
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA origami book biosensor for multiplex detection of cancer-associated nucleic acids†
Ivana
Domljanovic
a,
Morgane
Loretan
b,
Susanne
Kempter
c,
Guillermo P.
Acuna‡
 *b,
Samet
Kocabey‡
*a and
Curzio
Ruegg‡
*a
*b,
Samet
Kocabey‡
*a and
Curzio
Ruegg‡
*a
aLaboratory of Experimental and Translational Oncology, Department of Oncology, Microbiology and Immunology, Faculty of Science and Medicine, University of Fribourg, Chemin du Musée 18, PER17, 1700 Fribourg, Switzerland. E-mail: samet.kocabey@unifr.ch; curzio.ruegg@unifr.ch
bPhotonic Nanosystems, Department of Physics, Faculty of Science and Medicine, University of Fribourg, Chemin du Musée 3, PER08, 1700 Fribourg, Switzerland. E-mail: guillermo.acuna@unifr.ch
cDepartment of Physics, Ludwig-Maximilians-University, Geschwister-Scholl-Platz 1, 80539 Munich, Germany
First published on 11th October 2022
Abstract
DNA nanotechnology provides a promising approach for the development of biomedical point-of-care diagnostic nanoscale devices that are easy to use and cost-effective, highly sensitive and thus constitute an alternative to expensive, complex diagnostic devices. Moreover, DNA nanotechnology-based devices are particularly advantageous for applications in oncology, owing to being ideally suited for the detection of cancer-associated nucleic acids, including circulating tumor-derived DNA fragments (ctDNAs), circulating microRNAs (miRNAs) and other RNA species. Here, we present a dynamic DNA origami book biosensor that is precisely decorated with arrays of fluorophores acting as donors and acceptors and also fluorescence quenchers that produce a strong optical readout upon exposure to external stimuli for the single or dual detection of target oligonucleotides and miRNAs. This biosensor allowed the detection of target molecules either through the decrease of Förster resonance energy transfer (FRET) or an increase in the fluorescence intensity profile owing to a rotation of the constituent top layer of the structure. Single-DNA origami experiments showed that detection of two targets can be achieved simultaneously within 10 min with a limit of detection in the range of 1–10 pM. Overall, our DNA origami book biosensor design showed sensitive and specific detection of synthetic target oligonucleotides and natural miRNAs extracted from cancer cells. Based on these results, we foresee that our DNA origami biosensor may be developed into a cost-effective point-of-care diagnostic strategy for the specific and sensitive detection of a variety of DNAs and RNAs, such as ctDNAs, miRNAs, mRNAs, and viral DNA/RNAs in human samples.
1. Introduction
Cancer-associated nucleic acids, including circulating cell-free, tumor-derived DNA fragments (ctDNAs) carrying tumor-specific sequence alterations or circulating microRNAs (miRNAs), are increasingly considered as potential cancer biomarkers for use in non-invasive early cancer diagnostics and disease monitoring strategies.1 These nucleic acids are present in low abundance in the blood of cancer patients and their concentrations and profiles are significantly different in comparison to those of healthy individuals, making them ideal tumor biomarkers.2 Currently, ctDNA and miRNAs are typically detected by quantitative real-time PCR (qRT-PCR), next-generation sequencing (NGS), or microarray hybridization techniques. Regardless of their enormous analytical progress, these methods have some limitations. Most typically, they are complex, involve multiple steps, demand expensive instrumentation, and are time-consuming. In addition, methods based on direct oligonucleotide hybridization may be hampered by the difficulty to discriminate ctDNA fragments differing only by single (point) mutations. This is also the case for highly homologous miRNAs that differ by only one single base which makes it equally difficult to discriminate between different family members.3 Importantly, detection of multiple biological molecules in complex conditions is becoming increasingly frequent for early diagnosis and monitoring. Therefore, there is a growing and unfulfilled need for more efficient approaches and tools to detect cancer-associated nucleic acids with high sensitivity, sequence specificity, and high output for use in clinical and medical routines.4 These facts lead to a growing demand for point-of-care diagnostic nanoscale devices that are easy to use and cost-effective alternatives to expensive diagnostic devices, but also highly sensitive and specific.5–7DNA nanotechnology, especially DNA origami, enables the generation of nanoscale-sized structures with precisely defined geometries and functional modalities that can be engineered into highly specific, rapid, high throughput analytical devices with single-molecule sensitivity for biomedical applications.8,9 Importantly, DNA origami empowered the expansion of “dynamic” structures, in which their function depends on conformational changes induced by external stimuli such as DNA-based strand-displacement reactions.10,11 Conformational changes can generate a measurable signal that can be exploited to sense the external stimuli.12 The measurable signal can be detected by optical and electrochemical techniques.13,14 In the case of optical detection, for example, DNA origami structures can be functionalized with fluorophores.13 One intrinsic feature of DNA self-assembly is the ability to generate precise and predetermined three-dimensional arrangement of fluorophore networks to produce strong optical signals.15,16 This precise arrangement of multiple fluorophores can be used to create Förster resonance energy transfer (FRET) and/or quenching networks.17–19 These FRET-based DNA photonic networks are especially relevant for multiplex detection applications. One of the first DNA origami structures created to detect small oligonucleotides (viral RNA or miRNA) was reported by Andersen et al.20 With the advances in the field of nanotechnology, in particular, the development of the DNA origami method, more complex and advanced structures were generated, that were able to detect low concentrations of nucleic acids.21–25
Here, we presented a DNA origami book biosensor for the dual detection of specific target oligonucleotides. Our biosensor offers two different FRET-based mechanisms for the detection: (1) a FRET-based detection system that can reveal the presence of one target of interest and (2) a fluorescence quenching-based detection system that can simultaneously detect two different targets. Within our structure, we can incorporate 20 FRET and/or 20 quenching fluorophores pairs on each side of the book. In both FRET-based detection systems, our book biosensor has a high-intensity output signal at the single molecule level. We demonstrated that our device can detect target oligonucleotides at concentrations as low as 1–10 pM with a detection time of 10 min via both detection mechanisms. Importantly, our approach is highly versatile, as the locks can be easily redesigned to detect different oligonucleotides including different short tumoral ctDNA fragments or miRNAs, viral DNA, or RNA fragments (e.g. SARS-CoV-2, Zika, or HPV), or even longer mRNAs through the combination of multiple locks. When applied in a matrix form it can be expanded to detect hundreds of different targets simultaneously, possibly changing the way oligonucleotide-based diagnostics will be made in the future.
