
Looking into a crystal ball: printing and patterning self-assembled peptide nanostructures

Abstract
The solution processability of organic semiconductors and conjugated polymers along with the advent of nanomaterials as conducting inks have revolutionized next-generation flexible consumer electronics. Another equally important class of nanomaterials, self-assembled peptides, heralded as next-generation materials for bioelectronics, have a lot of potential in printed technology. In this minireview, we address the self-assembly process in dipeptides, their application in electronics, and recent progress in three-dimensional printing. The prospect of a generalizable path for nanopatterning self-assembled peptides using ice lithography and its challenges are further discussed.
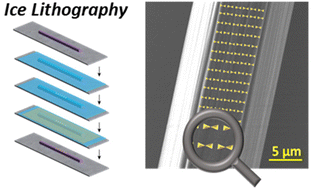
- This article is part of the themed collections: Nanomaterials for printed electronics and Recent Review Articles
 Please wait while we load your content...
Please wait while we load your content...

