
Wet flue gas CO2 capture and utilization using one-dimensional metal–organic chains†
 a
Ryan P.
Loughran,
a
Rebecca
Vismara,
c
Ah-Hyung Alissa
Park
b
and
Kyriakos C.
Stylianou
*a
a
Ryan P.
Loughran,
a
Rebecca
Vismara,
c
Ah-Hyung Alissa
Park
b
and
Kyriakos C.
Stylianou
*a
Abstract
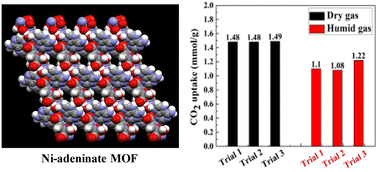
Herein, we describe the use of an ultramicroporous metal–organic framework (MOF) with a composition of [Ni3(pzdc)2(ade)2(H2O)1.5]·(H2O)1.3 (pzdc: 3,5-pyrazole dicarboxylic acid; ade: adenine), for the selective capture of carbon dioxide (CO2) from wet flue gas followed by its conversion to value-added products. This MOF is comprised of one-dimensional Ni(II)-pyrazole dicarboxylate-adenine chains; through pi–pi stacking and H-bonding interactions, these one-dimensional chains stack into a three-dimensional supramolecular structure with a one-dimensional pore network. Upon heating, our MOF undergoes a color change from light blue to lavender, indicating a change in the coordination geometry of Ni(II). Variable temperature ultraviolet-visible (UV/vis) spectroscopy data revealed a blue shift of the d–d transitions, suggesting a change in the Ni-coordination geometry from octahedral to a mixture of square planar and square pyramidal. The removal of the water molecules coordinated to Ni(II) leads to the generation of a MOF with open Ni(II) sites. Nitrogen isotherms collected at 77 K and 1 bar revealed that this MOF is microporous with a pore volume of 0.130 cm3 g−1. Carbon dioxide isotherms show a step in the uptake at low pressure, after which the CO2 uptake is saturated. The step in the CO2 uptake is likely attributable to the rearrangement of the three-dimensional supramolecular structure to accommodate CO2 within its pores. The affinity of this MOF for CO2 is 35.5 kJ mol−1 at low loadings, and it increases to 41.9 kJ mol−1 at high loadings. While our MOF is porous to CO2 and water (H2O) at 298 K, it is not porous to N2, and the CO2/N2 selectivity increases from 28.5 to 31.5 as a function of pressure. Breakthrough experiments reveal that this MOF can capture CO2 from dry and wet flue gas with uptake capacities of 1.48 ± 0.01 and 1.14 ± 0.06 mmol g−1, respectively. The MOF can be regenerated and reused at least three times, demonstrating consistent CO2 uptake capacities. Upon understanding the sorption behavior of this MOF, catalysis experiments show that the MOF is catalytically active in the fixation of CO2 into an epoxide ring for the formation of a cyclic carbonate. The turnover frequency for this reaction is 21.95 ± 0.03 h−1. The MOF showed no catalytic deterioration after two cycles and maintained comparable catalytic activity when dry and wet CO2/N2 mixtures were used. This highlights that both N2 and H2O do not dramatically affect the catalytic activity of our MOF toward CO2 fixation.
- This article is part of the themed collection: CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