2. Experimental section
2.1. Design and assembly of DNA origami book biosensor
The structure was designed using caDNAno software (https://cadnano.org/).26 The generated file from caDNAno software was submitted to CanDo (Computer-aided engineering for DNA origami, https://cando-dna-origami.org/) for further evaluation of the shape, flexibility, and dynamics. The assembly of the DNA origami book biosensor is achieved by mixing 10 nM single-stranded scaffold DNA (type: p8064, Europhins MWG Operon, Ebersberg, Germany) with unmodified staple strands (100 nM each) (HPSF purified, LubioScience-Switzerland IDT, Zurich, Switzerland) and 1 μM of modified staple strands with fluorophores (ddUTP-Cy3 and ddUTP-Cy5, Jena Bioscience, Germany), oligonucleotides with bh2 and bbq650q (Biomers, Ulm, Germany) and 1 μM of biotin modified oligonucleotides (Biomers, Ulm, Germany). All modified oligonucleotides were purified by HPLC. The sequences of all oligonucleotides used are given in Tables S1 and S2.† The annealing of the scaffold DNA and staple strands mix was done by using the temperature ramp method: heating the mix solution to 80 °C for 5 min, cooling to 65 °C during the first 15 min, and cooling further down to 4 °C for 16 h.2.2. Labeling of oligonucleotides with Cy3 and Cy5 fluorophores by terminal transferase (TdT)
Unmodified staple strands were purchased at a concentration of 100 μM. Selected ones were labelled with ddUTP-Cy3, and ddUTP-Cy5, using Terminal Transferase (TdT) (Roche, Switzerland). TdT reaction was performed in a total volume of 20 μL consisting of TdT reaction buffer (1×), CoCl2 (5 mM), 100 pmol unmodified staple strands and 500 pmol of modified ddUTP conjugates. The solution was mixed well and incubated with TdT (400 U per reaction) for 30 min at 37 °C. TdT reaction was stopped by heating the mixture at 70 °C for 10 min and the oligonucleotides were precipitated with NaOAc (0.1 M, pH 5.2) and 2.5 volumes of 96% EtOH for 1 h at −20 °C. Precipitation followed by centrifugation for 30 min at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g resulted in a pellet of the product, which was washed with 70% EtOH, dried and dissolved in water. These labelled oligonucleotides were analyzed on HPLC (Agilent Technologies 1260 Infinity II system) with an Agilent Bio SAX, non-porous 4.6 mm × 250 mm, 5 μm HPLC column, and then used for self-assembly (Fig. S11†).
000g resulted in a pellet of the product, which was washed with 70% EtOH, dried and dissolved in water. These labelled oligonucleotides were analyzed on HPLC (Agilent Technologies 1260 Infinity II system) with an Agilent Bio SAX, non-porous 4.6 mm × 250 mm, 5 μm HPLC column, and then used for self-assembly (Fig. S11†).
2.3. Agarose gel mobility shift assays
Assembled DNA origami book biosensors were loaded onto the 1.5% agarose gel containing 11 mM MgCl2 and let migrate for 2 hours at 70 V. To detect the migrated structures, gels were stained with 1× SYBR Gold for 30 min and visualized with BioRad Gel Doc XR+.2.4. Structure purification
DNA origami book biosensor structures were assembled with an excess of staple strands and purification of unincorporated staple strands was done with two different methods. The first method was based on 100 kDa Amicon Ultra-0.5 mL centrifugal filters (Millipore, Germany). Amicon filters were pre-wet by filling with annealing buffer (1× TAE/10 mM Mg2+) with 500 μL and centrifuged at 8000g for 10 min. Then, the samples were loaded onto the filter (100 μL), and completed to 500 μL with the annealing buffer. The sample was centrifuged at 8000g for 8 min, and washed 4 times with 400 μL of annealing buffer. The second method used for purification was agarose gel electrophoresis. The sample containing assembled book biosensor structure (100 μL) and 6× glycerol (20 μL) was loaded into 1% agarose gel. The gel was run on 70 V for 1 h and the corresponding band for fluorescently labelled structures was cut out from the gel using a razor blade and extracted from the gel by squeezing with a glass slide.2.5. AFM imaging
Freshly cleaved mica was incubated with 10 μL of annealing buffer containing 2 mM MgCl2 for 1 min and rinsed with ddH2O before deposition of the purified sample (5–10 μL of 0.8–1 nM sample) for 2 min. Imaging was performed using an AFM NT-MDT (NTEGRA II). All images were analyzed using Gwyddion analysis software.2.6. TEM Imaging
