
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOutstanding MRI contrast with dysprosium phosphate nanoparticles of tuneable size†
Elisabet
Gómez-González
 a,
Carlos
Caro
b,
María L.
García-Martín
b,
Ana Isabel
Becerro
*a and
Manuel
Ocaña
a
a,
Carlos
Caro
b,
María L.
García-Martín
b,
Ana Isabel
Becerro
*a and
Manuel
Ocaña
a
aInstituto de Ciencia de Materiales de Sevilla (ICMS) CSIC-Universidad de Sevilla, c/Américo Vespucio, 49, 41092 Seville, Spain. E-mail: anieto@icmse.csic.es
bInstituto de Investigación Biomédica de Málaga – Plataforma Bionand (IBIMA-Plataforma BIONAND) and CIBER-BBN, Málaga 29590, Spain
First published on 26th July 2022
Abstract
The use of high-field magnets for magnetic resonance imaging (MRI) is expected to experience the fastest growth rate during the present decade. Although several CAs for MRI scanners using high magnetic fields have been reported, they are mostly based on fluoride matrices, which are known for their low chemical stability in aqueous suspensions. Chemically stable MRI CAs for high-field magnets are therefore needed to enable the advances in MRI technique. Herein, we synthesized uniform DyPO4 nanoparticles (NPs) with tuneable sizes between 23 and 57 nm using homogeneous precipitation in butanol. The NPs were successfully functionalized with polyacrylic acid (PAA) and showed good colloidal stability in aqueous suspensions. Chemical stability was also assessed in PBS, showing negligible solubility. The effect of particle size on the transversal relaxivity value (r2) was further explored at 9.4 T, finding a clear increase in r2 with particle size. The r2 value found for the largest NPs was 516 mM−1 s−1, which is, to the best of our knowledge, the highest r2 value ever reported at 9.4 T for any Dy-based nanometric particles in the literature. Finally, the latter NPs were submitted to biosafety studies after polyethylene glycol (PEG) functionalization. Cell morphology, induction of necrotic/late apoptotic cells, and mitochondrial activity were thoroughly analyzed. The results clearly indicated negligible toxicity effects under the assayed conditions. Short- and long-term in vivo pharmacokinetics of the intravenously injected NPs were assessed by dynamic T2-weighted MRI and quantitative T2 mapping, revealing faster liver than spleen uptake, while no accumulation was observed in the kidneys. Finally, no histopathological changes were observed in any of the studied organs, including the liver, kidney, spleen, and lung, which provide further evidence of the biocompatibility of DyPO4 NPs and, therefore, their suitability as bioimaging probes.
1. Introduction
Magnetic Resonance Imaging (MRI) is a non-invasive imaging technology that produces three-dimensional detailed anatomical images thanks to the effect of an external magnetic field. It is very often used for biomedical research as well as for disease detection, diagnosis, and treatment monitoring. Compared to other imaging techniques, such as optical imaging, MRI has high spatial resolution but rather low sensitivity. Increasing magnetic field strength (B0) increases sensitivity and produces, therefore, a higher signal-to-noise ratio with shorter acquisition times. Although the so-called high-field scanners (1.0 T < B0 < 3.0 T) are the most popular MRI instruments used in clinics nowadays, the very high field (3.0 T < B0 < 7.0 T) and ultra-high field (UHF) (B0 > 7.0 T) segments are expected to experience the fastest growth rate during the present decade. In fact, some companies have already developed 7 T MRI machines that are installed in a few hospitals around the world.1,2 Preliminary MRI studies at 9.4 T in humans were reported as early as 2006,3 and have been further continued, demonstrating the feasibility of safe and successful human imaging.4 Even MRI instruments operating at fields as high as 21.1 T are currently being used in preclinical research. Safety studies have demonstrated that static exposure at UHF for 2 h does not have severe long-term effects on mice.5 The primary goal of UHF systems is to obtain structural and functional information about the brain with the highest possible spatial resolution.The contrast in MRI images is generated from differences in local water proton density and in the proton relaxation times (T1 and T2), which is a magnetic resonance characteristic of protons. In addition to using higher magnetic fields, the contrast in MRI can be further improved through the use of the so-called MRI contrast agents (CAs). MRI CAs are exogenous substances that decrease T1 and T2. The capacity of a CA to decrease T1 is known as longitudinal relaxivity (r1), while its capacity to shorten T2 is known as transversal relaxivity (r2). Gadolinium complexes and, to a lesser extent, iron-based CAs are currently used, respectively, as positive and negative CAs in clinics. Both show good contrast at low magnetic fields, but their efficiency drastically decreases at high fields. For example, iron oxide CAs are known to saturate their magnetization at around 1.5 T,6 while Gd-based CAs present their optimal contrast below 1 T.7 Suitable CAs for use in UHF MRI scanners are therefore urgently demanded.8,9
It is well known that paramagnetic ions other than Gd3+, with fast electronic relaxation rates,10 relax protons via a mechanism known as Curie spin relaxation that primarily affects T2.11 Curie-spin relaxation increases substantially with the strength of the magnetic field and is proportional to the square of the magnetic moment of the lanthanide (Ln) ion. Among lanthanides, Dy3+ shows the highest magnetic moment value, which has generated a number of studies on the use of Dy-complexes as T2 CAs for high field MRI.2,12–16 They exhibit moderate r2 values at very high (>3.0 T) magnetic fields although they suffer from short blood circulation times. At UHF, these complexes possess low r2 values, exemplified by Dy-DOTA with r2 = 10.02 mM−1 s−1 at 11.7 T (ref. 14) and Dy[EOB-DO3A] with r2 = 2.67 mM−1 s−1 at 9.4 T.15 In contrast to Dy-complexes, several studies on Dy-based inorganic nanoparticles (NPs) have been published showing r2 values as high as 559.37 mM−1 s−1 at 9.4 T, which demonstrate their potential as MRI CA at UHF.17–20 Ln-Based inorganic NPs show an additional and important advantage relative to organic complexes, consisting in their capacity to be surface functionalized. NPs functionalization consists of the attachment of functional groups to the NP surface to confer them with colloidal stability, which is an essential requirement for bioapplications. A frequently used additive for the functionalization of Ln-based NPs is polyacrylic acid (PAA), whose carboxylate groups can also be used as anchors for further binding different molecules. Among them, polyethylene glycol (PEG) is especially interesting as it allows for decreasing immunogenicity and increasing the NPs circulation time.21 Functionalized NPs can also be conjugated to biological molecules such as antibodies and peptides to provide them with targeting capacity, i.e., the NPs can be specifically directed to the region of interest, thus reducing the dose required.22 Most of the reported Dy-based inorganic NPs consist of fluoride matrices, which are known to exhibit poor stability in aqueous media,23,24 leading to the release of potentially toxic fluoride ions. This justifies the search for less soluble alternatives such as Ln-phosphates which show higher chemical stability in water.25 To the best of our knowledge, the only Dy phosphate-based NPs reported in the literature as MRI CAs are Dy3+-doped β-tricalcium phosphate NPs, with a low content in Dy and correspondingly low r2 value (3.43 mM−1 s−1 at 1.5 T),26 but their relaxivity at high fields is not reported.
