
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailored polyethylene glycol grafting on porous nanoparticles for enhanced targeting and intracellular siRNA delivery†
Jounghyun
Yoo‡
 a,
Kyunghwan
Kim‡
b,
Suhyun
Kim
a,
Hee Ho
Park
*c,
Heungsoo
Shin
*c and
Jinmyoung
Joo
*a
a,
Kyunghwan
Kim‡
b,
Suhyun
Kim
a,
Hee Ho
Park
*c,
Heungsoo
Shin
*c and
Jinmyoung
Joo
*a
aDepartment of Biomedical Engineering, Ulsan National Institute of Science and Technology, Ulsan 44919, Republic of Korea. E-mail: jjoo@unist.ac.kr
bDepartment of Chemistry, Ulsan National Institute of Science and Technology, Ulsan 44919, Republic of Korea
cDepartment of Bioengineering, Hanyang University, Seoul 04763, Republic of Korea. E-mail: hshin@hanyang.ac.kr; parkhh@hanyang.ac.kr
First published on 12th September 2022
Abstract
Surface functionalization of nanoparticles with polyethylene glycol (PEG) has been widely demonstrated as an anti-opsonization strategy to reduce protein corona formation which is one of the major concerns affecting target receptor recognition. However, excessive surface passivation with PEG can lead to the strong inhibition of cellular uptake and less efficient binding to target receptors, resulting in reduced potential of targeted delivery. To improve specific cell targeting while reducing the nonspecific protein adsorption, a secondary packaging of the nanoparticles with shorter PEG chains, making the targeting ligands densely stretched out for enhanced molecular recognition is demonstrated. Particularly, we report the tailored surface functionalization of the porous nanoparticles that require the stealth shielding onto the open-pore region. This study shows that, in addition to the surface chemistry, the conformation of the PEG layers controls the cellular interaction of nanoparticles. Since the distance between neighboring PEG chains determines the structural conformation of the grafted PEG molecules, tailored PEG combinations can efficiently resist the adsorption of serum proteins onto the pores by transitioning the conformation of the PEG chains, thus significantly enhance the targeting efficiency (>5-fold). The stretched brush PEG conformation with secondary packaging of shorter PEG chains could be a promising anti-opsonization and active targeting strategy for efficient intracellular delivery of nanoparticles.
Introduction
In targeted drug delivery systems, the surface of nanoparticles is typically functionalized with antibody, peptide, or nucleic acid that recognize specific receptors on the plasma membrane of the target cells.1 These targeting ligands enable the nanoparticles to selectively bind to the specific cells, resulting in selectively high accumulation in diseased tissues such as cancers or infected sites. Diagnostic sensitivity and therapeutic efficacy can be improved with the targeted delivery of nanoparticles while reducing the off-target accumulation in healthy tissues.2,3 In this regard, the dynamic interfacial phenomena occurring between the nanoparticles and biological components such as serum proteins or cells are important factors that determine the targeting efficacy.4–7 Particularly, serum proteins spontaneously interact with the surface of nanoparticles to form a protein corona when the nanoparticles are exposed to blood. The protein corona affects not only the pathophysiological properties of the nanoparticles during blood circulation but also targeting capability by masking the ligands, resulting in reduced specificity and affinity to the target cells.8To suppress the nonspecific protein adsorption onto nanoparticles either in in vitro or in vivo administration, antifouling polyethylene glycol (PEG) has been grafted on the nanoparticle surface.9–13 The PEG coating, PEGylation, is one of the most prominent shielding strategy to avoid rapid opsonization of the nanoparticles, thus not only prolonging the blood circulation time but also reducing nonspecific accumulation of the nanoparticles to off-target cells. However, excessive PEGylation substantially leads to less efficient binding affinity to the target receptor, resulting in the inhibition of intracellular uptake.14 Therefore, underlying details of the surface grafting, such as the amount of PEG or type of PEG on the nanoparticle surface are essential to effectively reduce the unwanted interactions between serum proteins and nanoparticles. For instance, it has been known that the molecular weight of PEG is an important determinant of effective surface shieling to evade the interactions with serum proteins. The total amount of serum proteins adsorbed onto the nanoparticle significantly decreased as PEG molecular weight increased up to 5 kDa while negligible improvement was seen with the longer chains.15
In addition to the molecular weight of PEG chains that is beneficial to form a sufficiently thick layer shielding the nanoparticle surface sterically, the grafting density or distance between neighboring PEG chains can further determine the structural conformation.16–18 Since the conformation of the attached PEG can critically affect the degree of protein corona formation, followed by cellular interaction, the regulation of PEG grafting to improve the specific binding affinity of nanoparticles to the targeted cell is essential. This is a particularly important issue for porous nanoparticles which are widely applied for drug delivery systems owing to their large surface area and pore volume for superior drug loading capacity. The pores are hardly shielded by PEG grafting, thus directly exposed to the serum proteins upon administration. Although the PEG conformation in terms of molecular weight and density has been extensively investigated to see how it affects protein corona composition and cellular uptake, most of the studies have focused on the nanoparticles composed of gold, silica, lipid and polymers that have a smooth surface.18–21 However, a systematic demonstration of PEG grafting on porous nanoparticles, which have promising potential as a drug delivery system due to the large pore volume for biological payloads, has not been investigated. In this study, we report a secondary packaging of the porous surface on nanoparticles by tailoring a densely grafted “brush” conformation of PEG. We then investigated whether the secondary packaging of the porous nanoparticles with combinational PEG chains could suppress the protein corona formation and enhance the specific targeting efficiency. Porous silicon nanoparticles (PSiNPs) were chosen because of their superior biocompatibility, robust surface chemistry, well-defined mesoporous structures, and intrinsic photoluminescence for spatiotemporal tracking.22–24 While 5 kDa PEG linkers were covalently attached to PSiNPs, the secondary packaging with shorter (e.g., 1 and 2 kDa) PEG chains leads additional coverage of unsaturated surfaces resulting in the transition to the stretched brush conformation from the mushroom structure. We establish a clear correlation of PEG conformation and targeting capability through precise surface passivation. The secondary packaging is suggested to improve the targeted binding of nanoparticles by overcoming the inhibitory effect of the protein corona, resulting in increased intracellular drug delivery activity.