TEM carbon grids (Formvar/carbon, 400 mesh, Cu, TedPella, Inc., USA) were plasma-exposed for 1 min (24 W). A sample (5 μL) was placed and incubated on the grid for 30 s and then removed with filter paper. Subsequently, samples were negatively stained with 1% Uranyl acetate aqueous solution (5 μL) containing 25 mM NaOH for 15 s and excess stain solution was removed with filter paper. Imaging was performed using a JEM-1011 transmission electron microscope (JEOL) operated at 80 kV.2.7. Fluorescence measurements of DNA origami book biosensors by spectrophotometry
A solution containing the self-assembled and purified DNA origami book biosensor structures (45 μL, 0.5 nM), were loaded into a cuvette with a final volume of 60 μL (Hellma). Fluorescence measurements was done by using a spectrofluorometer (Horiba, Duetta Fluorescence and Absorbance Spectrometer) and emission spectra of the different book biosensors were collected after excitation of donor fluorophores at 530 nm and acceptor fluorophores at 630 nm.2.8. Single-molecule measurements by wide-field fluorescence microscopy
The 12-well glass slide (Ibidi, Germany) with removable silicon chambers was incubated with 50 μL of 0.5 mg mL−1 biotinylated bovine serum albumin solution (Sigma-Aldrich, Buchs, Switzerland) for 15 min at room temperature, washed three times with 1× TAE/12 mM Mg2+, and incubated with 50 μL of 0.5 mg mL−1 neutravidin solution (Thermo Fisher Scientific, Basel, Switzerland) for 15 min and washed with 1× TAE/12 mM Mg2+. Biotinylated DNA origami book samples (100 pM in 1× TAE/12 mM Mg2+) were incubated for 15 min and washed three times with 1× TAE/12 mM Mg2+. The mix of the oxygen-scavenging system consisted of a 1:1:1 ratio of 1× PCA, 1× PCD, and 1× Trolox mix in 1× TAE/12 mM Mg2+ was added into the glass slide chambers where structures were functionalized. In particular, the oxygen-scavenging system was prepared from the stock solutions. The 100× Trolox stock solution contains 100 mg of Trolox, 430 μL of methanol, and 345 μL of NaOH (1 M) in 3.2 mL of water. The 40× PCA stock solution contains 154 mg of PCA in 10 mL of water (pH 9.0). The 100× PCD stock solution contains 9.3 mg of PCD and 13.3 mL of buffer (50% glycerol stock in 50 mM KCl, 1 mM EDTA, and 100 mM Tris–HCl, pH 8.0).
Experiments were performed on a home-built TIRF wide-field microscope, based on an inverted Olympus IX83 body. Laser lines of 532 and 640 nm were used (gem, Laser quantum), filtered by a clean-up filter (ZET532/640×). Dichroic Mirrors (DM) were used to join the path and Pol + λ/4 is included for circular polarization. The laser light then was focused by two lenses (AC508-100-A-ML and AC254-030-A-ML, Thorlabs, Germany) into the back focal plane of the UPLAPO100xOHR (1.5 NA, Olympus) objective. After, a Laser Dual Band Set (ET-532/640 nm, DM3) was used to excite the sample and filter the emission simultaneously. The emission was split into two channels using DM4 (T635lpxr) into an Optosplit II bypass (Cairn) system, to finally reach the chip of the CMOS camera (C14440 ORCA-Fusion, Hamamatsu). To image the structures with FRET-based detection, we used 532 nm laser at 1.0 mW for Cy3, and for the structures with a quenching-based detection, we used lasers 532/640 nm at 1.0 mW for Cy3/Cy5 with direct excitation. After excitation, images were taken every 2 min for 10 min, and data points were collected using two channels: the first one for Cy3 emission and the second one for Cy5 emission. For the quenching-based detection mechanism, the direct excitation of Cy3 or Cy5 was collected using the respective channel.
Data analysis was performed on a home-developed Python software. With this software, we were able to find an individual structure, fit them by using a 2D Gaussian model to the center of the section and extract its intensity from all the relevant pixels in each image taken. Energy-transfer efficiency for single-molecule analysis was calculated by:

For quenching-based detection, the increase of fluorescence intensity was calculated in the way that intensity at 0 min period is subtracted from 6 min. The difference between the two intensities was then divided by the 0 min of the same DNA origami book biosensor. The final data point was an increase in intensity over time.
