
Ru-promoted perovskites as effective redox catalysts for CO2 splitting and methane partial oxidation in a cyclic redox scheme†
 *a
*a
Abstract
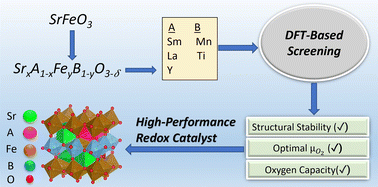
The current study reports AxA′1−xByB′1−yO3−δ perovskite redox catalysts (RCs) for CO2-splitting and methane partial oxidation (POx) in a cyclic redox scheme. Strontium (Sr) and iron (Fe) were chosen as A and B site elements with A′ being lanthanum (La), samarium (Sm) or yttrium (Y), and B′ being manganese (Mn) or titanium (Ti) to tailor their equilibrium oxygen partial pressures (PO2s) for CO2-splitting and methane partial oxidation. DFT calculations were performed for predictive optimization of the oxide materials whereas experimental investigation confirmed the DFT-predicted redox performance. The redox kinetics of the RCs improved significantly by 1 wt% ruthenium (Ru) impregnation without affecting their redox thermodynamics. Ru-impregnated LaFe0.375Mn0.625O3 (A = 0, A′ = La, B = Fe, and B′ = Mn) was the most promising RC in terms of its superior redox performance (CH4/CO2 conversion >90% and CO selectivity ∼95%) at 800 °C. Long-term redox testing over Ru-impregnated LaFe0.375Mn0.625O3 indicated a stable performance during the first 30 cycles followed by an ∼25% decrease in the activity during the last 70 cycles. Air treatment was effective to reactivate the redox catalyst. Detailed characterizations revealed the underlying mechanism of the redox catalyst deactivation and reactivation. This study not only validated a DFT-guided mixed oxide design strategy for CO2 utilization but also provides potentially effective approaches to enhance redox kinetics and long-term redox catalyst performance.
- This article is part of the themed collection: CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