Results and discussion
Porous silicon nanoparticle preparation
Porous silicon nanoparticles (PSiNPs) were prepared by electrochemical etching of highly doped p-type single crystalline silicon wafers in an aqueous electrolyte containing hydrofluoric acid and ethanol, followed by lift-off, ultrasonic fracture, and activation of photoluminescence (PL) as reported in the literature.25,26 The nominal pore diameter of the PSiNPs was ∼15 nm, and the pores were aligned in the perpendicular orientation (Fig. 1a and b). The narrow size distribution of PSiNPs with a mean diameter of ∼171 ± 23 nm was consistent in transmission electron microscopy (TEM) and dynamic light scattering (DLS) analysis (Fig. 1c), supporting the stable colloidal motion of the PSiNPs as a model drug carrier to study cellular uptake. Along with the mesoporous structure, the intrinsic PL displaying a relatively broad emission spectrum in the red to near-infrared region is a beneficial property as a drug carrier compared to other platforms such as mesoporous silica nanoparticles (MSN) or lipid nanoparticles (LNP) that require additional fluorescence labeling for spatiotemporal tracking in cellular uptake study (Fig. 1d). In particular, the intrinsic PL is useful to avoid the possible risk caused by the cleavage of the fluorescent dyes from the nanoparticles, causing mistracing in cellular trafficking.27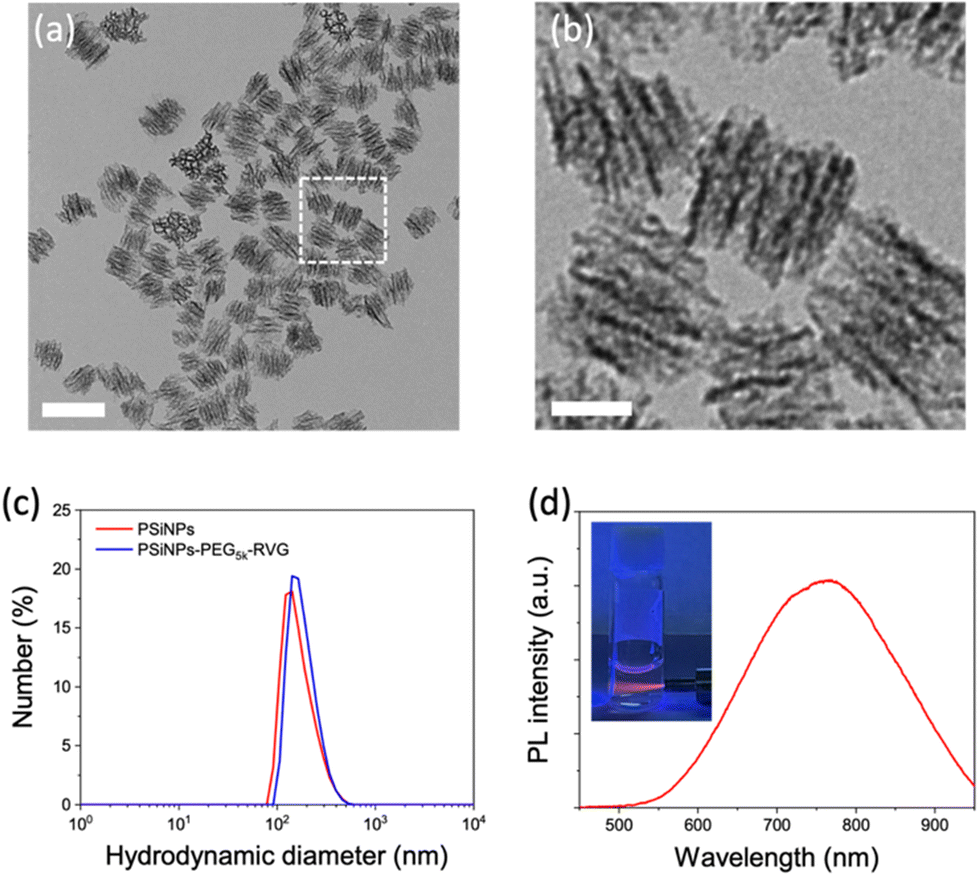 | ||
| Fig. 1 Characteristics of porous silicon nanoparticles (PSiNPs). (a and b) Transmission electron microscopy images of PSiNPs. Scale bar: (a) 200 nm, (b) 50 nm. (c) Hydrodynamic size distribution of PSiNPs before and after RVG peptide conjugation obtained from dynamic light scattering (DLS) analysis. (d) Photoluminescence (PL) spectrum of PSiNPs in water. Inset: photograph of aqueous PSiNP solution under UV irradiation (λex: 365 nm). | ||
Surface functionalization of PSiNPs
PSiNPs were further functionalized with a neuron-targeting peptide derived from rabies virus glycoprotein (RVG), which has been used for targeting neuronal cells in the central nervous system.28 The RVG peptide-decorated PSiNPs are capable of specific uptake into nerve cells by neuronal nicotinic acetylcholine receptor (AChR)-mediated endocytosis.29–31 To suppress the nonspecific adsorption of serum proteins upon administration, the RVG peptide was conjugated to PSiNPs via a PEG linker (5 kDa). The PSiNPs were firstly functionalized with primary amines on the oxide surface through the reaction with 3-(ethoxydimethylsilyl)propylamine, and then amine-reactive succinimidyl ester chemistry led to the conjugation of NHS–PEG5k–maleimide linkers. The RVG peptides with N-terminal cysteine residues were grafted with maleimide to form a stable conjugation (Fig. 2a and S1†). The hydrodynamic diameter of the PSiNPs after grafting of the PEG linkers and RVG peptides slightly increased to ∼184 ± 31 nm (Fig. 1c and 2b), and nanoparticles were stably suspended in aqueous media without aggregation (Fig. 1c and Fig. S2†). The surface of PSiNPs were supposed to be fully covered by the PEG linkers since the excessive amount (>20-fold higher than the theoretical number for complete coverage) of NHS–PEG5k–maleimide linkers were exposed to the amine-functionalized PSiNPs. However, it should be noted that the increase in the hydrodynamic radius of PSiNPs was limited to ∼6.5 nm, which is substantially less than the stretched chain length of PEG5k (∼30 nm). The results indicate that the neighboring PEG chains are in a “mushroom” conformational regime rather than stretching away to form a “brush” layer (Fig. 2a). The mushroom conformation of PEG chains certainly induces the random orientation of RVG peptides on PSiNPs, causing a reduced binding efficiency to the target receptors. Moreover, steric hinderance of the mushroom type PEG linkers is responsible for less coverage onto PSiNPs, possibly resulting in the inefficient prevention of nonspecific protein adsorption (discussed below). It should be also noted that the porous surface particularly allows the sparse coverage of the PEG linkers only at the pore wall while the pore region is readily exposed to serum proteins. Therefore, tailoring the surface-grafted PEG chains to be the stretched brush layer is necessarily required to improve the targeting capability to the specific cells.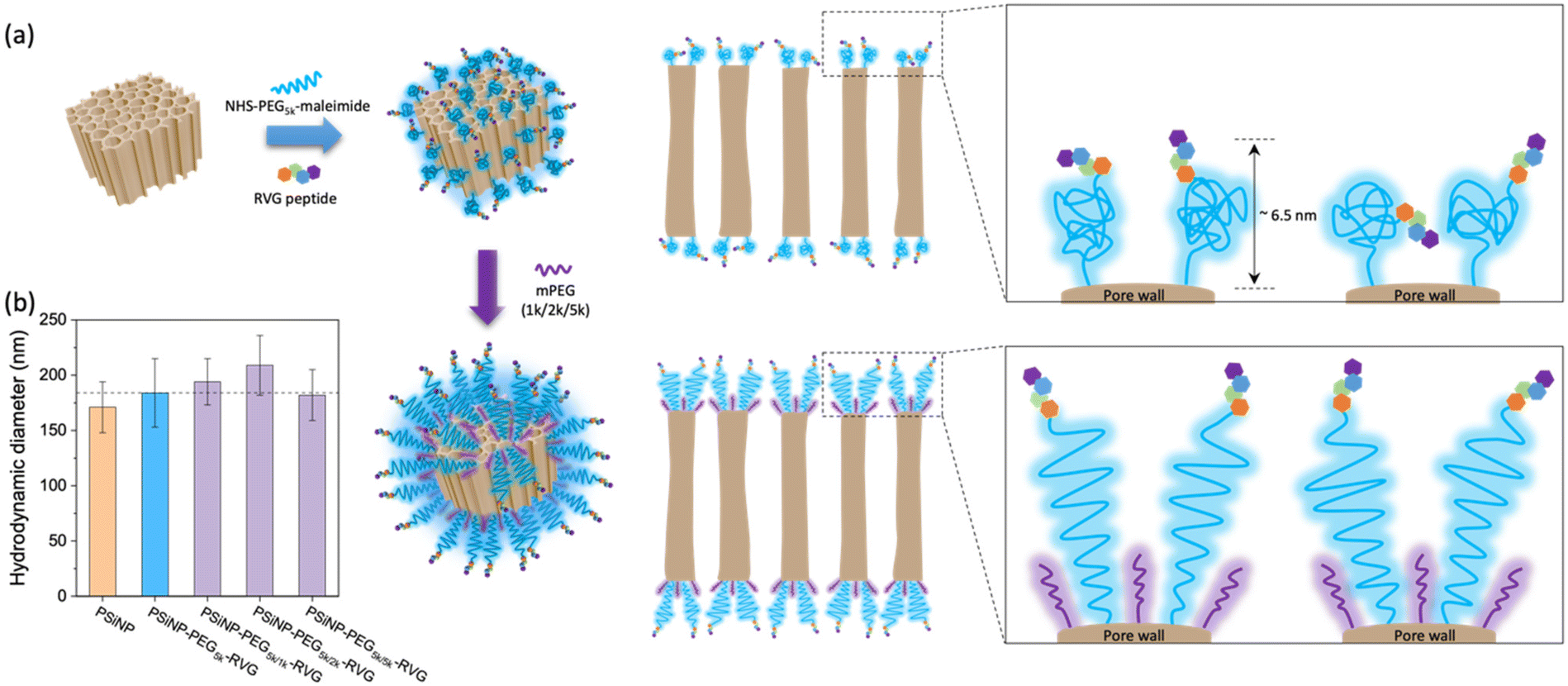 | ||