In this study, we have synthesized uniform DyPO4 nanoparticles with tuneable sizes from 23 nm to 57 nm. The NPs have been functionalized with polyacrylic acid (PAA) and the colloidal and chemical stability of the resulting NPs in phosphate buffered saline has been analyzed. The ability of the NPs to increase the contrast of MRI images at an ultra-high magnetic field has been evaluated through the measurement of the proton relaxation times in aqueous suspensions of the NPs at 9.4 T. Finally, in vitro cytotoxicity, in vivo pharmacokinetics, and biodistribution of PAA@PEG functionalized NPs have been studied to assess the suitability of the studied NPs as MRI contrast agents for in vivo use.
2. Results
2.1. NPs synthesis, morphology, and crystal phase
TEM micrographs in Fig. 1 show the NPs obtained after aging, at 150 °C for 1 hour in a microwave oven (MW), three butanol solutions containing dysprosium nitrate (0.02 M) and phosphoric acid (0.075 M, 0.15 M, and 0.30 M). All three micrographs showed uniform NPs whose size increased with increasing H3PO4 concentration, as observed in the histograms plotted in the same figure. Specifically, the use of very dilute (0.075 M) H3PO4 resulted in cube-like NPs with an average value of the face diagonal of 23 nm, while doubling H3PO4 concentration to 0.15 M gave also rise to cube-like NPs showing a larger face diagonal size (37 nm). Finally, when the concentration of H3PO4 was increased to 0.30 M, more elongated NPs were precipitated with a long axis length of 57 nm. The H3PO4 concentration allowed therefore tuning the NPs size from 23 nm to 57 nm while keeping their uniform shape. The NPs shown in Fig. 1 will from now on be called Dy23, Dy37, and Dy57, in reference to their size.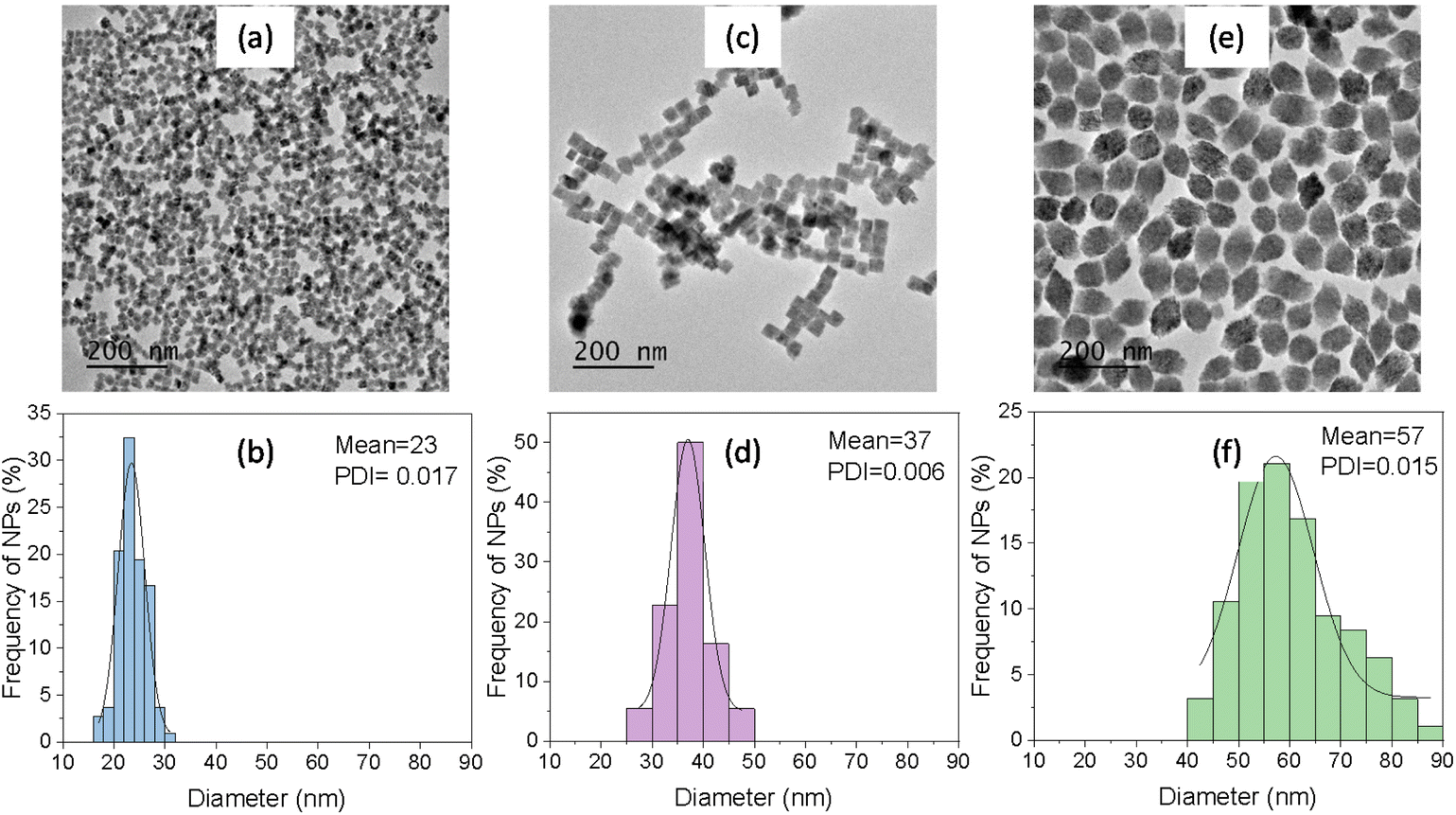 | ||
| Fig. 1 TEM micrographs and corresponding size distribution histograms of DyPO4 NPs synthesized by solvothermal reaction of butanol solutions of dysprosium nitrate (0.02 M) and different H3PO4 concentrations in a microwave oven at 150 °C for 1 h. [H3PO4] = 0.075 M (a and b, Dy23 NPs), 0.15 M (c and d, Dy37 NPs), 0.30 M (e and f, Dy57 NPs). PDI = Polydispersity index. | ||
The effect of other experimental parameters such as solvent nature, aging temperature, and heating source on the NPs morphology was subsequently analyzed. Fig. S1† shows the TEM micrographs of the precipitates obtained by changing just one of the experimental conditions used to synthesize the NPs shown in Fig. 1c (Dy37). It can be observed that the solvent nature produced a drastic change in the precipitate obtained, so that using octanol instead of butanol resulted in the precipitation of heterogeneous, fully aggregated tiny NPs (Fig. S1a†). Another key factor for the synthesis of uniform NPs was the reaction temperature. When the reaction was carried out at 120 °C (Fig. S1b†) the coexistence of both heterogeneous rods and cubic-like NPs was observed in the reaction product while increasing the temperature to 180 °C gave rise to uniform, cubic-like NPs with ∼70 nm edge size (Fig. S1c†). A minimum reaction temperature of 150 °C was, therefore, necessary to obtain homogeneous NPs, while increasing the temperature to 180 °C gave rise to an increase in particle size. Finally, when conventional heating was used instead of MW heating, uniform, cubic-like NPs with ∼65 nm edge size were observed (Fig. S1d†). Conventional heating resulted, therefore, in the precipitation of larger particles compared to MW heating. All these changes must be assigned to variations in the kinetics of precipitation which also affect the nucleation and growth processes.27
The crystal phase of the Dy23, Dy37, and Dy57 NPs was analyzed by XRD (Fig. 2). All three patterns exhibited the same set of reflections, compatible with tetragonal DyPO4 (PDF 00-026-0593). No additional reflections were observed in any of the patterns, indicating the absence of other crystalline phases. It can be noted that the width of the reflections decreased with increasing particle size, indicating that the crystallite size increased correspondingly. The crystallite size was estimated from the half-width at half maximum of the reflection located at ∼35° 2θ using the Scherrer formula. Crystallite size values of 26 nm, 34 nm, and 52 nm were obtained for the Dy23, Dy37, and Dy57 NPs, respectively. The value of the crystallite size, very close to the NPs size in each case, suggests that the NPs were single crystals in nature.