2.9. Isolation of miRNAs
MCF-7 human breast cancer cells were cultured at 37 °C, 5% CO2, and 95% humidity in Dulbecco's modified Eagle's medium (DMEM) supplemented with Glutamax, 10% fetal bovine serum (FBS), and 1% Penicillin and Streptomycin. All cell culture reagents were purchased from Thermo Fischer Scientific (Basel, Switzerland). MiRNAs were extracted from MCF-7 cells (25 ng μL−1 for 106 cells) using a commercial system (High Pure miRNA Isolation Kit; Roche, Switzerland). Extraction was done as described in the kit protocol.3. Results and discussion
3.1. Design of DNA origami book biosensor
Inspired by the DNA origami beacon design by Selnihhin et al.,27 we wanted to construct a biosensor capable of detecting multiple oligonucleotides and at the same time that has a high output signal to enable detection with low sensitivity, simple microscopy setup, for example smartphone-based portable devices.6 The DNA origami book was designed using caDNAno software.26,28 The rectangular structure consists of three layers of DNA helices packed on a square lattice with dimensions of 78.5 × 33.9 × 8 nm (Fig. 1a) such that the upper layer consists of two parts connected to the middle layer with a hinge from the centre, allowing the opening of the upper layer at both sides. Each part of the layer has an array of precisely positioned fluorophores (Fig. 1 and Fig. S1†). For FRET-based detection, two arrays of fluorophores were positioned on the upper and middle layers: one array with donor fluorophores on the middle layer facing the upper layer, and the other one on the upper layer with corresponding acceptor fluorophores (Fig. 1c). Each array consists of 4 columns of 5 precisely located fluorophores (Fig. 1d). For the quenching-based detection approach, the middle layer of the DNA origami book was decorated with an array of different types of fluorophores (Cy3 or Cy5), and the top layer of the book was decorated with quencher molecules (Black Hole Quencher-2 (bh2) and/or Black-Berry Quencher 650 (bbq650)) facing to the corresponding fluorophore in the middle layer (Fig. 1d). The distance between each fluorophore on one layer of DNA within a column is 5.8 nm and the distance between two columns is 6.6 nm, according to our design. The mapping of the whole structure can be found in Fig. S2.† Partially hybridized locks on both sides constitutively keep the layers in a closed state upon assembly (Fig. 1b). Each of the four locks on each side of the book forms 15 bp long duplex where one of the strands is elongated with 8 bases allowing the binding of the complementary target key of 23 bases through toehold-mediated strand displacement. Once bound to the toehold, the target oligonucleotide then displaces the shorter lock strand from the duplex.29 That promotes the opening of the layers due to the electrostatic repulsion of the DNA and the entropy of the device. The change between open and closed state results in increased distance between donor and acceptor fluorophores causing the decrease in the energy transfer between the fluorophore pairs in both mechanisms (Fig. 1c and d) and the increase in the fluorescence signal of the donor fluorophores by releasing the respective FRET pair. Transmission electron microscopy (TEM) images demonstrated the formation of both open and closed states of the DNA origami book biosensor (Fig. 1e, f and Fig. S3†). Furthermore, atomic force microscopy (AFM) images performed in scanning mode showed the formation of DNA origami book sensor in a closed state (Fig. S4†). To further demonstrate the opening of the DNA origami book biosensor, structures were assembled and mixed with one or two DNA oligonucleotide targets and then analysed by agarose gel electrophoresis (Fig. S5†). The differences in electrophoretic shift (mobility) between structures in closed or open states (one or two sides) were observed, thereby demonstrating the functionality of the DNA origami book biosensor in response to the presence of a corresponding oligonucleotide target. | ||
| Fig. 1 Characterization of the DNA origami book biosensor. (a) Representation of the dimensions of the book biosensor in its closed state. (b) Top view of the book biosensor with 4 locks on each side. (c) Side view of the FRET biosensor in the open state. Green and red spheres represent the donor and acceptor fluorophore arrays. In total, 20 FRET pairs were positioned on each side. (d) Side view of the quencher biosensor in the open state. Green, red and black spheres represent the arrays of donor and quencher groups. In total, 20 fluorophore/quencher pairs were positioned on each side. Electron micrographs of DNA origami structures assembled in the (e) closed conformation and (f) open conformation at 12 mM Mg2+ (scale bars 50 nm). Arrows show the opening of the upper layer at the sides of the structures. | ||
3.2. Optimization of the fluorescence output signal
To check the efficiency of incorporation of the labelled oligonucleotides within the structure, we imaged the DNA origami book structures with 1 column (5 Cy3 or Cy5), 2 columns (10 Cy3 or Cy5), and 4 columns (20 Cy3 or Cy5). The increase in the number of fluorophores incorporated within the structure resulted in a linear increase in the mean fluorescence intensity in the absence of self-quenching (Fig. S6†). The signal of the DNA origami book biosensor is based on distance-dependent energy transfer between fluorophores, actually between columns of donor and acceptor/quencher. Opening of the structure will result in a bigger distance between the columns of donor and acceptor/quencher pairs that is closer to the locks compared to the distance between the columns closer to the hinge. Our theoretical calculation of possible angles of opening and distances in nanometers between columns is presented in Fig. 2a.30 This calculation indicated that the distance between columns within the structure will be different, and that would result in variations of the FRET. First, we defined the optimal salt concentration for the maximum signal output of the book structure that was subsequently used for the further functional experiments using a spectrophotometer (Fig. S7†). Then, we studied the FRET efficiency for each column (consisting of 5 donor/acceptor pairs) in the open and close states using a spectrophotometer (Fig. 2b). Results confirmed the theoretical calculations that columns closer to the locks have the highest difference in FRET efficiency between open and closed states (30%) while columns close to the hinge have a very low difference in FRET efficiency (1.6%). To determine the reaction time of the DNA origami book biosensor to fully open, the structures were assembled in the close state with a single column of FRET pairs (column 1). To quantify the FRET efficiencies, fluorescence emission profiles of acceptors after excitation of donors of the biosensor device were recorded by spectrophotometer before and every 5 min after the addition of 1 μM target key over a period of 30 min, and FRET efficiency was calculated (Fig. 2c and d). Plotted FRET efficiency showed a decrease over time which indicated that our structure was opening within 10 min after the addition of the target. Longer incubation times did not improve the FRET efficiency significantly. These results were valuable for the subsequent study at the single-structure level. | ||