| Fig. 2 Conformational transition of PEG grafted on PSiNPs upon secondary packaging with shorter PEG chains. (a) Schematic illustrations of PEG conformation onto the PSNPs with RVG peptide conjugation. (b) Hydrodynamic diameters of PSiNPs after secondary packaging with shorter PEG chains. Note that the mushroom conformation of PEG5k is stretched to form a brush layer with the secondary packaging. | ||
As a means of secondary packaging, the shorter chains of methoxy-terminated PEG (mPEG) molecules were additionally grafted to the uncovered surface of PSiNPs. By adding the mPEG to the RVG-conjugated PSiNPs (PSiNPs–PEG5k–RVG), the unsaturated amine groups, which were not linked with the NHS–PEG5k–maleimide linkers due to steric hindrance, were further available to link with mPEG (Fig. 2a). Depending on the molecular weight of mPEG, the hydrodynamic diameters of PSiNPs slightly increased up to ∼38 nm (PSiNPs–PEG5k/2k–RVG), indicating the conformational transition of the PEG5k linkers from the mushroom to the brush (Fig. 2a and b). The resulting PEG chains were forced into a stretched brush conformation to minimize the PEG–PEG contacts and to maximize the PEG–water contacts at the elevated grafting density by secondary packaging with shorter mPEG chains. However, it should be noted that the secondary packaging with the same length of mPEG (5 kDa) has a negligible effect on the conformational transition of the PEG layers due to a lack of available grafting sites. The conformation of surface-bound PEG can be determined by the Flory radius (RF) of PEG, distance between adjacent PEG chains (D), and length of the grafted PEG layer (L) following the de Gennes’ model (Fig. S3 and Table S1†).32 The PEG molecules adopt a mushroom conformation at a low grafting density (RF/D < 1) and eventually reaches a more extended brush conformation with a higher grafting density (L/Rf > 2).33 Our results support that the increasing density obtained by the secondary packaging with the shorter mPEG chains between the grafted PEG5k leads the conformational transition since the PEG chains repel each other and adopt the more extended dense brush configuration (Fig. 2a).
Cellular targeting via enhanced receptor recognition
To verify the enhanced targeting efficacy resulting from the secondary packaging, we then incubated those nanoparticles with the AchR-expressing Neuro-2a cells, derived from mouse neuroblastoma. The confocal laser scanning microscopy (CLSM) images showed a strong binding affinity and intracellular uptake of PSiNPs–PEG5k–RVG to the Neuro-2a cells but not to the AchR-negative HeLa cells in accordance with the literature (Fig. 3a and S4†).28,34 Interestingly, the results emphasize that the secondary packaging with the shorter PEG chains tremendously improved the receptor targeting capability and cellular uptake (Fig. 3b). It has been generally accepted that nonspecific serum protein binding onto the bare nanoparticles can initiate the nucleation of the protein complexes and finally lead to the growth of the protein corona, which is typically reduced by PEG grafting.35 However, the surface of porous nanoparticles suffers from incomplete coverage of PEG molecules due to the naked porous structure that is potentially susceptible to protein corona nucleation (Fig. 4a). The secondary packaging with shorter PEG chains can cover the unsaturated surface, thus inducing the conformational transition from the mushroom to brush layer of the PEG5k. This effectively suppresses the nonspecific adsorption of serum proteins on the open-pore area (Fig. 4b). Enhanced receptor binding and intracellular uptake efficiency indicate that the serum proteins in the culture media are rapidly adsorbed to the nanoparticles, inhibiting the interaction of the RVG peptides with the AchR (Fig. 4c). The result emphasizes that the differences in the targeting efficiency, while the RVG peptide density on PSiNPs remains the same and are attributed to the PEG conformations related to the secondary packaging. We found superior targeting efficiency (>5-fold) of PSiNPs grafted with an additional 2 kDa mPEG (PSiNPs-PEG5k/2k-RVG), which is supposed to be the optimal chain length for the given PSiNP system not only to cover the unsaturated gap between the RVG-conjugated PEG linkers with minimal steric hinderance but also to fill the open-pore area by transitioning the PEG conformation (Fig. 4a and b). In addition, the stretched brush conformation of the PEG5k linkers can contribute to the outward orientation of the RVG peptide for a stronger association between the peptide and the AchR on the Neuro-2a cell membrane (Fig. 4d). Overall, the protein corona formation on PSiNPs is potentially responsible for reducing the cell binding affinity and targeting efficiency.