 | ||
| Fig. 2 XRD patterns of Dy23, Dy37, and Dy57 NPs synthesized in the conditions described in Fig. 1. Ticks at the bottom of the figure correspond to tetragonal DyPO4 (PDF 00-026-0593). | ||
2.2. Analysis of the colloidal and chemical stability
The colloidal stability of Dy23 and Dy37 (Fig. S2†) and of Dy57 NPs (Fig. 3a) suspended in water (native pH ∼ 3.5) was evaluated from DLS curves. Mean hydrodynamic diameters of 240 nm, 164 nm, and 142 nm, respectively, were obtained for Dy23, Dy37, and Dy57. These values, higher than the dimensions of the particles obtained from TEM micrographs, indicated that all three types of NPs were aggregated in water. Functionalization of the NPs with PAA was carried out to try to reduce such aggregation. The NPs obtained after such a functionalization process will be called Dy23@PAA, Dy37@PAA, and Dy57@PAA from now on. The success of the functionalization process was monitored through FTIR spectroscopy and thermogravimetry curves. Fig. 3b shows the FTIR spectra of Dy57 NPs before and after PAA coating. The corresponding FTIR spectra of Dy23 and Dy37 NPs are plotted in Fig. S3.† The spectra of the three NPs before functionalization showed the characteristic bands associated with the phosphate vibrational modes (located at wavenumbers <1250 cm−1)28 and adsorbed water (at 3400 cm−1 and 1625 cm−1) plus a low-intensity band at around 1400 cm−1 (marked with an asterisk) that can be assigned to the presence of residual solvent molecules on the NPs surface. On the other hand, the FTIR spectra of all three NPs after functionalization showed two additional bands at ∼1460 cm−1 and 1560 cm−1 (labeled with arrows) that can be assigned to the vibrations of the carboxylate groups of the PAA molecules.29 On the other hand, the TG curve of Dy57 NPs before PAA functionalization (Fig. 3c) showed a first weight loss of around 4% at T < 100 °C, corresponding to the removal of adsorbed water and a second, progressive one at T > 100 °C that could be assigned to the decomposition of the residual solvent molecules. The curve corresponding to the Dy57@PAA NPs showed, in addition to both weight losses, a steeper loss of around 4% at ∼300 °C that can be due to the decomposition of PAA entities. Similar TG curves were observed for Dy23 and Dy37 before and after functionalization (Fig. S4†). The FTIR and TG results demonstrate, therefore, that all three types of NPs were successfully coated with PAA. The colloidal stability of the functionalized NPs suspended in water (native pH ∼ 7.0) was then re-evaluated using DLS. The DLS curves of the functionalized NPs suspended in water significantly shifted towards lower hydrodynamic values with respect to those recorded before functionalization, as observed in Fig. 3a for Dy57@PAA NPs and in Fig. S2† for Dy23@PAA and Dy37@PAA. It can be therefore concluded that the functionalization process notably improved the colloidal stability of the NPs in water. However, all three types of NPs showed a certain degree of aggregation, as inferred from the higher values of their hydrodynamic size in water (86 nm, 85 nm and 105 nm for Dy23, Dy37, and Dy57 NPs, respectively) compared with their TEM size. | ||
| Fig. 3 (a) DLS size distribution curve in water, (b) FTIR spectra, and (c) thermogravimetry curves of Dy57NPs and Dy57@PAA NPs. Insets in (b) are magnifications of selected regions for clarity. | ||
Finally, the PAA-functionalized NPs were suspended in PBS (pH = 7.4) to assess their colloidal stability in a buffer solution commonly used in biological research to simulate physiological fluid. All three NPs functionalized with PAA could be easily re-dispersed in the buffer, as demonstrated by the DLS curves shown in Fig. 4, with hydrodynamic sizes similar to those in water (between 80 nm and 100 nm).
