
Designing optimal core–shell MOFs for direct air capture†
 a
Austin R.
Lieber,
c
Janice A.
Steckel,
d
Nathaniel L.
Rosi,
ab
Christopher E.
Wilmer
*ae
a
Austin R.
Lieber,
c
Janice A.
Steckel,
d
Nathaniel L.
Rosi,
ab
Christopher E.
Wilmer
*ae
Abstract
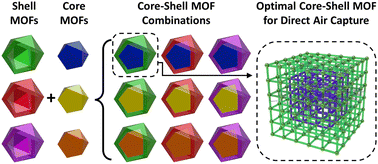
Metal–organic frameworks (MOFs), along with other novel adsorbents, are frequently proposed as candidate materials to selectively adsorb CO2 for carbon capture processes. However, adsorbents designed to strongly bind CO2 nearly always bind H2O strongly (sometimes even more so). Given that water is present in significant quantities in the inlet streams of most carbon capture processes, a method that avoids H2O competition for the CO2 binding sites would be technologically valuable. In this paper, we consider a novel core–shell MOF design strategy, where a high-CO2-capacity MOF “core” is protected from competitive H2O-binding via a MOF “shell” that has very slow water diffusion. We consider a high-frequency adsorption/desorption cycle that regenerates the adsorbents before water can pass through the shell and enter the core. To identify optimal core–shell MOF pairs, we use a combination of experimental measurements, computational modeling, and multiphysics modeling. Our library of MOFs is created from two starting MOFs-UiO-66 and UiO-67-augmented with 30 possible functional group variations, yielding 1740 possible core–shell MOF pairs. After defining a performance score to rank these pairs, we identified 10 core–shell MOF candidates that significantly outperform any of the MOFs functioning alone.
- This article is part of the themed collections: CO2 capture and conversion and Nanoscale Most Popular 2022 Articles
 Please wait while we load your content...
Please wait while we load your content...