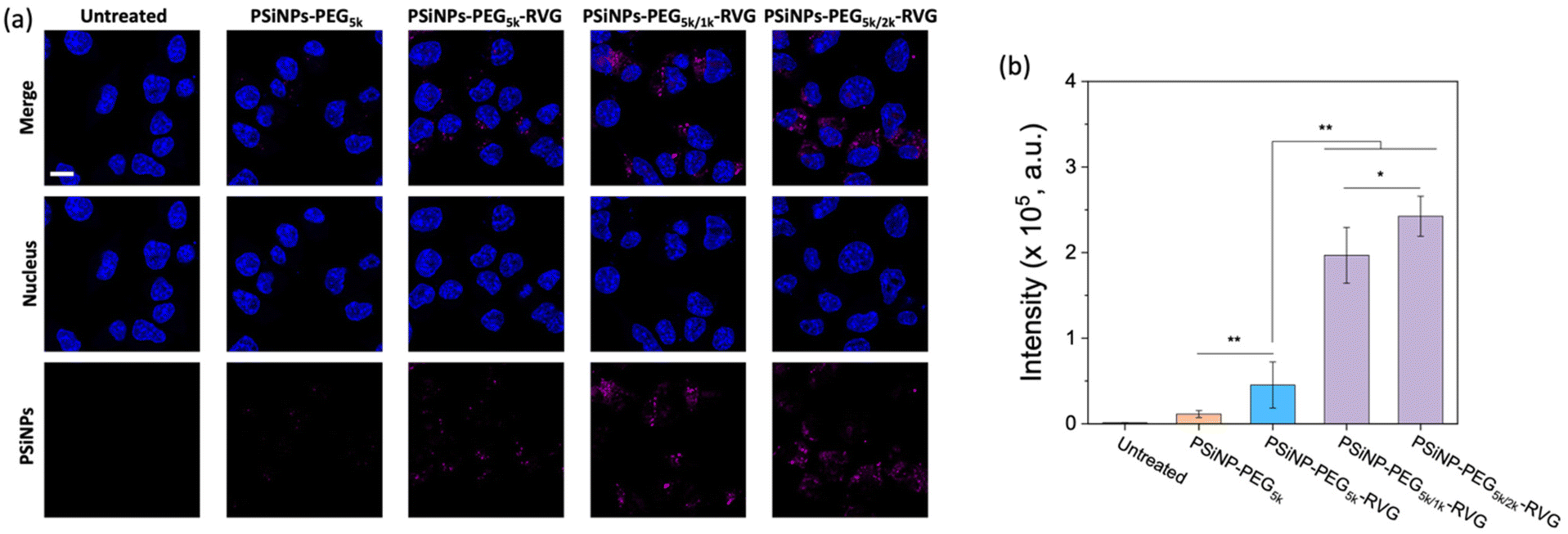 | ||
| Fig. 3 Influence of secondary packaging on PSiNPs for cellular uptake. (a) CLSM images of Neuro-2a cells treated for 2 h with PSiNPs containing various secondary PEG chains as indicated. Note that nuclei were stained with Hoechst 33342 (λex: 360 nm, λem: 495 nm), and the intrinsic photoluminescence of PSiNPs was imaged (λex: 385 nm, λem: 750 nm). Scale bar: 10 μm. (b) Quantitative analysis of the targeted PSiNPs obtained from the mean PL intensity (±standard deviation) per cell (n = 30). *p < 0.05, and **p < 0.01 from the two-tailed Student's t test. | ||
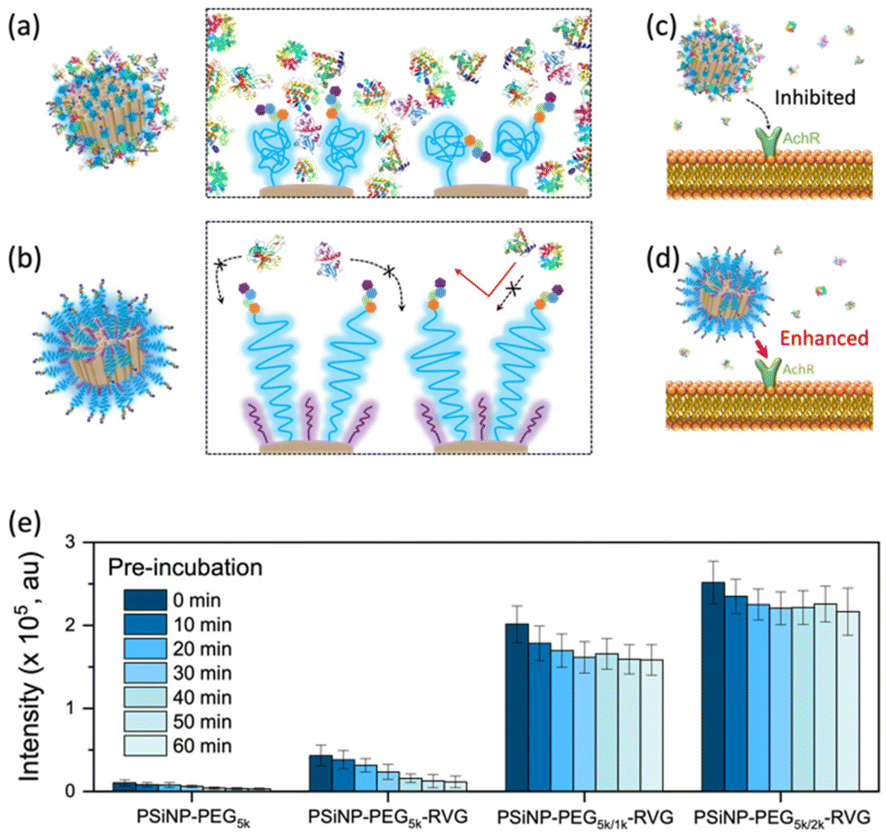 | ||
| Fig. 4 Schematic illustrations of the interactions between PSiNPs and serum proteins. (a) Protein corona formation initiated by nonspecific serum protein adsorption on the porous cavity of PSiNPs. (b) Stretched brush conformation of PEG inhibiting serum protein adsorption. (c) Protein corona is inhibiting the acetylcholine receptor (AchR) recognition. (d) Dense brush conformation of PEG is beneficial for the strong association of the RVG peptide to the AchR due to both the straight orientation of the peptide and inhibited protein corona formation. (e) Photoluminescence intensity (±standard deviation) of PSiNPs found in a cell (n = 50) after pre-incubation in serum-rich cell culture media for the designated time as indicated. | ||
We further confirmed that the protein corona formation on the PSiNPs is responsible for reducing the targeting efficiency. The PSiNPs were incubated with serum proteins prior to treatment with Neuro-2a cells. Correspondingly, a substantial decrease in the targeting efficiency compared to that of the nanoparticles which were not pre-exposed to serum proteins prior to incubation (Fig. 4e and Fig. S5†) was observed. The results indicate that the well-established protein corona during the pre-incubation in serum-rich media certainly reduces the interaction of the RVG-conjugated PSiNPs with the AchR. Particularly, ∼75% of PSiNP–PEG5k–RVG without the secondary packaging lost the targeting capability upon pre-incubation in serum-rich media, presumably due to protein corona formation despite conjugation of the PEG ligands on the nanoparticles. The limited PEG grafting density due to the porous surface should be responsible for unwanted protein corona formation in accordance with the above finding. However, the secondary packaging shows remarkably effective prevention of non-specific binding of serum proteins, followed by a noticeable cellular uptake. Although protein corona formation might not be completely inhibited, PSiNPs–PEG5k/2k–RVG maintained the cellular uptake efficiency to be ∼86% even after 60 min of pre-incubation. It should be noted that the interactions between PSiNPs and cells may still occur with alternative pathways mediated by the adsorbed protein corona, resulting in a certain degree of cell binding affinity. However, the secondary packaging with the shorter PEG chains (either 1 or 2 kDa) certainly contribute to not only improving the targeting efficiency but also preventing protein corona formation. This is attributed to the strong association between the RVG peptide and the AchR on the Neuro-2a cell membrane. In addition, the conformational transition of PEG ligands associated with secondary packaging showed no additional cytotoxicity to the cells, supporting that the biocompatible nature of PSiNPs was maintained regardless of the PEG conformation (Fig. S6†).36 Overall, the protein corona formation on PSiNPs is potentially responsible for reducing the cell binding affinity and targeting efficiency.