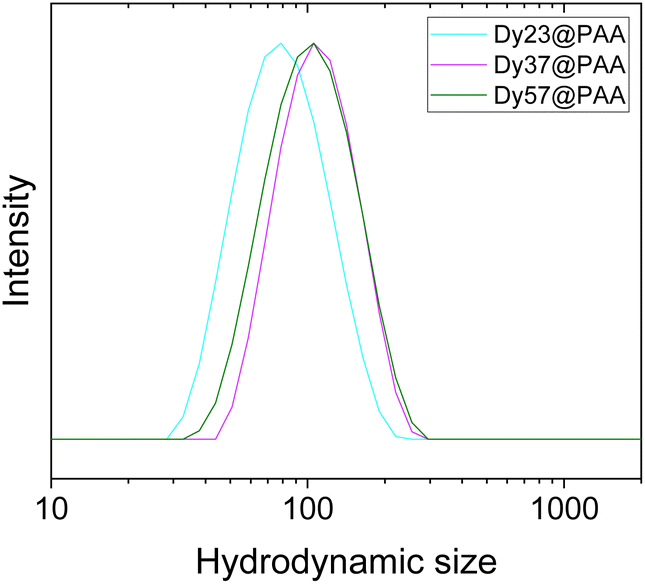 | ||
| Fig. 4 DLS curves of Dy23@PAA, Dy37@PAA, and Dy57@PAA NPs suspended in PBS. | ||
Finally, in order to analyze the chemical stability of DyPO4 NPs in physiological medium, we have selected the Dy57@PAA NPs as a proof of concept and studied their possible dissolution with time in PBS suspension (pH = 7.4) at 37 °C (physiological temperature). The value of Dy fraction dissolved after 1, 3, and 5 weeks of incubation was 0.15%, 0.10%, and 0.72%, respectively, which indicates a negligible degree of dissolution of DyPO4 in PBS. This value was, in addition, significantly lower than those reported for fluoride-based NPs (NaYF4 and KYF4) incubated in the same conditions for just 3 days, which showed 30% and 45%, respectively, of fluorine dissolved.23 In conclusion, DyPO4 NPs show an excellent degree of chemical stability under simulated physiological conditions.
2.3. Relaxivity study
The performance of the three different NPs@PAA as UHF MRI contrast agents was evaluated using a preclinical 9.4 T MRI scanner at room temperature (RT, 23 °C). Fig. 5a shows the T2-weighted MR images obtained in aqueous suspensions with different concentrations of Dy23@PAA, Dy37@PAA, and Dy57@PAA NPs. The images became darker as the NPs concentration increased for all three samples, and they turned practically black at concentrations as low as ∼0.3 mM Dy, which indicates that the NPs behaved as excellent negative MRI CAs. The inverse of the experimentally determined transverse relaxation rates (1/T2) of water protons was plotted against the molar concentration of Dy3+ in the aqueous suspensions (Fig. 5b). The r2 values, determined from the slope of the fitted line, were as high as 395, 432, and 516 mM−1 s−1 for Dy23@PAA, Dy37@PAA, and Dy57@PAA NPs, respectively. The r2 values increased, therefore, with increasing particle size. This trend agrees well with the literature for other nanoparticulated systems such as HoF3,16 NaHoF4,30 NaDyF4,17 Dy2O3,18 and HoPO4,29 where an increase in transversal relaxivity was observed with increasing nanoparticle size. This effect is due to the dependence of the proton relaxation enhancement on spins correlation times, as explained in detail by Yung.31 It is reported that NPs aggregation could also influence their MRI performance.32 However, in our case, the Dy37 NPs clearly showed a higher r2 value than the Dy23 NPs in spite of their very similar hydrodynamic diameters in water. Therefore, the r2 values are governed by the core size. The relaxivity values reported for our DyPO4@PAA NPs were, to the best of our knowledge, the highest r2 values ever reported at 9.4 T for any Dy-based nanometric particles in the literature, such as DyF3, NaDyF4, or Dy2O3, which showed r2 values between 100 and 380 mM−1 s−1.16–18 Sample Dy57@PAA, showing the highest r2 value (516 mM−1 s−1) among the three NPs of this study, was subsequently used for biocompatibility studies and for in vivo experiments. | ||
| Fig. 5 (a) Concentration-dependent T2 weighted phantom images of the Dy23@PAA, Dy37@PAA, and Dy57@PAA NPs at 9.4 T. (b) T2 relaxation rates as a function of Dy concentration of Dy23@PAA, Dy37@PAA, and Dy57@PAA aqueous suspensions at 9.4 T. | ||
2.4. Biocompatibility studies
Previous to biocompatibility studies, the Dy57@PAA NPs were coated with PEG, as described in the Experimental section, with the purpose of improving their systemic circulation time and avoiding their clearance by the immune system.21 The NPs were successfully PEGylated, as demonstrated by the FTIR spectra shown in Fig. S5a.† Fig. S5b† shows the DLS curve of the corresponding NPs suspended in PBS, exhibiting a hydrodynamic size of 164 nm. The PEGylated sample was submitted as well to magnetic resonance studies, obtaining a value for r2 of 438 mM−1 s−1, slightly lower than that of the NPs before PEGylation (516 mM−1 s−1) (Fig. S5c†). The surface coating is an important factor that influences transversal relaxivity. In general, according to a quantum mechanical outer-sphere theory, r2 decreases with increasing the thickness of the surface coating.33 In our case, the PEGylated sample, with a thicker coating than the non-PEGylated nanoparticles (just coated with PAA), gave a lower r2 value than the latter, in agreement with the theory. The corresponding phantom images also show a clear darkening with increasing NPs concentration (Fig. S5d†).Biosafety studies of Dy57@PAA@PEG NPs were evaluated in HFF-1 human foreskin fibroblasts. Several parameters were examined to get a complete assessment of the cytotoxic behavior, such as cell morphology, induction of necrotic/late apoptotic cells, and mitochondrial activity. When merging brightfield, DNA staining (Hoechst 33342), and nuclei staining (TO-PRO-3 Iodine) to get microscopy images, it was found that cells exposed to a NPs concentration up to 100 μg mL−1 (referred to Dy3+), did not show any appreciable morphological change (Fig. 6a–c).
 | ||
| Fig. 6 Optical microscopy images of HFF-1 fibroblasts resulting from the merge of bright field (grey), Hoechst 33342 (blue) and TO-PRO-3 Iodine (red) images: (a) negative control, (b) positive control, (c) cells exposed to 100 μg ml−1 of Dy57@PAA@PEG NPs. Scale bar corresponds to 50 μm. (d) Total number of cells per well after exposure to increasing concentration of Dy57@PAA@PEG NPs. (e) Percentage of dead cells after exposure to increasing concentration of Dy57@PAA@PEG NPs. (f) MTT assay of cells exposed to increasing concentration of Dy57@PAA@PEG NPs. | ||
On the other hand, cellular necrosis and apoptosis were assessed in the “live-dead” assay. The total number of cells per well remained unaltered for all the NPs concentrations tested (Fig. 6d) while these NP concentrations did not give rise to a relevant increase in cell death, which remained very close to zero in all cases (Fig. 6e). Finally, no statistically significant effect on mitochondrial activity was observed with the MTT assay, which showed a cell survival close to 100% for NPs concentrations up to 100 μg mL−1 Dy (Fig. 6f). These results clearly indicate negligible toxicity effects under the assayed conditions and, therefore, the suitability of the DyPO4 NPs as bioimaging probes.