| Fig. 2 Determination of FRET efficiency of the DNA origami book biosensor using fluorescence spectroscopy. (a) Calculations of the relative distance between FRET pairs at a theoretical angle of opening of the structure. (b) Fluorescence spectroscopy characterization of FRET performance of individual columns within the structure. (c) Donor excited fluorescence spectra of acceptors at closed and open states of DNA origami book biosensors with 5 FRET pairs. (d) Time-dependent opening of DNA origami book biosensors with 5 FRET pairs. | ||
3.3. Single-structure analysis of DNA origami book biosensors using wide-field fluorescence microscopy
Next, we tested the detection specificity of our book biosensor using different targets for the left side and the right side of the structure separately. We initially used DNA analogs of target miRNAs, ODN-153 (for miR-153) and ODN-342 (for miR-342). The structures with 3 columns (1–3) were imaged every 2 min for a total of 10 min with the target added after one minute. Then, the intensity of DNA origami structures was extracted, and FRET efficiency was calculated (Fig. S8†). As a control experiment, we imaged structures before adding the actual target in the same chamber but at different positions to assess how the laser was affecting the fluorophores and to observe the behaviour of the structures. Our results showed that when non-target DNA is added there is a small change in the mean FRET efficiency for the left (8%) and right (6%) sides of the structure due to exposure to the laser and photobleaching (Fig. 3). The addition of the DNA target (ODN-153) for the left side results in a significant change in the average FRET efficiency (26–30%) for concentrations in the range from 1 μM to 100 pM, while the change for 10 pM was around 10% (Fig. 3a). The addition of the DNA target (ODN-342) for the right side in high concentration triggered the opening of the structure faster than the DNA target for the left side. For the concentrations in the range of 1 μM to 100 pM change in FRET efficiency is approximately 20% while the addition of the 10 pM target resulted in a smaller change of FRET efficiency, 11% (Fig. 3b). The reason for the faster opening in higher concentration could be due to different GC% of the target sequence.31 ODN-342 has higher GC content (52.2%) in comparison to ODN-153 (40.9%) (Table S2†). We estimated the theoretical limit of detection (LoD) of both targets based on the measured normalized counts of the non-target oligonucleotide. By fitting all the measured data points (101 to 106 pM) to an asymmetric five-parameter curve, the LoDs of ODN-153 and ODN-342 were determined at 1.6 pM and 1 pM, respectively. As the DNA origami book biosensor has 4 locks on each side, which keep the structure in a close state, we tested the impact of the number and position of the locks on the detection of the lower concentration of the target of interest using the FRET mechanism of detection. Structures with 3 columns (1–3) were assembled with different numbers (2 or 3) and positions of locks followed by the addition of 100 pM of ODN-153. The FRET changes obtained showed that the incorporation of fewer locks or altering the positioning of the locks within the structure did not increase the mean FRET intensity at the 100 pM concentration of the target of interest (Fig. S9†). | ||
| Fig. 3 Detection limit of the book biosensor with 3 columns (1–3) on each side at a single DNA origami level. (a) Opening at the left side of the DNA origami book upon addition of ODN-153. (b) Opening at the right side of the DNA origami book upon addition of ODN-342. The red and green histograms represent FRET efficiency distribution before (0 min) and after (6 min) the addition of target and non-target (control) ODNs at different concentrations. | ||
Furthermore, we tested the detection ability of the book biosensor using the fluorescence quenching mechanism as an alternative FRET-based detection approach. For this, the structures were assembled with 2 columns (1–2) of Cy3 dyes and bh2 quenchers on the left side and imaged every 2 min for a total of 10 min, with the target DNA (ODN-153) added after one minute. The intensity of DNA origami structures was extracted and the change of fluorescence intensity between 0 min and 6 min was calculated. Upon binding of the target of interest, the upper layer opens and the fluorescence intensity of Cy3 increases due to the increased distance between Cy3 and bh2 quencher dyes. The results showed that the addition of ODN-153 in the range of 100 pM to 1 μM increased the fluorescence intensity (14 to 20%), while the addition of 10 pM increases the fluorescence intensity only by 7%.
The intensity change between closed and open structures can be seen in the histograms (Fig. 4a). The same experiment was repeated with book biosensors containing 2 columns (1–2) of Cy5 dyes and bbq650 quenchers on the right side of the structure. Our results showed the addition of ODN-342 in the range of 100 pM to 1 μM resulted in an increase in the Cy5 fluorescence intensity in the range of 11 to 20%, while the addition of 10 pM target ODN increased the fluorescence intensity by 8% (Fig. 4b). By fitting all the measured data points (101 to 106 pM) to an asymmetric five parameter curve, the LoDs of ODN-153 and ODN-342 were determined as 3.3 pM and 3.9 pM, respectively.
 | ||
| Fig. 4 Detection limit of the book biosensor with 2 columns (1–2) of dye-quencher pairs on each side at a single DNA origami device level. (a) Opening at the left side of the DNA origami book upon addition of ODN-153. (b) Opening at the right side of the DNA origami book upon addition of ODN-342. The red and green histograms represent fluorescence intensity increase after the addition of the target of interest due to the opening of the structure. A yellow histogram represents fluorescence intensity in the closed state. | ||
3.4. Multisensing of two target miRNAs and miRNAs extracted from cancer cells
Next, we performed the multiplex detection of two targets by adding both oligonucleotides simultaneously. The structures were assembled by including 2 columns (1–2) of Cy3/bh2 dye-quencher pairs on the left side and 2 columns (1–2) of Cy5/bbq650 dye-quenchers pairs on the right side. We applied the same procedure described above by adding two target DNAs (ODN-153 and ODN-342) at the same time in the range of 10 pM to 10 nM. Results showed that the fluorescence intensity of both Cy3 and Cy5 dyes were increased (12 to 20%) when targets were added in the concentration range of 100 pM to 10 nM, whereas the fluorescence intensity remained at 10% at the concentration of 10 pM (Fig. S10†).Furthermore, to test the RNA detection ability of our system we chose two highly expressed miRNAs (miR-21 and let-7a) by redesigning the locks that are responding to these targets respectively (Tables S1 and S2†). As above, the structures were assembled by including 2 columns (1–2) of Cy3/bh2 dye-quencher pairs on the left side and 2 columns (1–2) of Cy5/bbq650 dye-quenchers pairs on the right side. The locks on the left side of the biosensor were designed to detect miR-21 and the locks on the right side were designed to detect let-7a, so that increase in Cy3 fluorescence intensity reveals the detection of miR-21 while an increase in Cy5 fluorescence intensity reveals the detection of let-7a. Testing was done at miRNAs concentrations of 10 nM and 10 pM. Results showed that the fluorescence intensity of Cy3 and Cy5 after the addition of the 10 nM target was increased to 18% to 19% respectively, indicating the concomitant detection of both miRNAs at these concentrations (Fig. 5). The fluorescence intensities of both dyes after adding the 10 pM target were approximately 10%.