Intracellular delivery of siRNA
Next, we assessed the targeted delivery of siRNA using PSiNPs as a carrier. Since the naked siRNA has been hampered by poor intracellular delivery due to negative charge of the phosphate backbone despite the promising potential for RNA interference-based therapeutics, the siRNA was used as a model drug in this study for secure loading and intracellular transport. In addition, siRNA is highly susceptible to enzymatic degradation during delivery; therefore it requires a protective loading system (e.g., porous interior) isolated from the harsh environment upon administration. The siRNA payloads were readily packed into the pores, where the cationic (amine-terminated) surface of the interior pore walls can stably harbor the payloads via electrostatic interactions. The loading capacity of siRNA was ∼8% by mass, corresponding to ∼3.0 × 104 siRNA molecules per nanoparticle. Upon exposure to cell culture conditions, we found that the secondary packaging is beneficial for the delayed release of the siRNA cargo by capping the pore (Fig. 5a and b). The PSiNP–PEG5k–RVG linker exhibited a half-life for the release of the siRNA payloads of 2.3 h, whereas the secondary packaging with 2 kDa PEG (PSiNP–PEG5k/2k–RVG) significantly extended the half-life to 15 h. This is attributed to the prolonged retention of the siRNA payloads owing to the dense PEG brush layer that partially covers the entrance of the porous interior (Fig. 5b). The prompt leakage of siRNA can be remarkably prevented by the secondary packaging of the porous surface, and the sustained biodegradation of the porous silicon matrix leads to the gradual further release of the siRNA payloads. Overall, the PEG conformation mediates not only the targeting efficiency but also the release profile of drug payloads from the cavity as a means of molecular capping.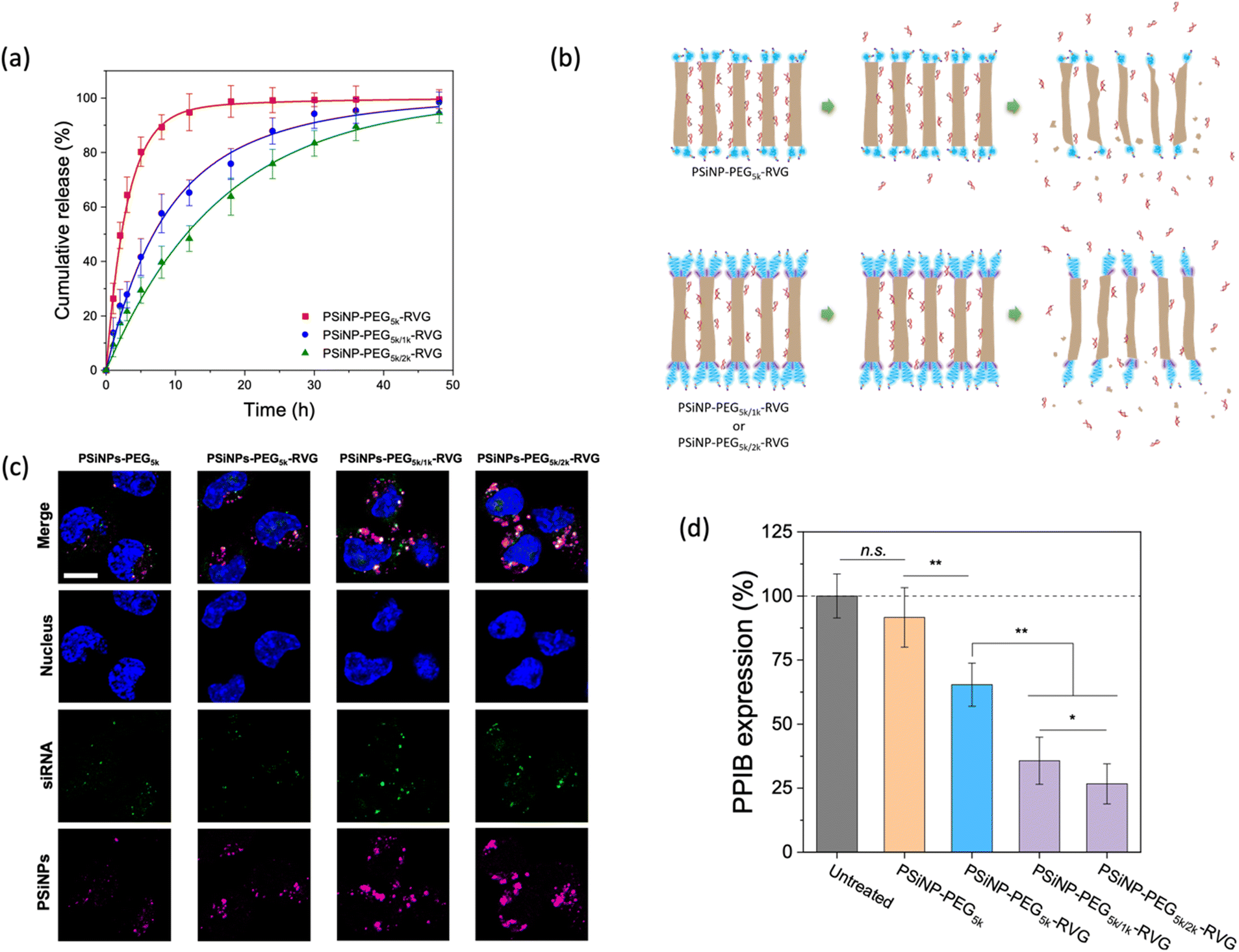 | ||
| Fig. 5 Release kinetics and intracellular delivery of siRNA. (a) Cumulative release profile of siRNA payloads from the PSiNPs incubated in phosphate-buffered saline (PBS) at 37 °C (n = 3 for each group). (b) Schematic illustrations of siRNA-loaded PSiNPs with or without secondary packaging of PEG under release conditions. Note that the stretched brush conformation of PEG ligands leads to the delayed release of siRNA payloads compared to the mushroom conformation. (c) CLSM images of Neuro-2a cells treated for 2 h with siRNA-loaded PSiNPs containing various secondary PEG chains as indicated. siRNA was labeled with a fluorescent dye (Dy547) for imaging. Nuclei and siRNA were measured via the fluorescence of Hoechst 33342 (λex: 360 nm, λem: 495 nm) and Dy547 (λex: 540 nm, λem: 570 nm), respectively. The photoluminescence of PSiNPs was used for quantitative visualization (λex: 385 nm, λem: 750 nm). Scale bar: 5 μm. (d) Relative PPIB gene silencing in Neuro-2a cells after treatment with the PSiNPs (n = 4 for each group). *p < 0.05, and **p < 0.01 from the two-tailed Student's t test (n.s., not statistically not significant). | ||
We then demonstrated the intracellular uptake of siRNA-loaded PSiNPs. When incubated with Neuro-2a cells, the CLSM showed significant uptake of PSiNPs–PEG5k/2k–RVG (Fig. 5c). Correspondingly, the siRNA payloads were also localized in the cytosol, coincident with the PSiNPs. Since we used siRNA against an endogenously expressed peptidylprolyl isomerase B (PPIB), the gene-specific knockdown was further evaluated. The mRNA expression level of PPIB in the Neuro-2a cells was quantitatively determined by reverse transcription polymerase chain reaction (qRT-PCR) after 48 h of co-incubation with the siRNA-loaded PSiNPs. The superior knockdown efficiency of the PPIB mRNA level was obtained with PSiNPs–PEG5k/2k–RVG while decreased gene silencing efficiency was found depending on the conformation of the grafted PEG layers (Fig. 5d). The PSiNPs showed no significant cytotoxicity and gene silencing compared to untreated cells, and the substantially less knockdown in PPIB expression for the RVG-omitted PSiNPs (PSiNP-PEG5k) supports that RVG-mediated targeting and cellular uptake are essential for effective gene delivery to Neuro-2a cells. Therefore, superior RNA interference efficacy can be achieved by the secondary packaging, which is particularly essential for porous nanoparticles for effectively utilizing their large pore volume with enhanced receptor recognition.