2.5. In vivo studies
The in vivo behavior of intravenously injected Dy57@PAA@PEG NPs was assessed by dynamic T2-weighted MRI and quantitative T2 mapping. Dynamic MRI was used to obtain short-term pharmacokinetics (Fig. 7), which revealed fast liver and spleen uptake, while no accumulation was observed in the kidneys. Interestingly, liver uptake was notably higher and faster than splenic uptake, agreeing with uptake pharmacokinetics reported for PEGylated iron oxide NPs in the same size range.34 This behavior can be explained by a higher endocytic/phagocytic affinity for nanomaterials from Kupffer cells compared to splenic macrophages, as reported by Tsoi et al.35 | ||
| Fig. 7 Short-term pharmacokinetics of intravenously injected Dy57@PAA@PEG NPs (5 mg Dy kg−1 animal) determined by dynamic T2-weighted MRI. (a) liver, (b) spleen, and (c) kidney. Muscle (grey) is plotted as a reference in all cases. | ||
Additionally, long-term pharmacokinetics, up to 24 h, were also studied by quantitative T2 mapping. As expected, a drastic T2 decay (or R2 increase) was observed in the liver and spleen 1 hour after the injection of Dy57@PAA@PEG NPs, with ΔR2 of 24 s−1 and 14 s−1, respectively (Fig. 8a and b). At 24 h, a considerable number of NPs was still retained in the liver, as evidenced by its low T2 values (ΔR2 = 6.6 s−1, while T2 values in the spleen were almost the same as after 1 hour (ΔR2 = 12.3 s−1). These quantitative results agree well with the short-term pharmacokinetics discussed above, showing higher uptake/affinity of NPs by the liver. Post-mortem quantification of Dy57@PAA@PEG NPs content in tissue extracts by ICP-MS (Fig. 8c) showed high concentrations of Dy in the liver (115 g Dy kg−1 of tissue) and much lower in the spleen (29 g Dy kg−1 of tissue), which corresponded to ≈23% and ≈0.6% respect of the injected dose, respectively. This result might seem controversial with the MRI data; they could be explained by the total or partial degradation of NPs in the liver, leading to a decay in the R2 values, even with a high concentration of Dy in the tissue, while much less NPs degradation occurred in the spleen. Therefore, hepatobiliary elimination, which has been proclaimed as one of the main mechanisms of removing NPs from the bloodstream,36 can be described as the main excretion pathway for these NPs.
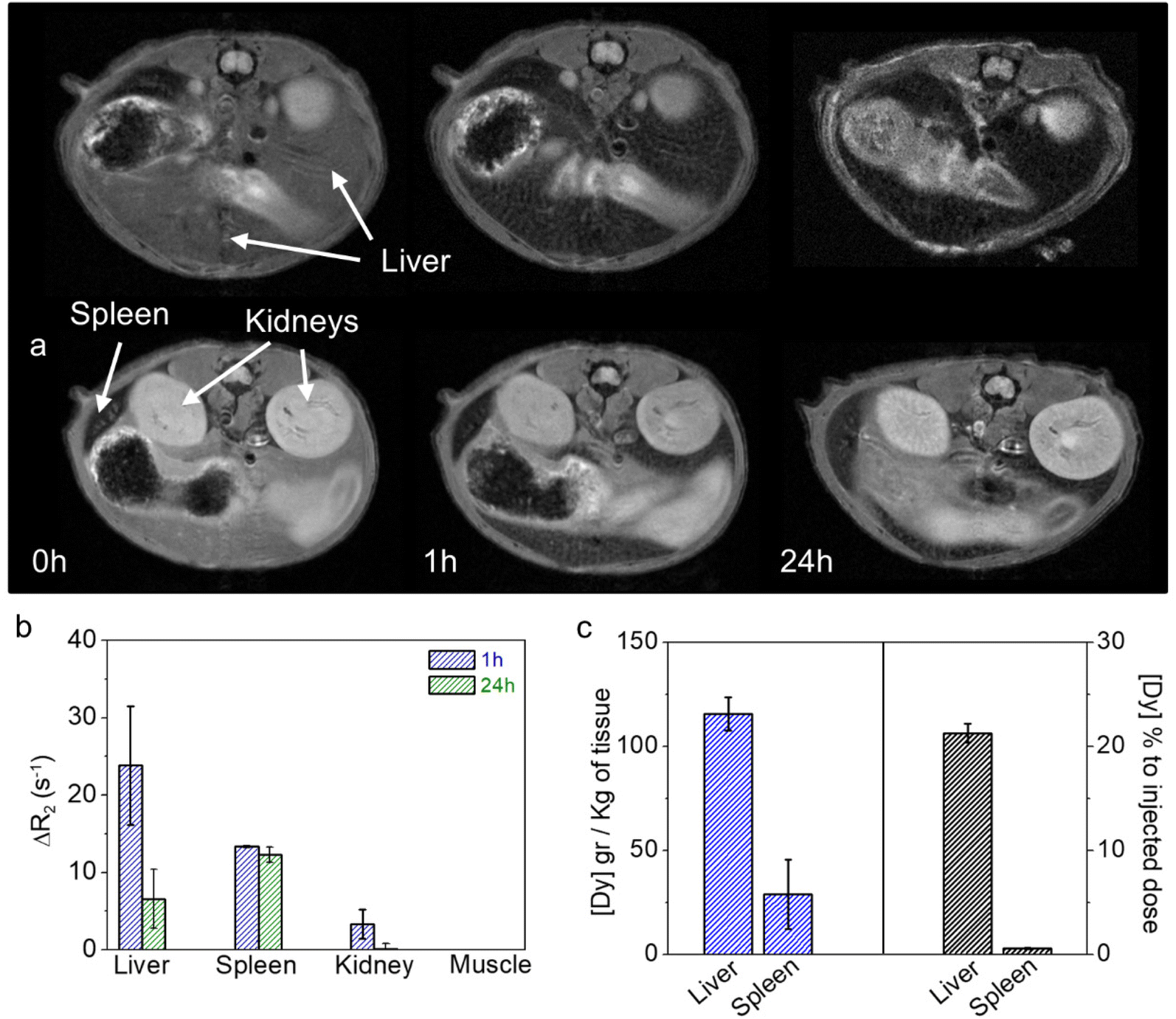 | ||
| Fig. 8 Representative T2-weighted MR images at different time points after the intravenous injection of Dy57@PAA@PEG NPs (5 mg Dy kg−1 animal) (a). ΔR2 changes in liver, spleen, kidney, and muscle calculated from MRI T2 maps at different time points (b). Absolute and relative concentrations of Dy (g Dy kg−1 of tissue and % to the injected dose) in liver and spleen at 24 h post-injection of NPs (c). | ||
As for the kidneys, a slight T2 decay (ΔR2 = 4 s−1) was observed 1 h after NPs injection, which can be attributed to the passage of the remaining NPs in the bloodstream. As expected, kidney T2 values returned to normality at 24 hours, consistent with the absence of NPs in the bloodstream at that time.
Finally, tissue sections of the main organs were analyzed histologically to assess the in vivo toxicity of Dy57@PAA@PEG NPs (Fig. 9). All organs analyzed, including liver, kidney, spleen, and lung, exhibited normal cytoarchitecture, comparable to controls (injected with saline), with no observable histopathological changes, such as necrosis or inflammatory cell infiltrations, indicative of tissue damage.37 These findings provide further evidence of the biocompatibility of Dy57@PAA@PEG NPs.
 | ||
| Fig. 9 H&E staining of tissue sections of liver, kidney, spleen, and lung injected with saline (Ctrl) and with Dy57@PAA@PEG NPs. | ||
3. Conclusions
DyPO4 nanoparticles were synthesized using a homogeneous precipitation method in butanol at 150 °C from dysprosium nitrate and phosphoric acid in a microwave oven. NPs size could be tuned from 23 nm to 57 m by varying the phosphoric acid concentration in the starting solution. The experimental conditions that were found necessary to synthesize uniform NPs were quite restrictive, as their variation gave rise to either heterogeneous or aggregated NPs. After functionalization with PAA, the NPs showed good colloidal stability in both water and PBS suspensions. The high chemical stability was also checked in a simulator of physiological medium; the NPs remained unaltered after five weeks at 37 °C. Transversal relaxivity values (r2) of 395, 432, and 516 mM−1 s−1 were found for the 23, 37, and 57 nm sized NPs at 9.4 T, the latter being the highest r2 value ever reported for Dy-based NPs at that magnetic field. The biocompatibility of the PEG-functionalized NPs was assessed, exhibiting negligible toxicity effects. Moreover, pharmacokinetic assays indicated faster liver than spleen uptake, while no accumulation was observed in the kidneys. Finally, histological studies showed no tissue damage in any of the studied organs, providing further evidence on the biocompatibility of DyPO4 NPs and, therefore, their suitability as bioimaging probes.4. Experimental section
4.1. Reagents
Dysprosium nitrate hydrate (Dy(NO3)3·5H2O, Sigma Aldrich, 99.99%), phosphoric acid (Sigma Aldrich, 85%), butanol (Sigma Aldrich, >99.5%), and 1-octanol (Merck, >99%) were used for NPs synthesis. Poly(acrylic acid) (PAA, average Mw ∼ 1800, Sigma-Aldrich) was used for NPs functionalization with PAA. For NPs functionalization with PEG, the following reagents were used as received: 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide (EDC – Sigma Aldrich, USA), N-hydroxysulfosuccinimide (s-NHS -Sigma Aldrich, USA) and α-methoxy-ω-amino PEG of 5000 Da (PEG, RappPolymer, Germany). Colloidal stability of NPs was evaluated using an aqueous solution of Phosphate Buffered Saline (PBS, Sigma Aldrich) that was prepared as follows: one tablet of PBS was dissolved in 200 mL water to obtain 137 mM NaCl, 2.7 mM KCl and 10 mM phosphate buffer, pH 7.4 at 25 °C. An aqueous solution of sodium tetraborate decahydrate (Sigma Aldrich, >99.5%) 10 mM was prepared and its pH was adjusted to 9 with NaOH. We call this solution borate buffer from now on.4.2. NPs synthesis
For the synthesis of uniform dysprosium phosphate nanoparticles, we used a homogeneous precipitation reaction. Essentially, the method consists of aging a mixture of Dy(NO3)3 with phosphoric acid in butanol at 150 °C in a microwave oven (Sineo MDS-8, Microwave Chemistry Workstation). The standard procedure is as follows: dysprosium nitrate was dissolved in 30 mL of butanol at 50 °C for 1 h under magnetic stirring to facilitate dissolution in order to get a 0.02 M dysprosium nitrate solution. Once dissolved, the solution was admixed with 616 microliters of H3PO4 to obtain a final solution with 0.30 M H3PO4 concentration. The mixture was aged in tightly closed test tubes using a microwave oven at 150 °C for 1 h. The resulting dispersion was cooled down to RT, centrifuged to remove the supernatants, and washed twice with ethanol and once with double distilled water. The NPs were finally stored either in distilled water or dried at 50 °C for some analyses. Additional experiments were conducted to analyze the effect of synthesis parameters (solvent nature, H3PO4 concentration, aging temperature, and heating source) on the characteristics of the precipitated nanoparticles.4.3. NPs functionalization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 EDC:15 NHS (COOH refers to the carboxylic groups in PAA attached to the NPs). The resultant suspension was stirred at RT for 2 h in the end-over-end roller and admixed with 1.56 ml of a PEG solution in borate buffer (10 mg mL−1) (in accordance with the ratio 1 COOH:2 PEG). Finally, after 45 minutes of stirring at RT in the end-over-end roller, NPs were separated by centrifugation at 14500 rpm for 5 minutes. The resulting NPs were washed twice with borate buffer and they were suspended in 1 mL of fresh borate buffer and stored at 4 °C. This protocol was repeated ten times to have enough NPs for the rest of the analyses.
:10 EDC:15 NHS (COOH refers to the carboxylic groups in PAA attached to the NPs). The resultant suspension was stirred at RT for 2 h in the end-over-end roller and admixed with 1.56 ml of a PEG solution in borate buffer (10 mg mL−1) (in accordance with the ratio 1 COOH:2 PEG). Finally, after 45 minutes of stirring at RT in the end-over-end roller, NPs were separated by centrifugation at 14500 rpm for 5 minutes. The resulting NPs were washed twice with borate buffer and they were suspended in 1 mL of fresh borate buffer and stored at 4 °C. This protocol was repeated ten times to have enough NPs for the rest of the analyses.
4.4. Chemical stability studies
NPs functionalized with PAA were suspended in PBS (pH = 7.4) and the suspension was magnetically stirred at 37 °C for several weeks. A portion of the suspension was extracted and centrifuged after 1, 3, and 5 weeks and the supernatants were analyzed by ICP to determine the fraction of dissolved dysprosium.4.5. Characterization techniques
Transmission electron microscopy (TEM, JEOL 2100Plus) was used to determine particles shape and size. To prepare the sample for the TEM analyses, a droplet of the NPs aqueous suspension was deposited on a copper grid coated with a transparent polymer. Particle size distribution curves were obtained by counting about one hundred of particles on the TEM micrographs using the ImageJ open-source image processing program.The colloidal stability of the NPs before and after functionalization was assessed by Dynamic Light Scattering (DLS, Malvern Zetasizer Nano-ZS90). DLS curves were obtained both in aqueous and PBS suspensions with ∼0.5 mg NPs mL−1.
Ion Coupling Plasma (ICP) analyses of NPs were carried out in an iCAP 7200 ICP-OES Duo (ThermoFisher Scientific) apparatus. ICP analyses on tissues were carried using a PerkinElmer NexION installed at the Central Research Services (University of Malaga, Spain). Tissue samples digestion was performed in a microwave digestion system (Milestone). In brief, 0.1 mg of tissue was digested with 6 mL of nitric acid at 240 °C and 40 bar for 50 min. Then, Milli-Q water was added up to 25 mL.
X-ray powder diffraction (XRD) was used for phase identification. The XRD patterns were recorded with a Panalytical X'Pert Pro diffractometer (Cu Kα) with an X-Celerator detector in the angular range of 10° < 2θ < 80° using a 2θ step width of 0.02° and 10 s counting time. The crystallite size was estimated using the Scherrer formula from the full width at half maximum (FWHM) of a single reflection. A shape factor of k = 0.9 and an instrumental broadening factor of 0.112 were used for the calculation.
The success of the functionalization processes was proved using Fourier Transform Infrared (FTIR) spectroscopy and Thermogravimetric analyses (TGA). FTIR (Jasco FT/IR-6200) was conducted on NPs pellets diluted in KBr while TGA curves were recorded in a Q600TA Instrument at a heating rate of 10 °C min−1 in air.
4.6. Magnetic relaxivity
Transverse magnetic relaxivity (r2) was measured at 1.44 T on a Bruker Minispec (TD-NMR) system and at 9.4 T on a Bruker Biospec MRI system. T2 values were determined in aqueous suspensions of NPs with concentrations of Dy ranging from 0.01 to 0.70 mM, using spectroscopic or imaging sequences based on the Carl-Purcell-Meiboom-Gill (CPMG) scheme. r2 values were obtained from the slope of the linear fit of 1/T2 to the concentration of Dy expressed in mM.4.7. Cytotoxicity
Cytotoxicity was determined in human fibroblasts (HFF-1) using Live–Dead and MTT assays. Fibroblasts were used in this work with the purpose of testing unspecific cytotoxicity in normal cells. The Live–Dead assay was evaluated on a PerkinElmer Operetta High Content Imaging System. Cell viability markers to study mitochondrial activity, cell morphology, and plasma membrane integrity, were assessed in HFF-1 following the protocols described previously by some of us.394.8. In vivo MRI
All animal studies were performed complying with the Spanish and European Guidelines for Care and Use of Laboratory Animals (R.D. 53/2013 and 2010/62/UE) and approved by the Local Animal Ethics Committee and the Highest Institutional Ethics Committee (Andalusian Government, accreditation number 14/09/2021/130).Mice were anesthetized with isoflurane (0.5–1% in oxygen at 1 L min−1) and their tail vein cannulated for NPs administration. Then, mice were placed in the magnet, where the vital constants, respiration, and body temperature, were monitored throughout the experiment. NPs were administered intravenously at a concentration of 5 mg Dy kg−1 animal, which was enough to observe a good contrast.
MR images were acquired on the Bruker Biospec 9.4 T system mentioned above, equipped with a 40 mm quadrature bird-cage resonator. Both acquisition and image analysis were conducted as previously described elsewhere.40 The acquisition scheme consisted of high-resolution T2-weighted images, T2 parametric images (quantitative), intravenous injection of the NPs, dynamic T2-weighted sequence, and again high-resolution and parametric T2 images. High-resolution T2-weighted images were acquired using a turbo-RARE sequence with respiratory gating (TE = 16 ms, TR = 1000 ms, 4 averages, FOV = 4 cm, matrix size = 384 × 384, slice thickness = 1 mm). T2 maps were acquired using a multi-echo spin-echo sequence (TEs ranging from 7 ms to 448 ms, TR = 3500 ms, FOV = 4 cm, matrix size = 128 × 128, slice thickness = 1 mm). Short-term pharmacokinetics (first 30 min) were obtained from the semi-quantitative analysis of the dynamic T2-weighted sequence. Quantitative T2 images were acquired at 0, 1, and 24 h to assess the long-term pharmacokinetics and biodistribution of NPs. After 24 h of NPs injection, mice were sacrificed and tissue samples collected for histological evaluation and quantification of the Dy content.
4.9. Histology
Kidney, spleen, and liver tissue sections were stained with Haematoxylin and Eosin (H&E) to assess tissue architecture by light microscopy. The tissues were fixed in 4% formaldehyde (Panreac, pH 7 buffered) for 48 h, changing the 4% formaldehyde after 24 h. Then, the samples were dehydrated through graded ethanol, and embedded in paraffin (temperature 56° C for 2 h under stirring and vacuum). The detailed procedures are described below. Haematoxylin and Eosin (H&E): paraffin-embedded samples were sectioned at 7 μm thickness, then deparaffinized, rehydrated, and stained with H&E, and then dehydrated in ascending concentrations of ethanol, cleared in xylene, and mounted on commercial glass slides.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Financial support was provided by Junta de Andalucía (P20_00182). This publication is part of the I + D + I Grants RTI2018-094426-B-I00 and CTQ2017-86655-R funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”. Grant PRE2019-090170 is funded by MCIN/AEI/10.13039/501100011033 and by “ESF Investing in your future”. The in vitro and the in vivo experiments have been performed by the ICTS “NANBIOSIS”, more specifically by the nanoimaging unit (U28) of IBIMA-Plataforma BIONAND.References
- J. H. Duyn, NeuroImage, 2012, 62, 1241–1248 CrossRef PubMed.
- H. K. Kim, A. R. Baek, G. Choi, J. J. Lee, J. U. Yang, H. Jung, T. Lee, D. Kim, M. Kim, A. E. Cho, G. H. Lee and Y. Chang, Biomaterials, 2020, 243, 119939 CrossRef CAS PubMed.
- T. Vaughan, L. DelaBarre, C. Snyder, J. F. Tian, C. Akgun, D. Shrivastava, W. Z. Liu, C. Olson, G. Adriany, J. Strupp, P. Andersen, A. Gopinath, P. F. van de Moortele, M. Garwood and K. Ugurbil, Magn. Reson. Med., 2006, 56, 1274–1282 CrossRef PubMed.
- N. J. Shah, Brain Struct. Funct., 2015, 220, 1867–1884 CrossRef PubMed.
- X. F. Tian, D. M. Wang, S. Feng, L. Zhang, X. M. Ji, Z. Wang, Q. Y. Lu, C. Y. Xi, L. Pi and X. Zhang, NeuroImage, 2019, 199, 273–280 CrossRef PubMed.
- M. Harris, S. Biju and T. N. Parac-Vogt, Chem. – Eur. J., 2019, 25, 13838–13847 CrossRef CAS PubMed.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- A. Gautam, P. Komal and R. S. Singh, J. Chem. Sci., 2019, 131 CrossRef CAS.
- A. Banerjee, B. Blasiak, A. Dash, B. Tomanek, F. C. J. M. van Veggel and S. Trudel, Chem. Phys. Rev., 2022, 3, 011304 CrossRef CAS.
- B. M. Alsaadi, F. J. C. Rossotti and R. J. P. Williams, J. Chem. Soc., Dalton Trans., 1980, 2147–2150 RSC.
- G. Pintacuda, M. John, X. C. Su and G. Otting, Acc. Chem. Res., 2007, 40, 206–212 CrossRef CAS PubMed.
- S. Aime, M. Botta, L. Barbero, F. Uggeri and F. Fedeli, Magn. Reson. Chem., 1991, 29, S85–S88 CrossRef CAS.
- P. Caravan, M. T. Greenfield and J. W. M. Bulte, Magn. Reson. Med., 2001, 46, 917–922 CrossRef CAS PubMed.
- M. Harris, C. Henoumont, W. Peeters, S. Toyouchi, L. Vander Elst and T. N. Parac-Vogt, Dalton Trans., 2018, 47, 10646–10653 RSC.
- X. Y. Zheng, J. Pellico, A. A. Khrapitchev, N. R. Sibson and J. J. Davis, Nanoscale, 2018, 10, 21041–21045 RSC.
- A. Baek, H. K. Kim, J. U. Yang, G. Choi, M. Kim, A. E. Cho, Y. H. Kim, S. Kim, B. Sung, B. W. Yang, H. Seo, G. H. Lee, H. K. Ryeom, H. Jung, T. Lee and Y. Chang, J. Ind. Eng. Chem., 2020, 85, 297–307 CrossRef CAS.
- D. Gonzalez-Mancebo, A. I. Becerro, T. C. Rojas, M. L. Garcia-Martin, J. M. de la Fuente and M. Ocana, Part. Part. Syst. Charact., 2017, 34, 1700116 CrossRef.
- G. K. Das, N. J. J. Johnson, J. Cramen, B. Blasiak, P. Latta, B. Tomanek and F. van Veggel, J. Phys. Chem. Lett., 2012, 3, 524–529 CrossRef CAS PubMed.
- M. Norek, E. Kampert, U. Zeitler and J. A. Peters, J. Am. Chem. Soc., 2008, 130, 5335–5340 CrossRef CAS PubMed.
- Y. Li, Y. Y. Gu, W. Yuan, T. Y. Cao, K. Li, S. P. Yang, Z. G. Zhou and F. Y. Li, ACS Appl. Mater. Interfaces, 2016, 8, 19208–19216 CrossRef CAS PubMed.
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernandez, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- J. Nam, N. Won, J. Bang, H. Jin, J. Park, S. Jung, Y. Park and S. Kim, Adv. Drug Delivery Rev., 2013, 65, 622–648 CrossRef CAS PubMed.
- D. Lisjak, O. Plohl, J. Vidmar, B. Majaron and M. Ponikvar-Svet, Langmuir, 2016, 32, 8222–8229 CrossRef CAS PubMed.
- M. I. Saleh, B. Ruhle, S. Wang, J. Radnik, Y. You and U. Resch-Genger, Sci. Rep., 2020, 10, 19318 CrossRef CAS PubMed.
- F. H. Firsching and S. N. Brune, J. Chem. Eng. Data, 1991, 36, 93–95 CrossRef CAS.
- R. Meenambal, P. Poojar, S. Geethanath and K. Sanjeevi, J. Am. Ceram. Soc., 2017, 100, 1831–1841 CrossRef CAS.
- V. K. Lamer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- Y. Gao, M. Fan, Q. Fang, B. Song and W. Jiang, J. Nanosci. Nanotechnol., 2013, 13, 6644–6652 CrossRef PubMed.
- A. I. Becerro and M. Ocana, RSC Adv., 2015, 5, 34517–34524 RSC.
- D. L. Ni, J. W. Zhang, W. B. Bu, C. Zhang, Z. W. Yao, H. Y. Xing, J. Wang, F. Duan, Y. Y. Liu, W. P. Fan, X. Y. Feng and J. L. Shi, Biomaterials, 2016, 76, 218–225 CrossRef CAS PubMed.
- K. T. Yung, Magn. Reson. Imaging, 2003, 21, 451–463 CrossRef CAS PubMed.
- H. T. Ta, Z. Li, Y. Wu, G. Cowin, S. Zhang, A. Yago, A. K. Whittaker and Z. P. Xu, Mater. Res. Express, 2017, 4, 116105 CrossRef.
- W. Zhang, L. Liu, H. Chen, K. Hu, I. Delahunty, S. Gao and J. Xie, Theranostics, 2018, 8, 2521–2548 CrossRef CAS PubMed.
- M. Pernia Leal, C. Caro and M. L. García-Martín, Nanoscale, 2017, 9, 8176–8184 RSC.
- K. M. Tsoi, S. A. MacParland, X.-Z. Ma, V. N. Spetzler, J. Echeverri, B. Ouyang, S. M. Fadel, E. A. Sykes, N. Goldaracena, J. M. Kaths, J. B. Conneely, B. A. Alman, M. Selzner, M. A. Ostrowski, O. A. Adeyi, A. Zilman, I. D. McGilvray and W. C. W. Chan, Nat. Mater., 2016, 15, 1212–1221 CrossRef CAS PubMed.
- W. Poon, Y.-N. Zhang, B. Ouyang, B. R. Kingston, J. L. Y. Wu, S. Wilhelm and W. C. W. Chan, ACS Nano, 2019, 13, 5785–5798 CrossRef CAS PubMed.
- K. E. Ibrahim, M. G. Al-Mutary, A. O. Bakhiet and H. A. Khan, Molecules, 2018, 23, 1848 CrossRef PubMed.
- Z. Grabarek and J. Gergely, Biophys. J., 1988, 53, A392 Search PubMed.
- E. Gomez-Gonzalez, C. Caro, D. Martinez-Gutierrez, M. L. Garcia-Martin, M. Ocana and A. I. Becerro, J. Colloid Interface Sci., 2021, 587, 131–140 CrossRef CAS PubMed.
- C. Caro, M. C. Muñoz-Hernandez, M. Pernia Leal and M. L. García-Martín, Preclinical Mri: Methods and Protocols, 2018, vol. 1718, pp. 409–419 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr02630a |
| This journal is © The Royal Society of Chemistry 2022 |