
Photocatalytic CO2 conversion: from C1 products to multi-carbon oxygenates
 a
Gengfeng
Zheng
*a
a
Gengfeng
Zheng
*a
Abstract
Photocatalytic CO2 conversion into high-value chemicals has been emerging as an attractive research direction in achieving carbon resource sustainability. The chemical products can be categorized into C1 and multi-carbon (C2+) products. In this review, we describe the recent research progress in photocatalytic CO2 conversion systems from C1 products to multi-carbon oxygenates, and analyze the reasons related to their catalytic mechanisms, as the production of multi-carbon oxygenates is generally more difficult than that of C1 products. Then we discuss several examples in promoting the photoconversion of CO2 to value-added multi-carbon products in the aspects of photocatalyst design, mass transfer control, determination of active sites, and intermediate regulation. Finally, we summarize perspectives on the challenges and propose potential directions in this fast-developing field, such as the prospect of CO2 transformation to long-chain hydrocarbons like salicylic acid or even plastics.
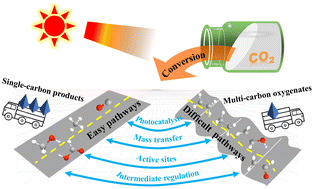
- This article is part of the themed collections: CO2 capture and conversion, Recent Review Articles and 2022 Nanoscale HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