The effects of PEG length and density grafted to nanoparticles for protein adsorption and cellular targeting efficiency have been extensively investigated over decades.16–19,37–40 While it is widely accepted that the thicker and densely packed PEG layer is more effective for reducing the nonspecific interactions of nanoparticles with the cells, a better understanding of the PEG grafting onto the porous surface is essential to ensuring the appropriate surface modification of drug carriers that contain a large volume to securely load therapeutic agents inside the pores. The secondary packaging with shorter PEG chains can provide a strategic tailoring method of the porous nanoparticle surfaces by reducing the number of serum proteins that sterically block the binding of the biorecognition molecules to the target receptor as well as by preventing the burst release of drug payloads. SDS-PAGE analysis further revealed that a reduced amount of proteins adsorbed on PSiNPs, which is potentially responsible for forming the protein corona. While maintaining protein profiles adsorbed on PSiNPs, the secondary packaging seemed to lead to less protein adsorption (Fig. S7†). Extending the incubation time up to 60 min in serum containing culture media, the amount of proteins that non-specifically bound to PSiNPs gradually decreased. This study provides certainly limited information on the adsorbed protein profile; therefore it should be further analyzed using quantitative analytical tools such as mass spectrometry to obtain detailed information on the composition and preferential binding affinity of serum proteins on different surface characteristics of nanoparticles as a follow-up study. Despite the unclear protein interactions with the conformational transition of PEG ligands, secondary packaging on the mesoporous surface certainly improves the specificity of the drug nanocarrier and allows to be used for highly targeted nanomedicine in a serum-rich environment. Although the present study is limited to a proof-of-concept demonstration on cellular uptake and targeted delivery efficiency in vitro, the tailored nanoparticles are expected to achieve prolonged blood circulation and targeting efficiency in vivo.
Conclusions
The effect of secondary packaging with shorter PEG chains to cover the unsaturated area on the PEG-grafted porous nanoparticle surface was investigated as a means of enhanced cellular targeting and uptake. Overall, an increased surface coverage with lower molecular weight mPEG is beneficial for suppressing nonspecific protein adsorption and further inducing conformational transition of PEG linkers to be stretched, thus improving the target recognition followed by intracellular delivery of drug payloads (e.g., siRNA). The secondary packaging contributes to forming not only a dense PEG packing but also a brush conformation rather than mushroom layer. Although the effects of PEG length and density on protein adsorption onto the nanoparticles have been extensively investigated over a decade, the conformational transition of the PEG chains induced by secondary packaging is reported for the first time in this study. In addition, our study shows that the conformation of grafted PEG molecules on porous nanoparticles can regulate the release of the drug payloads by capping the open-pore area. This is an important step toward not only understanding and controlling the stealth effect of the various types of porous nanoparticles but also improving the targeted delivery of nanomedicine.Experimental section
Preparation of PSiNPs
Highly boron-doped p++-type silicon wafer (∼1 mΩ cm resistivity, Virginia Semiconductor, Inc.) was anodically etched in a Teflon cell containing electrolyte composed of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (by volume) of 48% aqueous HF:ethanol.25 The backside of the silicon wafer was contacted with aluminum foil while the polished surface was exposed to the electrolyte using a coil of platinum wire as a counter electrode. The etching waveform consisted of a square wave in which a lower current density of 50 mA cm−2 was applied for 1.8 s, followed by a higher current density pulse of 400 mA cm−2 applied for 0.36 s for 140 cycles to generate a stratified porous silicon film with alternating layers of high and low porosity.26 The resulting porous silicon films were removed from the silicon substrate by applying a current density of 3.7 mA cm−2 for 250 s in a solution containing 1:30 (v:v) of 48% aqueous HF:ethanol. The freestanding porous silicon film was fractured by ultrasonication for 16 h and the resulting nanoparticles are isolated by centrifugation at 15000 rpm for 12 min.
:1 (by volume) of 48% aqueous HF:ethanol.25 The backside of the silicon wafer was contacted with aluminum foil while the polished surface was exposed to the electrolyte using a coil of platinum wire as a counter electrode. The etching waveform consisted of a square wave in which a lower current density of 50 mA cm−2 was applied for 1.8 s, followed by a higher current density pulse of 400 mA cm−2 applied for 0.36 s for 140 cycles to generate a stratified porous silicon film with alternating layers of high and low porosity.26 The resulting porous silicon films were removed from the silicon substrate by applying a current density of 3.7 mA cm−2 for 250 s in a solution containing 1:30 (v:v) of 48% aqueous HF:ethanol. The freestanding porous silicon film was fractured by ultrasonication for 16 h and the resulting nanoparticles are isolated by centrifugation at 15000 rpm for 12 min.
Preparation of RVG-functionalized PSiNPs
2 mg of PSiNPs in ethanol was mixed with 20 μl of 3-(ethoxydimethyl)-propylamine silane (Sigma-Aldrich) by vortexing overnight for amine-functionalization. The amine-terminated PSiNPs were rinsed with ethanol and further reacted with NHS–PEG5k–maleimide (10 mg ml−1, Nanocs) for 5 h to completely cover the nanoparticle surface with thiol-reactive maleimide. The succinimidyl carboxymethyl ester group of NHS reacts with the primary amine group on PSiNPs. Unbound PEG linkers were removed by centrifugation, and the maleimide-activated PSiNPs were further mixed with 500 μl of RVG peptide solution (1 mg ml−1) for 3 h to conjugate the RVG peptide via a free cysteine residue at the terminal group.24 The RVG peptide with N-terminal cysteine residues, (CCGG)YTIWMPENPRPGTPCDIFTNSRGKRASNG, was obtained from CPC Scientific, Inc.Secondary packaging of PSiNPs with mPEG
10 mg ml−1N-hydroxylsuccinimide functionalized methoxy PEG (NHS-mPEG, Nanocs) was added to PSiNP–PEG5k–RVG for secondary packaging with unreactive amine groups on the surface. To ensure sufficient conjugation, the reaction was carried out for 3 h, and PSiNP–PEG5k/(2k or 1k)–RVG was purified from unbound PEG by centrifugation.Characterization
Transmission electron microscope (TEM) images were obtained on a Tecnai G2 X-Twin operated at 200 kV (FEI, Netherlands). A Zetasizer Nano ZS (Malvern, UK) was used to determine the hydrodynamic size of the nanoparticles. Photoluminescence spectra were obtained using an Ocean Optics QE spectrophotometer. Concentration of siRNA was determined by measuring absorbance at 260 nm using a spectrometer (NanoDrop, Thermo, USA) based on the OD260 standard curve of siRNA. For the cellular uptake study, cells were cultured in a glass-bottomed dish and observed over a conformal laser scanning microscope (LSM 880, Carl Zeiss, Germany).Cellular uptake
Neuro-2a cells (CCL-131, ATCC, USA) were cultured in Eagle's Minimum Essential Medium (EMEM) containing 10% fetal bovine serum (FBS, Thermo, USA). Cells were seeded on a glass-bottomed culture dish (Nunc, Thermo, USA), and incubated at 37 °C with 5% CO2 overnight. Nanoparticles were injected into the cells, and imaged using a LSM 880 after 2 h of treatment. To quantitatively demonstrate the cellular uptake of PSiNPs with different surface characteristics, intrinsic photoluminescence signals of PSiNPs obtained from individual cells were collected and statistically analyzed. For the serum protein pre-exposure to demonstrate protein corona formation and the associated cellular targeting inhibition, the PSiNP formulations were exposed to cell culture media (EMEM containing 10% FBS) at 37 °C for a designated time in dark using Protein Lobind tubes (Eppendorf, Germany). At each time point, the PSiNPs–protein complexes were isolated from unbound proteins by centrifugation at 16000g for 20 min. The PSiNPs–protein complexes were resuspended in cell culture media (for cell targeting efficiency test) or PBS (electrophoresis). Serum protein pre-exposed PSiNPs were then injected into the cells for targeting capability study.
Electrophoresis analysis
The protein-coated PSiNPs in PBS were further incubated at 37 °C for 60 h to completely degrade the PSiNPs. Eluted proteins were diluted with PBS to adjust the sample buffer and separated on a 12% SDS–polyacrylamide gel at a constant voltage of 160 V. The protein bands were detected by Imperial Protein stain (Coomassie Brilliant Blue, Thermo, USA).siRNA loading and release
The small interfering RNA (siRNA) against peptidylprolyl isomerase B were purchased from Dharmacon, Inc. 1:1 mixtures of siPPIB(1) and siPPIB(2) were used for gene silencing. The sequences for the sense strands were
siPPIB(1): 5′-CAAGUUCCAUCGUGUCAUCdTdT-3′ and
siPPIB(2): 5′-GAAAGAGCAUCUAUGGUGAdTdT-3′.
The RVG-functionalized PSiNPs were mixed with siRNA in Rnase free water under sonication for 5 min, which enabled the facile penetration of siRNA by the temporal opening of the brushed PEG covered on pores. The amine-terminated pore interior harbored the siRNA via electrostatic interaction. The concentration of residual siRNA after centrifugation that isolated the siRNA-loaded PSiNP pellet was determined by measuring the absorbance. Release kinetics of the siRNA-loaded PSiNPs were obtained in phenol red-free cell culture media containing 10% FBS at 37 °C over time.
In vitro gene silencing
Neuro-2a cells were cultured in EMEM containing 10% fetal bovine serum. Cells were seeded in 24-well plates at 6 × 104 cell per well, and the PSiNPs were administered the next day. After 48 h of co-incubation, the cells were harvested and lysed to extract total RNA using a RNeasy mini kit (Qiagen, USA) according to the manufacturer's protocol. Reverse transcription reaction was performed using an iScript cDNA synthesis kit (Bio-Rad, USA). 1 μg of extracted RNA was reverse-transcribed into cDNA according to the manufacturer's protocol, and real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis was performed to examine the PPIB mRNA expression with the Mx3005P qPCR system (Agilent). The cDNA was subjected to RT-qPCR using the SYBR Green PCR Mater mix (Bio-Rad). Samples were normalized to the levels of housekeeping hypoxanthine phosphoribosyltransferase (HPRT) for qPCR analysis. Primer sequences used for qPCR werePPIB forward: GGAAAGACTGTTCCAAAAACAGTG
PPIB reverse: GTCTTGGTGCTCTCCACCTTCCG
HPRT forward: GTCAACGGGGGACATAAAAG and
HPRT reverse: CAACAATCAAGACATTCTTTCCA.
Cell viability
The cytotoxicity of PSiNPs was evaluated using the MTT assay against the cultured Neuro-2a cells. Briefly, the cells were seeded in 96-well plates at a density of 1 × 104 cells per well and allowed to attach overnight. Each well was treated for 24 h with PSiNPs containing different surface characteristics, and then washed twice with phenol red-free culture medium. Thereafter, 3-((4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to the culture medium and incubated at 37 °C for another 4 h. The solution was aspirated from the wells, and 200 μL of DMSO was added to each well and gently shaken to fully dissolve the colored formazan. The absorbance of the solution in each well was measured at 490 nm using a plate reader. The cell viability was expressed as the percentage of viable cells compared with the cells treated with PBS.Statistical analysis
All data represents mean value ± standard deviation. Statistical significance was evaluated using the two-tailed heteroscedastic Student's t test. Unless otherwise stated, the threshold of statistical significance was set to p < 0.05.Author contributions
YJ, KK, SK, and JJ performed the investigation including experiments and data collection. YJ and KK participated in validation and data curation. YJ, KK, HHP, HS, and JJ wrote, reviewed, and edited the manuscript. HHP, HS and JJ supervised the research with conceptualization and funding acquisition.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) and Korea Dementia Research Center (KDRC) funded by the Ministry of Health & Welfare and Ministry of Science and ICT, Korea (HU20C0094). This work was also supported in part by the Basic Science Research Program (2020R1A4A3078645 and 2020R1C1C1006081) through the National Research Foundation of Korea (NRF) funded by the Korean Government (MSIT), and further supported by the Future-Leading Project Research Fund (1.220023.01) of UNIST and POSCO Science Fellowship of the POSCO TJ Park Foundation.References
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef.
- K. Ulbrich, K. Hola, V. Subr, A. Bakandritsos, J. Tucek and R. Zboril, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed.