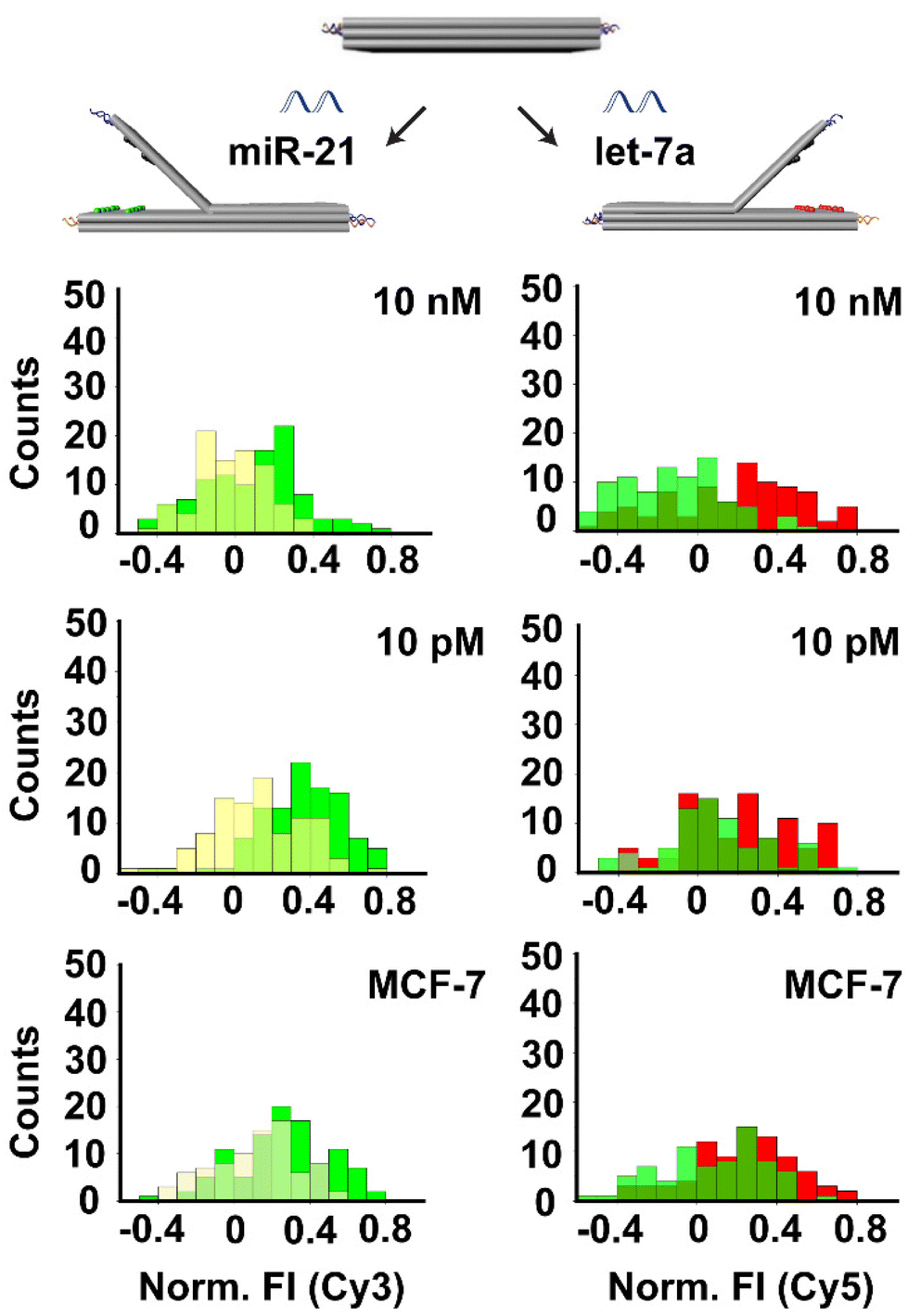 | ||
| Fig. 5 Simultaneous detection of miR-21 and let-7a using book biosensor with 2 columns (1–2) of dye-quencher pairs on each side. Left: detection of miR-21 at 10 nM and 10 pM concentrations. Right: detection of let-7a at 10 nM and 10 pM concentrations. Top and middle: detection of synthetic miR-21 and let-7a. Bottom: detection of miR-21 and let-7a from MCF-7 cell extract. The red and green histograms represent fluorescence intensity increase after the addition of the target of interest due to the opening of the structure. A yellow histogram represents fluorescence intensity in the closed state. | ||
Finally, in order to validate the capability of our assay to detect cell-derived miRNA in biological samples, we studied with miRNAs from MCF-7 breast cancer cell extracts. MiRNA was extracted from MCF-7 cells (25 ng μL−1 for 106 cells) using a commercial system. Then, we added 100 ng of the isolated RNA to our biosensor platform containing single DNA origami structures with locks designed to detect miR-21 and let-7a. Results showed an increase in fluorescence intensity of around 10% for Cy3 and 11% for Cy5, which is in the detection range of our system and above our system`s LoD.
4. Conclusions
In this study, we designed and constructed a dynamic DNA origami book biosensor that can specifically detect cancer-associated oligonucleotides based on the modulation of a strong fluorescence signal. The designed sensor can be operated in a one-step reaction in 10 min and without the need for multi-step manipulations or signal amplification. We demonstrated two different approaches for the detection of targets of interest: FRET and quenching, by incorporating 20 FRET pairs or quenching pairs on each side of the DNA origami book biosensor. Using a quenching-based detection system, we were able to test the option of multisensing capability by concomitantly detecting two distinct targets. The structure signal was characterized by single-molecule analyses as well as detection of all our targets of interest. Our theoretical limit of the detection is in the range of 1–10 pM based on the target sequence tested. In the future, to detect miRNAs in clinical samples, such as miRNAs extracted from serum samples of breast cancer patients, we are aiming to decrease the LoD into a low fM range (1–100 fM). One approach would be the immobilization of these structures into microfluidic chips with a closed circuit pumping system.32 With that, the target contained sample can be flushed on the immobilized origami sensors in a constant and continuous flow to increase binding events. Micro and nanopatterning of the surfaces by the controlled placement of DNA origami sensors in a single-molecule nanoarray platform can also increase the sensitivity of the system and maximize the yield of the generated signal by reducing the false positive counts arising from random immobilization of single structures.33–35Moreover, by using artificial catalysts (macrocrowders such as polyethylene glycol (PEG) or diethylene glycol (DEG)) the sensitivity of the biosensor could be further improved and the detection limit of our structure could be lowered.36,37 Modification of the locks with LNA could also potentially improve further binding of low concentration targets since LNA modification is known as a modification that increases stability and reduces the off-target effect.38 Furthermore, to increase the multiplexing capability and fluorescence intensity of the system, alternative DNA origami sensor designs can be generated by reconfiguring the recent book biosensor. For example, using two hinges on both sides of the DNA origami sheet (instead of one central hinge) where DNA helices on the upper layer are partially connected to each other at the center. In this way, up to 4 targets can be detected simultaneously.
Overall, here we generated a DNA origami book biosensor that can specifically detect target oligonucleotides using fluorescence-based optical readout with low pM sensitivity. With the amplified signal arising from decorated fluorophore arrays on our DNA origami sensor and by possibly integrating the approaches mentioned above into our system, we foresee that we will be able to detect target biomarkers with even higher sensitivity using a portable smartphone microscopy setup,6,39 bringing this biosensor technology one step closer to a point-of-care diagnostics device.
Author contributions
S. Ko. conceptualized the project and designed the DNA origami book biosensor. I. D. and M. L. performed the microscopy experiments, acquired, and analysed the data. S. Ke. performed TEM imaging of DNA origami structures. All of the authors interpreted and discussed the acquired data. I. D. wrote and prepared the original draft. S. Ko. and C. R. reviewed and edited the manuscript. S. Ko., G. P. A., and C. R. supervised the project. C. R. and G. P. A. received the funding. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Centre of Competence in Research Bioinspired Materials (NCCR) to C. R. and G. P. A. S. Ko. acknowledges funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 889031. G. P. A. acknowledges funding from the Swiss National Science Foundation (200021_184687).We thank Dr Maria Taskova for the help with HPLC characterization of oligonucleotide labeling and Dr Mathias Lakatos for the help with AFM measurements and helpful discussions.
References
- T. Ouyang, Z. Liu, Z. Han and Q. Ge, Anal. Chem., 2019, 91, 3179–3186 CrossRef CAS PubMed.
- P. S. Mitchell, R. K. Parkin, E. M. Kroh, B. R. Fritz, S. K. Wyman, E. L. Pogosova-Agadjanyan, A. Peterson, J. Noteboom, K. C. O'Briant, A. Allen, D. W. Lin, N. Urban, C. W. Drescher, B. S. Knudsen, D. L. Stirewalt, R. Gentleman, R. L. Vessella, P. S. Nelson, D. B. Martin and M. Tewari, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10513–10518 CrossRef CAS PubMed.
- T. K. Kamanu, A. Radovanovic, J. A. Archer and V. B. Bajic, Sci. Rep., 2013, 3, 2940 CrossRef PubMed.
- E. Gasperskaja and V. Kucinskas, Acta Med. Litu., 2017, 24, 1–11 Search PubMed.
- Y. Li, H. Du, W. Wang, P. Zhang, L. Xu, Y. Wen and X. Zhang, Sci. Rep., 2016, 6, 26879 CrossRef CAS PubMed.
- C. Vietz, M. L. Schutte, Q. Wei, L. Richter, B. Lalkens, A. Ozcan, P. Tinnefeld and G. P. Acuna, ACS Omega, 2019, 4, 637–642 CrossRef CAS PubMed.
- F. Wang and J. Liu, Nanoscale, 2015, 7, 919–923 RSC.
- L. Olejko, P. J. Cywinski and I. Bald, Angew. Chem., Int. Ed., 2015, 54, 673–677 CAS.
- S. Su, Q. Sun, X. Gu, Y. Xu, J. Shen, D. Zhu, J. Chao, C. Fan and L. Wang, TrAC, Trends Anal. Chem., 2019, 119, 115610 CrossRef CAS.
- H. Ijas, S. Nummelin, B. Shen, M. A. Kostiainen and V. Linko, Int. J. Mol. Sci., 2018, 19, 2114 CrossRef PubMed.
- R. Niu, C. Song, F. Gao, W. Fang, X. Jiang, S. Ren, D. Zhu, S. Su, J. Chao, S. Chen, C. Fan and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 11695–11701 CrossRef CAS PubMed.
- M. A. Goetzfried, K. Vogele, A. Muckl, M. Kaiser, N. B. Holland, F. C. Simmel and T. Pirzer, Small, 2019, 15, e1903541 CrossRef PubMed.
- M. Loretan, I. Domljanovic, M. Lakatos, C. Ruegg and G. P. Acuna, Materials, 2020, 13, 2185 CrossRef CAS PubMed.
- R. M. Zadegan, M. D. Jepsen, K. E. Thomsen, A. H. Okholm, D. H. Schaffert, E. S. Andersen, V. Birkedal and J. Kjems, ACS Nano, 2012, 6, 10050–10053 CrossRef CAS PubMed.
- P. D. Cunningham, A. Khachatrian, S. Buckhout-White, J. R. Deschamps, E. R. Goldman, I. L. Medintz and J. S. Melinger, J. Phys. Chem. B, 2014, 118, 14555–14565 CrossRef CAS PubMed.
- M. del Pilar Rodriguez-Torres and K. Pal, in Bio-manufactured Nanomaterials: Perspectives and Promotion, ed. K. Pal, Springer International Publishing, Cham, 2021, pp. 253–278 Search PubMed.
- F. Nicoli, A. Barth, W. Bae, F. Neukirchinger, A. H. Crevenna, D. C. Lamb and T. Liedl, ACS Nano, 2017, 11, 11264–11272 CrossRef CAS PubMed.
- I. H. Stein, V. Schuller, P. Bohm, P. Tinnefeld and T. Liedl, ChemPhysChem, 2011, 12, 689–695 CrossRef CAS PubMed.
- S. Buckhout-White, C. M. Spillmann, W. R. Algar, A. Khachatrian, J. S. Melinger, E. R. Goldman, M. G. Ancona and I. L. Medintz, Nat. Commun., 2014, 5, 5615 CrossRef CAS PubMed.
- E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73–76 CrossRef CAS PubMed.
- W. Xu, P. Yin and M. Dai, Angew. Chem., Int. Ed., 2018, 57, 14075–14079 CrossRef CAS PubMed.
- T. Funck, F. Nicoli, A. Kuzyk and T. Liedl, Angew. Chem., Int. Ed., 2018, 57, 13495–13498 CrossRef CAS PubMed.
- S. E. Ochmann, C. Vietz, K. Trofymchuk, G. P. Acuna, B. Lalkens and P. Tinnefeld, Anal. Chem., 2017, 89, 13000–13007 CrossRef CAS PubMed.
- D. Wang, Y. Fu, J. Yan, B. Zhao, B. Dai, J. Chao, H. Liu, D. He, Y. Zhang, C. Fan and S. Song, Anal. Chem., 2014, 86, 1932–1936 CrossRef CAS PubMed.
- D. Zhu, J. Huang, B. Lu, Y. Zhu, Y. Wei, Q. Zhang, X. Guo, L. Yuwen, S. Su, J. Chao and L. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 20725–20733 CrossRef CAS PubMed.
- S. M. Douglas, A. H. Marblestone, S. Teerapittayanon, A. Vazquez, G. M. Church and W. M. Shih, Nucleic Acids Res., 2009, 37, 5001–5006 CrossRef CAS PubMed.
- D. Selnihhin, S. M. Sparvath, S. Preus, V. Birkedal and E. S. Andersen, ACS Nano, 2018, 12, 5699–5708 CrossRef CAS PubMed.
- Y. Ke, S. M. Douglas, M. Liu, J. Sharma, A. Cheng, A. Leung, Y. Liu, W. M. Shih and H. Yan, J. Am. Chem. Soc., 2009, 131, 15903–15908 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- N. Stephanopoulos, ChemBioChem, 2019, 20, 2191–2197 CrossRef CAS PubMed.
- D. Y. Zhang and E. Winfree, J. Am. Chem. Soc., 2009, 131, 17303–17314 CrossRef CAS PubMed.
- R. Bruch, J. Baaske, C. Chatelle, M. Meirich, S. Madlener, W. Weber, C. Dincer and G. A. Urban, Adv. Mater., 2019, 31, e1905311 CrossRef PubMed.
- R. M. Shetty, S. R. Brady, P. W. K. Rothemund, R. F. Hariadi and A. Gopinath, ACS Nano, 2021, 15, 11441–11450 CrossRef CAS PubMed.
- I. V. Martynenko, V. Ruider, M. Dass, T. Liedl and P. C. Nickels, ACS Nano, 2021, 15, 10769–10774 CrossRef CAS PubMed.
- A. Gopinath and P. W. Rothemund, ACS Nano, 2014, 8, 12030–12040 CrossRef CAS PubMed.
- B. Feng, K. Frykholm, B. Norden and F. Westerlund, Chem. Commun., 2010, 46, 8231–8233 RSC.
- C. Shi, Y. Wang, M. Zhang and C. Ma, Sci. Rep., 2017, 7, 6818 CrossRef PubMed.
- O. Mook, J. Vreijling, S. L. Wengel, J. Wengel, C. Zhou, J. Chattopadhyaya, F. Baas and K. Fluiter, Artif. DNA PNA XNA, 2010, 1, 36–44 CrossRef PubMed.
- K. Trofymchuk, V. Glembockyte, L. Grabenhorst, F. Steiner, C. Vietz, C. Close, M. Pfeiffer, L. Richter, M. L. Schutte, F. Selbach, R. Yaadav, J. Zahringer, Q. Wei, A. Ozcan, B. Lalkens, G. P. Acuna and P. Tinnefeld, Nat. Commun., 2021, 12, 950 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr03985k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |