
Bimetallic RuNi-decorated Mg-CUK-1 for oxygen-tolerant carbon dioxide capture and conversion to methane†

Abstract
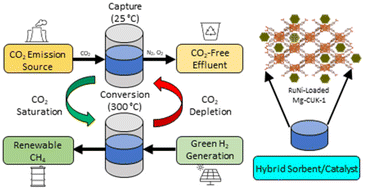
The development of hybrid sorbent/catalysts for carbon capture and conversion to chemical fuels involves several material and engineering design considerations. Herein, a metal–organic framework (MOF), known as Mg-CUK-1, is loaded with Ru and Ni nanoparticles and assessed as a hybrid material for the sequential capture and conversion of carbon dioxide (CO2) to methane (CH4). Low nanocatalyst loadings led to enhanced overall performance by preserving more CO2 uptake within the Mg-CUK-1 sorbent. Low temperature CO2 desorption from Mg-CUK-1 facilitated complete CO2 release and subsequent conversion to CH4. The influence of oxygen exposure on catalyst performance was assessed, with Ru-loaded Mg-CUK-1 exhibiting oxygen tolerance through sustained CH4 generation of 1.40 mmol g−1 over ten cycles. In contrast, Ni-loaded Mg-CUK-1 was unable to retain initial catalytic performance, reflected in an 11.4% decrease in CH4 generation over ten cycles. When combined, 0.3Ru2.7Ni Mg-CUK-1 yielded comparable overall performance to 3Ru Mg-CUK-1, indicating that Ru aids the re-reduction of NiO to Ni after O2 exposure. By combining multiple catalyst species within one hybrid sorbent/catalyst material, greater catalyst stability is achieved, resulting in sustained overall performance. The introduced strategy provides an approach for fostering resilient hydrogenation catalysts upon exposure to reactive species often found in real-world point source CO2 emissions.
- This article is part of the themed collection: CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