- N. Bertrand, P. Grenier, M. Mahmoudi, E. M. Lima, E. A. Appel, F. Dormont, J. M. Lim, R. Karnik, R. Langer and O. C. Farokhzad, Nat. Commun., 2017, 8, 777 CrossRef PubMed.
- F. Chen, G. Wang, J. I. Griffin, B. Brenneman, N. K. Banda, V. M. Holers, D. S. Backos, L. Wu, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2017, 12, 387–393 CrossRef CAS.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- Z. Mazidi, S. Javanmardi, S. M. Naghib and Z. Mohammadpour, Chem. Eng. J., 2022, 433, 134569 CrossRef CAS.
- K. E. Wheeler, A. J. Chetwynd, K. M. Fahy, B. S. Hong, J. A. Tochihuitl, L. A. Foster and I. Lynch, Nat. Nanotechnol., 2021, 16, 617–629 CrossRef CAS PubMed.
- L. Shi, J. Zhang, M. Zhao, S. Tang, X. Cheng, W. Zhang, W. Li, X. Liu, H. Peng and Q. Wang, Nanoscale, 2021, 13, 10748–10764 RSC.
- Q. Chen, S. Yu, D. Zhang, W. Zhang, H. Zhang, J. Zou, Z. Mao, Y. Yuan, C. Gao and R. Liu, J. Am. Chem. Soc., 2019, 141, 16772–16780 CrossRef CAS PubMed.
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed., 2011, 50, 1980–1994 CrossRef CAS PubMed.
- L. Zhou, L. S. Tang, X. F. Tao, J. Yang, M. B. Yang and W. Yang, Chem. Eng. J., 2020, 396, 125206 CrossRef CAS.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Laganà, Nanoscale, 2014, 6, 2782–2792 RSC.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernandez, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- J. L. Perry, K. G. Reuter, M. P. Kai, K. P. Herlihy, S. W. Jones, J. C. Luft, M. Napier, J. E. Bear and J. M. DeSimone, Nano Lett., 2012, 12, 5304–5310 CrossRef CAS.
- M. Y. Li, S. Jiang, J. Simon, D. Passlick, M. L. Frey, M. Wagner, V. Mailander, D. Crespy and K. Landfester, Nano Lett., 2021, 21, 1591–1598 CrossRef CAS PubMed.
- Q. Dai, C. Walkey and W. C. W. Chan, Angew. Chem., Int. Ed., 2014, 53, 5093–5096 CrossRef CAS PubMed.
- L. W. C. Ho, W. Y. Yung, K. H. S. Sy, H. Y. Li, C. K. K. Choi, K. C. F. Leung, T. W. Y. Lee and C. H. J. Choi, ACS Nano, 2017, 11, 6085–6101 CrossRef CAS.
- J. H. Park, J. A. Jackman, A. R. Ferhan, J. N. Belling, N. Mokrzecka, P. S. Weiss and N. J. Cho, ACS Nano, 2020, 14, 11950–11961 CrossRef CAS PubMed.
- Y. Park, J. Yoo, M. H. Kang, W. Kwon and J. Joo, J. Mater. Chem. B, 2019, 7, 6271–6292 RSC.
- Y. S. Jin, D. Kim, H. Roh, S. Kim, S. Hussain, J. Y. Kang, C. G. Pack, J. K. Kim, S. J. Myung, E. Ruoslahti, M. J. Sailor, S. C. Kim and J. Joo, Adv. Mater., 2018, 30, 1802878 CrossRef PubMed.
- S. Hussain, J. Joo, J. Kang, B. Kim, G. B. Braun, Z. G. She, D. Kim, A. P. Mann, T. Molder, T. Teesalu, S. Carnazza, S. Guglielmino, M. J. Sailor and E. Ruoslahti, Nat. Biomed. Eng., 2018, 2, 95–103 CrossRef CAS PubMed.
- J. Joo, X. Y. Liu, V. R. Kotamraju, E. Ruoslahti, Y. Nam and M. J. Sailor, ACS Nano, 2015, 9, 6233–6241 CrossRef CAS PubMed.
- J. Joo, J. F. Cruz, S. Vijayakumar, J. Grondek and M. J. Sailor, Adv. Funct. Mater., 2014, 24, 5688–5694 CrossRef CAS.
- L. Petrizza, M. Collot, L. Richert, Y. Mely, L. Prodi and A. S. Klymchenko, RSC Adv., 2016, 6, 104164–104172 RSC.
- P. Kumar, H. Q. Wu, J. L. McBride, K. E. Jung, M. H. Kim, B. L. Davidson, S. K. Lee, P. Shankar and N. Manjunath, Nature, 2007, 448, 39–43 CrossRef CAS PubMed.
- J. Joo, E. J. Kwon, J. Y. Kang, M. Skalak, E. J. Anglin, A. P. Mann, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nanoscale Horiz., 2016, 1, 407–414 RSC.
- C. Lee, H. S. Hwang, S. Lee, B. Kim, J. O. Kim, K. T. Oh, E. S. Lee, H. G. Choi and Y. S. Youn, Adv. Mater., 2017, 29, 1605563 CrossRef PubMed.
- Y. K. Gao, Z. Y. Wang, J. H. Zhang, Y. X. Zhang, H. Huo, T. Y. Wang, T. Y. Jiang and S. L. Wang, Biomacromolecules, 2014, 15, 1010–1018 CrossRef CAS PubMed.
- A. R. Nicholas, M. J. Scott, N. I. Kennedy and M. N. Jones, Biochim. Biophys. Acta, 2000, 1463, 167–178 CrossRef CAS.
- V. B. Damodaran, C. J. Fee, T. Ruckh and K. C. Popat, Langmuir, 2010, 26, 7299–7306 CrossRef CAS PubMed.
- A. Shah, M. S. Malik, G. S. Khan, E. Nosheen, F. J. Iftikhar, F. A. Khan, S. S. Shukla, M. S. Akhter, H. B. Kraatz and T. M. Aminabhavi, Chem. Eng. J., 2018, 353, 559–583 CrossRef CAS.
- L. W. Shi, J. Q. Zhang, M. Zhao, S. K. Tang, X. Cheng, W. Y. Zhang, W. H. Li, X. Y. Liu, H. S. Peng and Q. Wang, Nanoscale, 2021, 13, 10748–10764 RSC.
- J.-H. Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, H. B. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- Q. J. He, J. M. Zhang, J. L. Shi, Z. Y. Zhu, L. X. Zhang, W. B. Bu, L. M. Guo and Y. Chen, Biomaterials, 2010, 31, 1085–1092 CrossRef CAS PubMed.
- C. Sacchetti, K. Motamedchaboki, A. Magrini, G. Palmieri, M. Mattei, S. Bernardini, N. Rosato, N. Bottini and M. Bottini, ACS Nano, 2013, 7, 1974–1989 CrossRef CAS.
- P. del Pino, F. Yang, B. Pelaz, Q. Zhang, K. Kantner, R. Hartmann, N. M. de Baroja, M. Gallego, M. Moller, B. B. Manshian, S. J. Soenen, R. Riedel, N. Hampp and W. J. Parak, Angew. Chem., Int. Ed., 2016, 55, 5483–5487 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr02995b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |