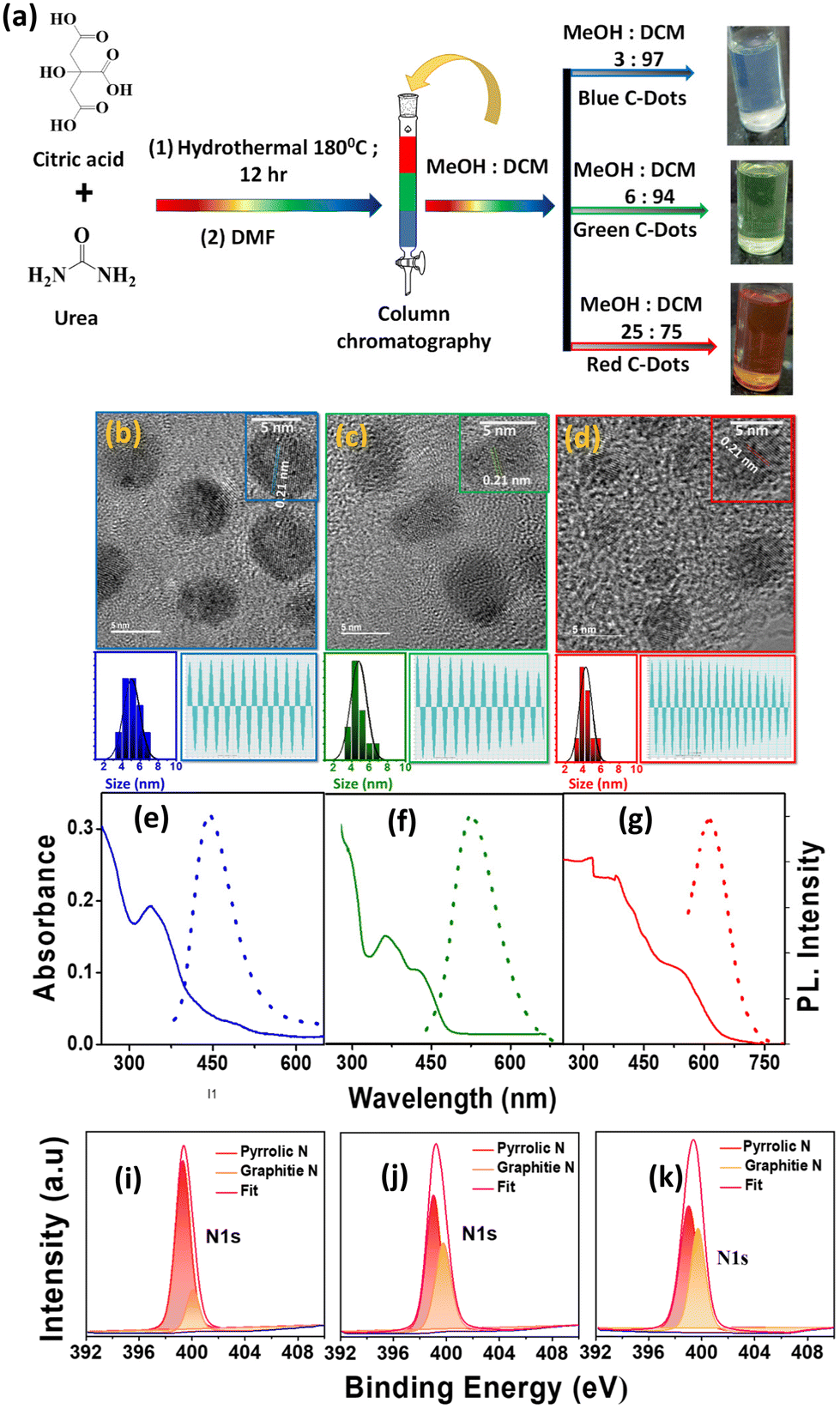 | ||
| Fig. 1 First panel: (a) schematic representation of the synthesis procedure and separation of the B-C-Dots, G-C-Dots, and R-C-Dots from a mixture of the C-Dots. Second panel: (b–d) TEM image and size distribution histogram and line profiles of the (b) B-C-Dots, (c) G-C-Dots, and (d) R-C-Dots. Third panel: (e–g) absorption (solid) and normalised emission (dot) spectra of the (e) B-C-Dots, (f) G-C-Dots, and (g) R-C-Dots after excitation at 340, 420, and 500 nm, respectively. Fourth panel: (i–k) high-resolution XPS spectra for the N 1s signal of the (i) B-C-Dots (j) G-C-Dots, and (k) R-C-Dots. | ||
The absorption spectra (solid line) and emission spectra (dotted line) for the three different colour-emitting C-Dots are shown in Fig. 1e–g, where the number of peaks increased from the B-C-Dots to R-C-Dots in the UV–vis region. The B-C-Dots exhibited a single absorption band at 340 nm, whereas the G-C-Dots and R-C-Dots displayed two and three distinct peaks at 340 and 420 nm and 345, 425, and 540 nm, respectively. This result indicated that additional light-absorbing materials were formed in the latter two substances. A strong blue emission peaking at 450 nm was observed for all the C-Dots upon excitation at 300–400 nm, which was independent of the excitation wavelength. However, upon excitation above 400 nm, we observed an excitation wavelength dependency of the emission from the C-Dots, whereby the emission maximum shifted towards higher wavelengths with the increase of the excitation wavelength (Fig. S3a–c†). Upon exciting at 340, 420, and 500 nm, all the C-Dots exhibited the PL peak maximum at ∼440 nm (2.75 eV), ∼520 nm (2.36 eV), and ∼610 nm (2.03 eV), respectively, as shown in Fig. 1e–g. These results indicate that the three different emissions from the C-Dots originated from different states of the same particle. We may state intuitively that the high energy emission (blue emission) originated from the core state, i.e. the carbon π–π* transitions in the sp2 domain, which was inherent to all the C-Dots. The other two-emission peaks, due to the lower energy transition (green and red emissions), commonly originated from the functional groups of the C-Dots attached at the edge of the carbon core, called the surface states.
High-resolution X-ray photoelectron spectroscopy (XPS) measurements were applied to reveal the internal structure and surface functional groups of the differently emitting C-Dots. Fig. 1(i–k) and Fig. S4† show the presence of a predominant C 1s peak at ∼286 eV, N 1s peak at ∼400 eV, and O 1s peak at ∼532 eV. As shown in Fig. 1(i–k), the high-resolution XPS spectra of N 1s could be fitted with two Gaussian peaks at around 399.8 eV (pyrrolic N) and 401.0 eV (graphitic N), where the intensity of graphitic N increased from the B-C-Dots to G-C-Dot to R-C-Dots. The high-resolution XPS spectra of C 1s and O 1s are shown in Fig. S4,† where the spectra of C 1s and O 1s were fitted with three and two Gaussian peaks, respectively. In the C 1s spectra, the peaks at 284.1, 286.7, and 288.8 eV were assigned as –CC, –C–O/C–N, and –COOH, respectively. In O 1s spectra, the peaks at 531.5 and 532.5 eV were assigned as CO and C–OH/C–O–C, respectively. Here, we observe that the relative content of O increased from the B-C-Dots to G-C-Dot to R-C-Dots. Thus, XPS analysis confirmed that the core of the C-Dots contained graphitized carbon, where the major contributions were graphitic N and pyrrolic N, and oxygen-containing surface functional groups have great importance for regulating the origin of fluorescence in C-Dots.35,36
The presence of different surface functional groups was investigated using the Fourier transform infrared (FTIR) spectra of the samples, as shown in Fig. S5.† The observations revealed that all the C-Dots contained different oxygen-containing functional groups on the surface, such as O–H (3200 cm−1) and –COOH/–CO (1703 cm−1), which make the C-Dots soluble in water.37,38 Moreover, the stretching frequencies of CN at 1627 cm−1, CC at 1445 cm−1, and C–N at 1139 cm−1 indicated the presence of graphitic nitrogen in the sp2 network of the C-Dots.37 Here, Raman spectroscopy was performed for all the C-Dots to prove the existence of the sp2 network with graphitic nitrogen in the core of the C-Dots, as shown in Fig. S6.† The Raman spectra of all the C-Dots exhibited both D and G bands at ∼1354 and ∼1581 cm−1, respectively, which indicated the presence of the sp3 defects and sp2 carbon network system. The ratio of ID/IG suggested that the core state was similar for all the emitting C-Dots. A closer look at the FTIR spectra shows an enhancement of the C–H stretching signal at 2885 cm−1 upon moving from the R-C-Dots to the B-C-Dots. This clearly indicated that the surface of the B-C-Dots contained more hydrogens at the periphery compared to the other C-Dots. This result supports the existence of more functional groups on the surface of the R-C-Dots than for the G-C-Dots and B-C-Dots and hence it is understandable that these functional groups play important roles in the change in colour from blue to red upon increasing the polarity of the eluting solvents in column chromatography. Moreover, we carried out absorption and emission measurements of the differently emitting C-Dots under different pH conditions to further confirm the effects of the surface functional groups (Fig. S7†). In the case of the B-C-Dots, a broad new absorption band grew at longer wavelengths alone with the absorbance peak at 340 nm. Moreover, the characteristic absorbance of the G-C-Dots (420 nm) and R-C-Dots (540 nm) also showed some changes with changing the pH of the medium. The pH-induced PL studies were carried out after exciting at 340 nm for a better understanding of the pH-dependent phenomenon. It was observed that the emission intensity also changed with the varying pH level of the medium. These results suggest that the nature of the energy levels and electronic transitions were influenced by the different pH conditions due to the protonation and deprotonation of the oxygen-containing functional groups present on the surfaces of the C-Dots.
We used time-resolved PL measurements to explore the excited-state dynamics of all the C-Dots after exciting them at 340 nm and 405 nm and by monitoring the emissions at 440 nm and 530 nm (Fig. 2). To explore the carrier dynamics of the core state in a longer time scale (ns), all the C-Dots were excited by a 340 nm light source, and the emission decay kinetics were recorded at 440 nm, as shown in Fig. 2a. The decay time constants were obtained by fitting the raw data with a bi-exponential routine, as provided in Table S1.† It was noticed that the fast decay component of the B-C-Dots (1.5 ns) became faster than those of the G-C-Dots (1.8 ns) and R-C-Dots (2.0 ns) (Fig. 2a and Table S1†), which was indicative of an e–h recombination process in the core state. The slower decay component represented the emission from the trap states created in the core carbon sp2 network. We found a clear correlation between the two distinct time constants, whereby the amplitude of the fast component was reduced from 30% for the B-C-Dots to 7% for the R-C-Dots, and those of the slower one increased from the B-C-Dots to the R-C-Dots. This is an indication of the presence of more surface defects on the R-C-Dots than on the others. Furthermore, the decay kinetics of all the C-Dots were recorded at 530 nm after exciting at 340 nm (Fig. 2b) and the results were fitted bi-exponentially, yielding the time constants shown in Table S1.† Interestingly, a growth component evolved for all the C-Dots due to solvatochromism, where the R-C-Dots showed the major contribution. These results also provide clear indications that the R-C-Dots contained more polar surface functional groups than the G- and B-C-Dots, which was in line with a previous study.33 To follow the surface dynamics in the C-Dots, we further excited the samples at 405 nm and monitored them at 440 nm and 530 nm (Fig. 2c and d). Similar decay kinetics was observed at 440 nm after exciting the samples at 405 nm. The data indicate that the carrier-relaxation pathways at the core state are similar for all the C-Dots when the excitation is at the band edge of the core state. This result supports the existence of similar core states in all the emitting C-Dots. Moreover, the radiative recombination (τ2) process of all the C-Dots became faster after excitation at 405 nm as compared to excitation at 340 nm when the monitoring wavelength was 440 nm. To follow the solvation dynamics of the surface state, the decays of all the C-Dots were recorded at 530 nm after exciting at 405 nm and the results fitted with a single-exponential growth and bi-exponential decay recovery time constants (Table S1†). While the growth component was observed for the R- and G-C-Dots (∼500 ps) due to solvent relaxation of the surface functional groups in the vicinity of the carbon-like peripheral structures, it was absent for the B-C-Dots (<200 ps).
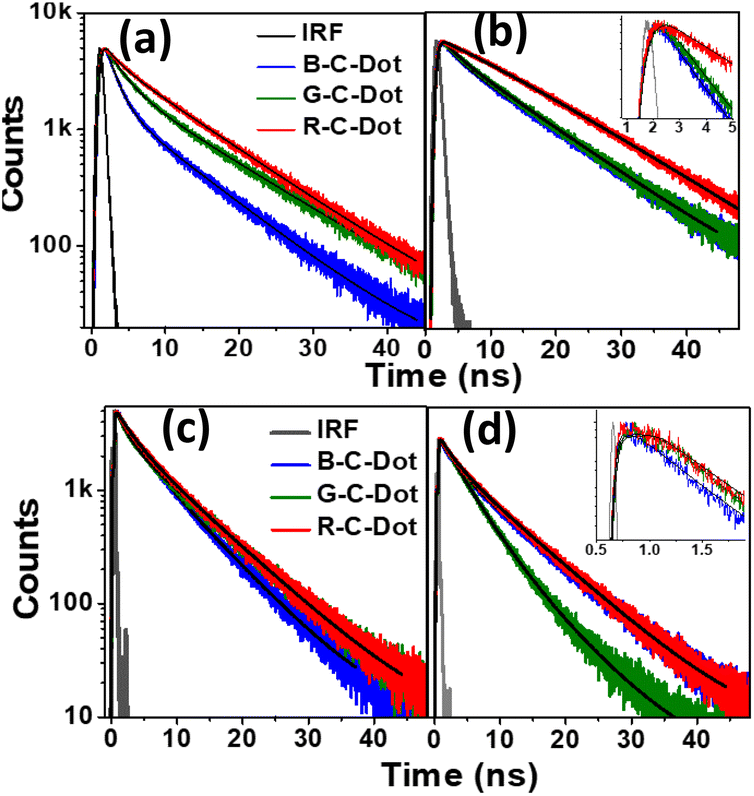 | ||
| Fig. 2 Time-resolved PL decay of the C-Dots monitored at (a) 440 nm and (b) 530 nm after excitation at 340 nm, and at (c) 440 nm and (d) 530 nm after 405 nm excitation (inset: zoomed data at the earlier time scale). | ||
Furthermore, the R-C-Dots were excited at 605 nm and the 630 nm emission was probed, as shown in Fig. S8.† The decay curve was fitted bi-exponentially with time constants of τ1 = 0.54 ns and τ2 = 1.07 ns (Table S1†), which indicated that the relaxation process was happening via two different channels and the red emission of the C-Dots originated from the highly polar functional groups of the surface state. We can thus infer that the blue emission originated from the pure core state, where the dynamics were controlled by the sp2 network and the defect states, while the red and green emissions occurred from the surface states with different polarities, along with the blue emission, where the surface functional groups played the major role. The origins of the different emissions (440, 520, and 610 nm) from the core and the surface states upon excitation at 340, 420, and 540 nm are shown diagrammatically in Scheme 1 where T and S represent the trap and surfsce state respectively. To investigate the solvatochromism effect, we measured the PL of the differently emitting C-Dots in different solvents, such as water, dimethyl sulfoxide (DMSO), and dimethylformamide (DMF) as shown in Fig. S9.† In Fig. S9a–c,† the emission maximum showed a gradual blue-shift as we moved from polar solvents (DMSO, water) to a non-polar solvent (THF) and the extent of the shift changed for the differently emitting C-Dots. Interestingly, the maximum blue-shift was observed for the R-C-Dots (24 nm), whereas the G-C-Dots and B-C-Dots showed shifts of 16 and 8 nm, respectively, which indicated the presence of more polar surface functional groups on the surface of the R-C-Dots. A time-resolved PL study was carried out to confirm the solvent effect in the C-Dots after exciting at 340 and 420 nm and by monitoring the emissions at 440 and 530 nm (Fig. S9d–i†). Here, a pulse-width-limited growth component was observed at 440 nm, but the kinetics at 520 nm was fitted with a rise component (Table S2†) and this rise time was slower in polar solvents (DMSO, water), while a very fast rise time was observed for the non-polar solvent (DMF). Interestingly, we observed the growth time was slower in DMSO solvent than water, which may be due to the effect of viscosity or hydrogen bonding. The solvent-dependent rise time indicated that the growth component of the differently emitting C-Dots was due to a solvatochromism effect, rather than other carrier-relaxation processes.
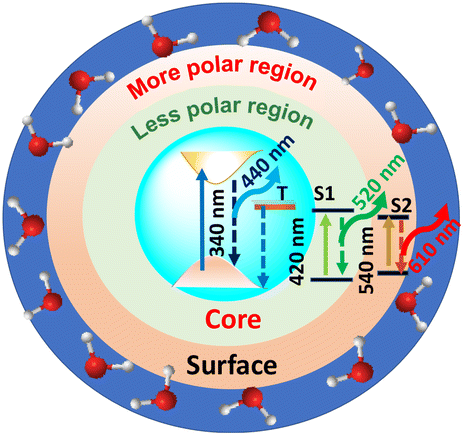 | ||
| Scheme 1 Schematic representation of the different states and radiative processes in the C-Dots. | ||
To understand the carrier dynamics of the differently emitting C-Dots in detail, we performed ultrafast TA measurements after selectively exciting the C-Dots at 320 and 380 nm and then probing the output signal in the UV–Vis range (Fig. 3). The TA spectra at different time delays are shown in Fig. 3a–c after 320 nm excitation. Upon excitation at 320 nm, i.e. exciting the core sp2 carbon domain of the C-Dots, we obtained two negative absorption bands at 360 and 440 nm, which could be attributed to the transient ground state bleach (GSB) due to the state-filling transition of the excitons, and stimulated emission (SE) due to the exciton depopulation. Interestingly, all the C-Dots showed similar spectral features for the GSB (360 nm) and the SE (430 nm) peaks upon excitation at 320 nm, indicating the existence of similar types of energy levels at the core of the different emitting C-Dots. It was also observed that the spectral shape and the position of the GSB and SE signals remained unchanged during the decay, which suggested the existence of a confined core state and the absence of a solvent effect therein. Moreover, to explore the role of the surface functional groups on the carrier dynamics at the surface states of the differently emitting C-Dots, we carried out TA measurements after selectively exciting the C-Dots at 380 nm and then probing them in the UV–vis range (Fig. 3d–f). The TA spectra at different time delays are shown in Fig. 3d–f after 380 nm excitation. Two negative absorption bands at 400–500 and 540 nm were observed, which were attributed to the GSB and SE of the surface states. The broad negative signal at 400–500 nm indicated an overlap of the surface state GSB (420 nm) and the core state SE (450 nm).
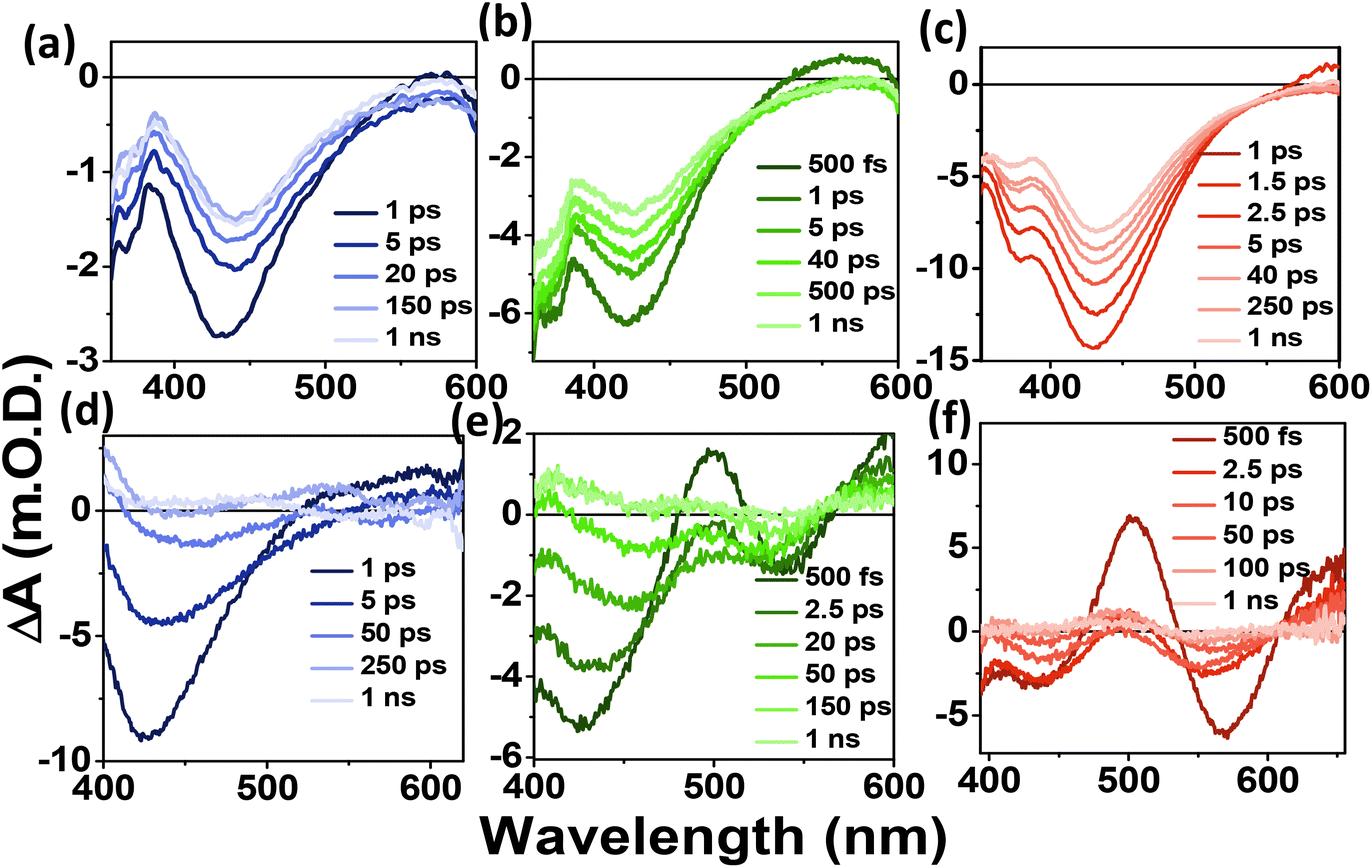 | ||
| Fig. 3 TA spectra of the (a) B-C-Dots, (b) G-C-Dots, and (c) R-C-Dot at different time delays after 320 nm excitation. TA spectra of the (d) B-C-Dots, (e) G-C-Dots, and (f) R-C-Dots at different time delays after 380 nm excitation. | ||
Hence, the existence of the two GSB, one at the core state (340 nm) and another at the surface state (420 nm), in all the C-Dots supported the occurrence of fluorescence at the blue and the green regions from the two different states.
We followed the decay kinetics at 340 nm (GSB) of all the C-Dots to explore the charge-carrier dynamics (Fig. 4a) and all the decays were fitted with a single-exponential growth and tri-exponential decay and the measured parameters are provided in Table 1. It was observed that the relaxation dynamics were significantly different for the B-, G-, and R-C-Dots. The bleach at 350 nm fitted with a single-exponential pulse-width-limited growth (<100 fs), which suggests that the hot carrier cooling time was very fast. The GS bleach recovery (τ1) could be assigned to non-radiative carrier trapping, which occurred due to the disordered and ordered carbon atom arrangements in the sp2 network of the core state. The second component (τ2) was due to the carrier relaxation from the trap state.
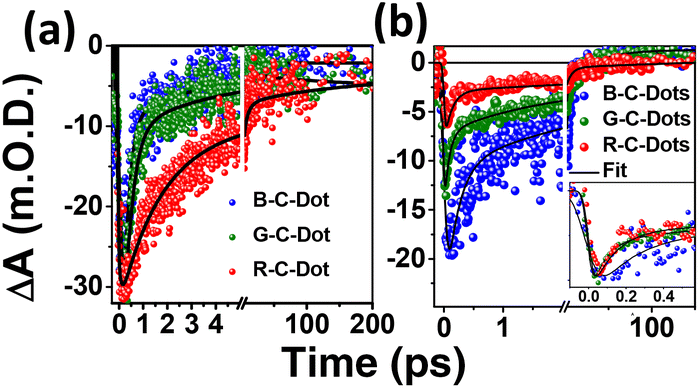 | ||
| Fig. 4 Kinetics at (a) 350 nm and (b) 420 nm after excitation at 320 and 380 nm, respectively (inset: normalized kinetics at 420 nm). | ||
| Sample | τ g (fs) (ag%) | τ 1d (ps) (a1d%) | τ 2d (ps) (a2d%) | τ 3d (ps) (a3d%) |
|---|---|---|---|---|
| a Values in parentheses indicate the percentage of amplitude (a) of the particular time constant (τ). | ||||
| B-C-DOT | <100 (+100%) | 0.25 ± 0.06 (−75%) | 2.0 ± 0.17 (−17%) | >100 (−08%) |
| G-C-DOT | <100 (+100%) | 0.55 ± 0.05 (−72%) | 2.5 ± 0.33 (−12%) | >100 (−16%) |
| R-C-DOT | <100 (+100%) | 1.10 ± 0.04 (−64%) | 7.5 ± 0.23 (−22%) | >100 (−14%) |
The carrier decay of the B-C-Dots (0.25 ± 0.06 ps, 2.0 ± 0.17 ps) was much faster than for the G-C-Dots (0.55 ± 0.05 ps, 2.5 ± 0.33 ps) and the R-C-Dot (1.10 ± 0.04 ps, 7.5 ± 0.23 ps), which indicated that formation of a deep trap state increased the amount of graphitic nitrogen atom in the sp2 network of the core state. The decay kinetics of GSB at 420 nm of all the emitting C-Dots can be helpful for exploring the charge-carrier dynamics (Fig. 4b) of the surface state. The GSB decays (420 nm) could be fitted tri-exponentially with a single-exponential pulse-width-limited growth (<100 fs) and multiexponential decay, as shown in Table 2. It is expected that the excitons were generated at a lower energy level since the excitation energy (380 nm) was higher than the GSB of the surface state (420 nm). Thus, the fast component (τ1) of the GSB recovery dynamics represents the non-radiative electron relaxation process between the trap states of the core to the surface states which becomes faster from the B- to G- to the emitting-C-Dots. The intermediate lifetime (τ2) implied a non-radiative carrier-relaxation process in the surface states and the longer lifetime (τ3) was most likely due to the recombination process related to the oxygen-containing functional groups on the surfaces of the C-Dots. Interestingly, we observed that τ1 became faster and τ2 got slower for the R-C-Dots (0.12 ± 0.044 ps, 5.7 ± 0.486 ps) and G-C-Dots (0.17 ± 0.025 ps, 5.6 ± 0.351 ps) compared to the B-C-Dots (0.23 ± 0.041 ps, 3.6 ± 0.293 ps) due to increase in the oxygen-containing functional groups on the surface of the R-C-Dots compared to the G- and the B-C-Dots. These functional groups can act as electron trap states on the surfaces of the C-Dots.
| Sample | GSB (420 nm) | |||
|---|---|---|---|---|
| τ g (fs) (ag%) | τ 1d (ps) (a1d%) | τ 2d (ps) (a2d%) | τ 3d (ps) (a3d%) | |
| B-C-DOT | <100 (+100%) | 0.23 ± 0.041 (−53%) | 3.6 ± 0.293 (−46%) | >50 (−1%) |
| G-C-DOT | <100 (+100%) | 0.17 ± 0.025 (−50%) | 5.6 ± 0.351 (−47%) | >50 (−3%) |
| R-C-DOT | <100 (+100%) | 0.12 ± 0.044 (−56%) | 5.7 ± 0.486 (−39%) | >50 (−5%) |
To examine the origin of the red emission and the carrier dynamics in the C-Dots, TA measurements were carried out selectively by exciting the R-C-Dots at 500 nm and probing in the visible range at different time delays, as shown in Fig. 5. Two negative signals could be observed for the R-C-Dots due to GSB and SE at 520 nm and a broad peak at 540–620 nm, corresponding to the energy band structure in the steady-state absorption and PL spectra (Fig. 5a). Here the presence of another GSB (520 nm), along with the GSB at 340 nm and 420 nm, was responsible for the red emission from the C-Dots. To follow the carrier-relaxation dynamics of the R-C-Dots, we monitored the GSB kinetics (520 nm), as shown in Fig. 5b. Interestingly, the GSB decay could be fitted bi-exponentially with time constants of τ1 = 10.5 ps (53%), and τ2 = >100 ps (5%). The 10.5 ps component could be attributed to the electron relaxation from the functional groups with less oxygen-rich surface functional groups and the longer components (>100 ps) could be assigned to the carrier relaxation occurring from a hybrid state. It was clear from the findings that the highly oxygen-rich surface functional groups were responsible for the red emission in the C-Dots. The different excited-state carrier-relaxation processes of the R-C-Dots are shown in Scheme 2.
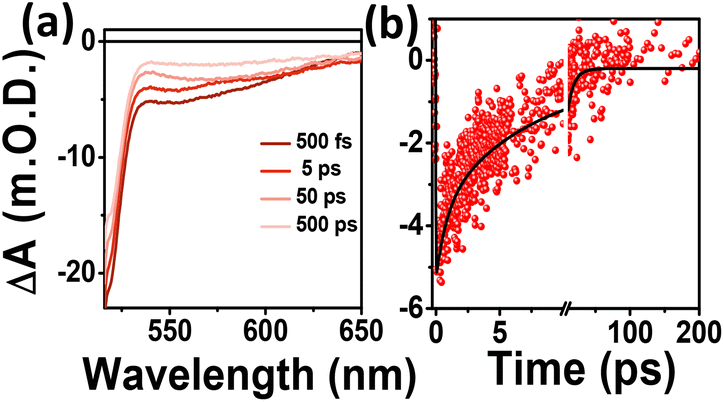 | ||
| Fig. 5 (a) TA spectra and (b) kinetics of GSB (525 nm) of R-C-Dots after excitation at 500 nm. | ||
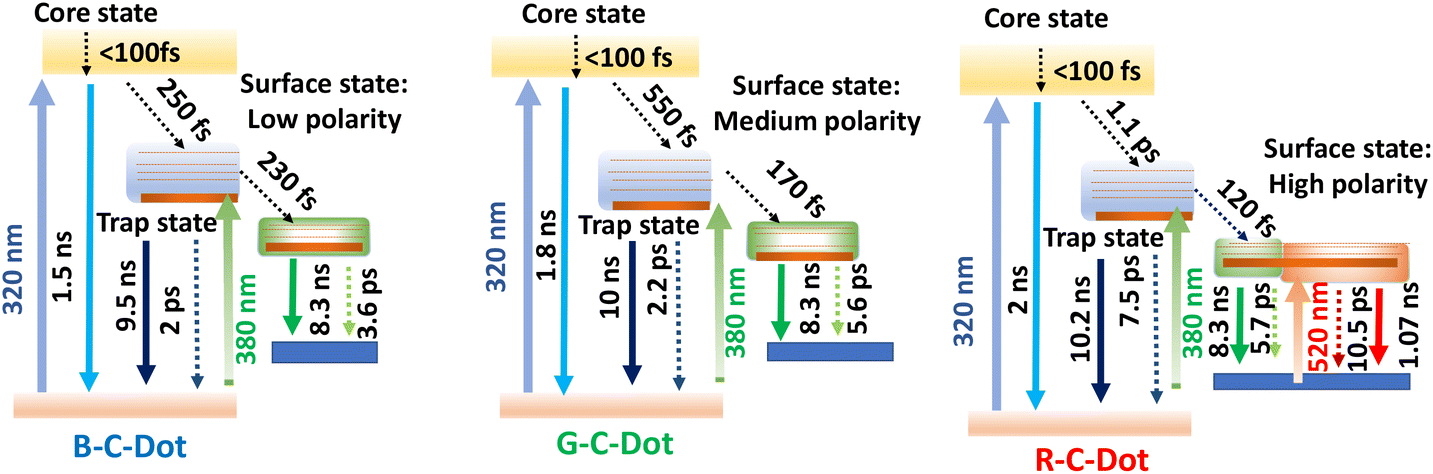 | ||
| Scheme 2 Schematic representation of different relaxation pathway with time scale after selective exaction at core and surface state. | ||
The stability of the different C-Dots was measured by monitoring the steady-state fluorescence intensity at the emission maximum (440 nm) when excited at 340 nm after irradiation with a constant power (∼75 Watt mercury lamp), as shown in Fig. S10a.† In addition, the steady-state PL luminescence intensity was measured before and after the TA to observe the stability of these C-Dots (Fig. S10b†). Both results showed that there were no changes in the emission wavelength and intensity of the sample, which clearly indicated that there was no deformation before or after the TA measurements.
All our results here imply there were different paths of carrier relaxation in the emitting C-Dot-variants. The observations regarding the carrier-relaxation time via different radiative and non-radiative processes are depicted in Scheme 2. The steady-state absorbance and PL measurements in this work indicated that the three different PL maxima for the B-, G-, and R-C-Dots originated from the same particulates. Time-resolved PL kinetics indicated that the excitons were recombined via two different radiative channels in the core state: one is direct e–h recombination and the other one is trap-mediated e–h recombination. It was observed that the polarity of the surface states increased from the B-C-Dots (less polarity) to the G-C-Dots (medium polarity) to the R-C-Dots (high polarity) due to the presence of the different oxygen-containing functional groups in the surface states. The fact that the dynamics of the core states on the trap states strongly depends on the presence of graphitic nitrogen, clearly indicating the existence of similar type of core state (sp2 network) for all the emitting C-Dots. The ultrafast carrier dynamics in the core state indicate that the excited carriers were relaxed from core state to the surface states via the trap states, as shown in Scheme 2, which strongly depended on the hybrid states that were formed by mixing the graphitic nitrogen in the core state and the different oxygen-containing functional groups on the surface of the C-Dots. In the surface states, a rise component was observed in the PL decay upon excitation at 420 nm, which indicated the occurrence of slow solvent relaxation in the picosecond time scale. Moreover, the solvent relaxation time progressively increased from the B-C-Dots to the R-C-Dots due to the presence of more polar surface states on the R-C-Dots, which ensured that the origin of the green and red fluorescence was from the dipole emission centre, i.e. dipole–dipole interactions between C-Dots and solvent molecules.
Conclusion
The excited-state carrier dynamics of differently emitting C-Dots were explored for the first time to find the origin of their fluorescence using steady-state, ultrafast time-resolved absorption, and emission techniques. Therefore, we synthesized pure blue-, green-, and red-emitting C-Dots using the same starting materials and separated them by column chromatography. In the steady-state PL studies, we observed three different emission maxima for all the emitting C-Dots after purification. Time-resolved PL measurements showed that all the emitting C-Dots contained two different emissive centres, namely, the core and surface states, where the core state was the same for all the emitting C-Dots with different extents of defects. Thus, the graphitic carbon core was responsible for blue emission for all the emitting C-Dots, which is an inherent property of all C-Dots. A growth component was observed in the time-resolved PL measurements when the 530 nm emission was monitored after excitation at 340 and 405 nm. This indicated that the surface states, containing different oxygen-containing functional groups, underwent solvation and controlled the emission maximum via creating different centres of fluorescence in the C-Dots. Ultrafast TA measurements showed two separated GSB and SE bands after the excitation at 320, 380, and 500 nm, clearly indicated that three different states were responsible for the fluorescence. The dynamics at the core state strongly depended on the graphitic nitrogen through the formation of new hybrid states. The dynamics of the surface states indicated that the green and red emissions originated from the oxygen-containing functional groups of the C-Dots, which acted as trapping states. Moreover, the dynamics of the surface states strongly depended on the oxygen-rich functional groups, which made the surface more polar. Here, we suggested three different models to explain the relaxation pathways in the B-, G-, and R-C-Dots within their time scales. Table S3† shows the origin of the emission centre and the novelty of our work, where the same kind of source was used to synthesize the different-colour-emitting C-Dots (B-C-Dots, G-C-Dots, and R-C-Dots). This new finding will help to develop new C-Dot-based systems for significantly serving the areas of photocatalysis, photosensitization, and for preparing optoelectronic devices.Experimental section
Materials
Citric acid and urea were obtained from Sigma-Aldrich, USA, and used without further purification. Water of high purity was used for all the experiments.Synthesis of the C-Dots
Here, 1.0 g of citric acid and 2.0 g of urea (1:2 molar ratio) were dissolved in 10 mL of DMF. The solution was then sonicated until it became clear, and was then poured into a Teflon-lined stainless-steel autoclave and heated at 180 °C for 12 h. The black product was filtered using Whatman filter paper to remove unwanted particles. The samples were purified by silica column chromatography (100–200 mesh) using dichloromethane (DCM) and methanol (MeOH) as the eluents. The B-, G-, and R-C-Dots were collected at different polarities. The polarity ratios of the MeOH and DCM mixtures (eluants) were 3%–5%, 6%–8% and 25%–30% for the B-, G-, and R-C-Dots, respectively.19
Microscopic measurements
The size of the C-Dots was analyzed by transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HR-TEM). A JEOL-2100F microscope, equipped with a 200 kV electron source, was used to take the images. The samples were prepared by simply drop-casting on carbon-coated copper grids followed by overnight drying in air. The size distribution was extracted from the TEM images by ImageJ software. The surface characterizations were done using a Bruker ALPHA FTIR spectrometer and HR-800, HORIBA JOBIN YVON Micro Raman spectrometer.Spectroscopic measurements
The absorption spectra of the samples were measured using a Hitachi U-2900 spectrophotometer. A PTI QM-40 spectrofluorometer was used for taking the emission spectra at room temperature (25 °C). The time-resolved fluorescence experiments were performed using a Horiba Jobin Yvon Fluoroscope instrument with 340 nm and 408 nm diode lasers (with a temporal resolution of <200 ps) as the excitation sources and the data were collected by using a time-correlated single-photon-counting method. The χ2 values of the fits signify the fitting quality.The following equation was used for the multiexponential decays to calculate the average lifetime from the fitted data,
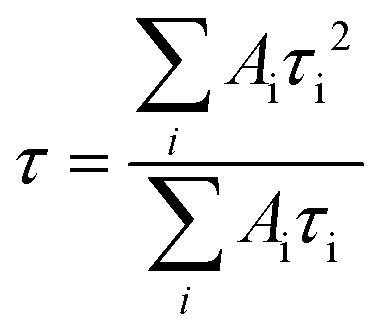
A K-Alpha X-ray photoelectron spectrometer system with an Al K micro focus monochromator with changeable operating energy in the range of 100–4000 eV was used for the X-ray photoelectron spectroscopy (XPS) measurements.
Femtosecond pump–probe-based TA studies were done using a HELIOS TA spectrometer (Ultrafast Systems). The laser system was based on a regenerative amplifier (Spitfire Ace Ti:Sapphire Amplifier, Spectra-Physics) with a pulse duration of ∼80 fs and repetition rate of 1 kHz, seeded by a Ti:Sapphire oscillator 800 nm. The output of the amplifier was split into pump and probe beam lines, where the pump beam was produced by passing part of the regenerative amplifier through an optical parametric amplifier (TOPAS, Light Conversion), altered to the required wavelength and then fed into the spectrometer through a synchronized chopper, thus generating 500 Hz synced signals. The probe beam path was followed through a computer-managed delay stage onto a CaF2 crystal to achieve white light continuum. The data analysis was carried out using Surface Xplorer software. The TA experiments were done using a Quartz cell with a 2 mm sample path length.39
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. thanks the DST-SERB (grant no SRG/2020/000936) for financial support. S. B. and N. G. acknowledge the Council for Scientific and Industrial Research and Indian institute of Science Education and Research-Kolkata and Institute of Nano science and Technology, Mohali, India for research fellowship.References
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- S. Mondal, N. Ghorai, S. Bhunia, H. N. Ghosh and N. Amdursky, Chem. Sci., 2021, 12, 8731–8739 RSC.
- K. Yuan, X. Zhang, R. Qin, X. Ji, Y. Cheng, L. Li, X. Yang, Z. Lu and H. Liu, J. Mater. Chem. C, 2018, 6, 12631–12637 RSC.
- Y. Liu, H. Gou, X. Huang, G. Zhang, K. Xi and X. Jia, Nanoscale, 2020, 12, 1589–1601 RSC.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- A. Sharma, T. Gadly, A. Gupta, A. Ballal, S. K. Ghosh and M. Kumbhakar, J. Phys. Chem. Lett., 2016, 7, 3695–3702 CrossRef CAS PubMed.
- A. Das, V. Gude, D. Roy, T. Chatterjee, C. K. De and P. K. Mandal, J. Phys. Chem. C, 2017, 121, 9634–9641 CrossRef CAS.
- A. Das, D. Roy, C. K. De and P. K. Mandal, Phys. Chem. Chem. Phys., 2018, 20, 2251–2259 RSC.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- G. A. M. Hutton, B. Reuillard, B. C. M. Martindale, C. A. Caputo, C. W. J. Lockwood, J. N. Butt and E. Reisner, J. Am. Chem. Soc., 2016, 138, 16722–16730 CrossRef CAS PubMed.
- C. Ding, A. Zhu and Y. Tian, Acc. Chem. Res., 2014, 47, 20–30 CrossRef CAS PubMed.
- F. R. Baptista, S. A. Belhout, S. Giordani and S. J. Quinn, Chem. Soc. Rev., 2015, 44, 4433–4453 RSC.
- P. Miao, K. Han, Y. Tang, B. Wang, T. Lin and W. Cheng, Nanoscale, 2015, 7, 1586–1595 RSC.
- X. T. Zheng, A. Ananthanarayanan, K. Q. Luo and P. Chen, Small, 2015, 11, 1620–1636 CrossRef CAS PubMed.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- P. Yu, X. Wen, Y.-R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 25552–25557 CrossRef CAS.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS PubMed.
- L. Wang, S.-J. Zhu, H.-Y. Wang, S.-N. Qu, Y.-L. Zhang, J.-H. Zhang, Q.-D. Chen, H.-L. Xu, W. Han, B. Yang and H.-B. Sun, ACS Nano, 2014, 8, 2541–2547 CrossRef CAS PubMed.
- V. Strauss, J. T. Margraf, C. Dolle, B. Butz, T. J. Nacken, J. Walter, W. Bauer, W. Peukert, E. Spiecker, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2014, 136, 17308–17316 CrossRef CAS PubMed.
- N. Soni, S. Singh, S. Sharma, G. Batra, K. Kaushik, C. Rao, N. C. Verma, B. Mondal, A. Yadav and C. K. Nandi, Chem. Sci., 2021, 12, 3615–3626 RSC.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Adv. Mater., 2018, 30, 1704740 CrossRef PubMed.
- K. Holá, M. Sudolská, S. Kalytchuk, D. Nachtigallová, A. L. Rogach, M. Otyepka and R. Zbořil, ACS Nano, 2017, 11, 12402–12410 CrossRef PubMed.
- Z. Tian, X. Zhang, D. Li, D. Zhou, P. Jing, D. Shen, S. Qu, R. Zboril and A. L. Rogach, Adv. Opt. Mater., 2017, 5, 1700416 CrossRef.
- S. Mondal, A. Yucknovsky, K. Akulov, N. Ghorai, T. Schwartz, H. N. Ghosh and N. Amdursky, J. Am. Chem. Soc., 2019, 141, 15413–15422 CrossRef CAS PubMed.
- S. Bhunia, N. Ghorai, S. Burai, P. Purkayastha, H. N. Ghosh and S. Mondal, J. Phys. Chem. C, 2021, 125, 27252–27259 CrossRef CAS.
- L. Wang, W. Li, L. Yin, Y. Liu, H. Guo, J. Lai, Y. Han, G. Li, M. Li, J. Zhang, R. Vajtai, P. M. Ajayan and M. Wu, Sci. Adv., 2020, 6, eabb6772 CrossRef CAS PubMed.
- H. A. Nguyen, I. Srivastava, D. Pan and M. Gruebele, ACS Nano, 2020, 14, 6127–6137 CrossRef CAS PubMed.
- S. Gavalas and A. Kelarakis, Nanomaterials, 2021, 11, 2089 CrossRef CAS PubMed.
- S. Deshmukh, A. Deore and S. Mondal, ACS Appl. Nano Mater., 2021, 4, 7587–7606 CrossRef CAS.
- B. Zhao, H. Ma, M. Zheng, K. Xu, C. Zou, S. Qu and Z. Tan, Carbon Energy, 2022, 4, 88–114 CrossRef CAS.
- F. Mocci, L. V. Engelbrecht, C. Olla, A. Cappai, M. F. Casula, C. Melis, L. Stagi, A. Laaksonen and C. M. Carbonar, Chem. Rev., 2022, 122, 13709–13799 CrossRef CAS PubMed.
- M. Langer, M. Paloncýová, M. Medveď, M. Pykal, D. Nachtigallová, B. Shi, A. J. A. Aquino, H. Lischka and M. Otyepk, Appl. Mater. Today, 2021, 22, 100924 CrossRef.
- S. Lu, L. Sui, J. Liu, S. Zhu, A. Chen, M. Jin and B. Yang, Adv. Mater., 2017, 29, 1–6 Search PubMed.
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan, G. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J.-J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844–849 CrossRef CAS PubMed.
- H. Ding, S.-B. Yu, J.-S. Wei and H.-M. Xiong, ACS Nano, 2016, 10, 484–491 CrossRef CAS PubMed.
- J. Zhou, Y. Yang and C. Zhang, Chem. Commun., 2013, 49, 8605–8607 RSC.
- S. Ghosh, A. Ghosh, G. Ghosh, K. Marjit and A. Patra, J. Phys. Chem. Lett., 2021, 12, 8080–8087 CrossRef CAS PubMed.
- N. Ghorai and H. N. Ghosh, J. Phys. Chem. C, 2021, 125, 11468–11477 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr04642c |
| ‡ Both authors have equal contributions. |
| This journal is © The Royal Society of Chemistry 2022 |