
Coordinated regulation of phosphorus/nitrogen doping in fullerene-derived hollow carbon spheres and their synergistic effect for the oxygen reduction reaction†
Fancang
Meng‡
a,
Suwei
Wang‡
b,
Bohong
Jiang
a,
Li
Ju
a,
Haijiao
Xie
c,
Wei
Jiang
 *b and
Qingmin
Ji
*a
*b and
Qingmin
Ji
*a
aHerbert Gleiter Institute for Nanoscience, School of Materials Science and Engineering, Nanjing University of Science & Technology, 200 Xiaolingwei, Nanjing, 210094, China. E-mail: jiqingmin@njust.edu.cn
bNational Special Superfine Powder Engineering Technology Research Center, Nanjing University of Science and Technology, 200 Xiaolingwei, Nanjing, 210094, China. E-mail: superfine_jw@126.com
cHangzhou Yanqu Information Technology Co., Ltd., Building 2, Xixi Legu Creative Pioneering Park, No. 712 Wen'er West Road, Hangzhou, 310003, China
First published on 24th June 2022
Abstract
Fullerene-derived carbons have been demonstrated as effective electrode materials for electrocatalytic reactions. The heteroatoms in the carbon matrix are essential to enhance their electrocatalytic performance but are still challenging for effective doping strategies and understanding their synergistic effect. Herein, we regulate the phosphorus/nitrogen (P/N) doping in the carbon structure based on the control mixing of pyritic acid (PA) with the assembled diamine-C60 hollow spheres (N@FHS). After pyrolysis, the carbon spheres are shown to have a homogenous distribution of N and P (NP@CHS). The structural and molecular analysis reveals that the doping of P may facilitate the formation of graphitic N in the carbon framework. When used as electrocatalysts for the oxygen reduction reaction (ORR), NP@CHSs exhibit superior oxygen reduction reaction (ORR) performance in contrast to those of fullerene-derived carbon with single N doping and the commercial Pt/C (20 wt%) catalyst. Density functional theory (DFT) studies indicate that P/N-doping promotes the charge transfer in the carbon structure owing to its strong electronegativity. The enhanced ORR activity should be mainly due to the P- and N-coordinated neighboring C sites with the defective fullerene pentagon ring.
1. Introduction
Owing to their large surface area, good conductivity, tunable morphology, facile preparation, and economic viability, metal-free carbon materials attract great attention to replace Pt as effective electrocatalysts for the oxygen reduction reaction (ORR), the oxygen evolution reaction (OER), the hydrogen evolution reaction (HER), the carbon dioxide reduction reaction (CO2RR) and the nitrogen reduction reaction (NRR).1–4 To boost their electrochemical performance, it is essential to manipulate heteroatom doping in the carbon matrix.5,6 Various strategies based on post-treatment, in situ doping, template-mediated synthesis, etc., have been developed to fabricate single and multiple heteroatom-doped carbons for catalytic applications.7–9 However, comprehensive effects between carbon defects or functions and doping distribution are still unclear. More rational design for effective regulation of heteroatom doping in carbon hosts remains challenging.Fullerenes are an important group of carbon allotropes. The presence of a closed carbon skeleton with pentagon and hexagon rings distinguishes them from other conventional carbon materials, enabling easy chemical modification with functions and unique electronic properties.10,11 They may also act as building blocks to construct supramolecular assemblies with multi-dimensional architectures.12,13 Recently, unfolding fullerenes have been proved to generate a carbon matrix with rich topological defects and active sites, which may facilitate binding with other functional heteroatoms.14,15 These characteristics make them have great potential to impart fullerene-derived carbons with high catalytic activity.
Among various heteroatoms, N-doped fullerene-derived carbons are the most optimal type for electrocatalysis.16,17 A similar atomic radius of N with C makes it relatively easier to incorporate into the carbon framework. The doped N may provide a positive charge density for easier oxygen adsorption and reduce the bandgap between the HOMO and the LUMO for electron transfer. The intrinsic pentagon carbon in fullerenes with N doping is suggested to possess higher electrochemical reactivity than that of N-doped hexagon carbons.18 The compressive surface strain induced by the curved carbon structure may also enhance their catalytic activity.19 With the additional doping of other atoms, the doped fullerene carbons can further boost the catalytic activity due to their synergistic effects.20,21 But until now reports about co-doped fullerene-derived carbon electrocatalysts are still rare. Besides exploring fabrication strategies for precise control of the doped species, the synergistic effect between incorporated elements is also expected for a better understanding to achieve the best catalytic performance from fullerene carbons.
Herein, we study the co-doping of N and P in C60-derived carbons for ORR application through regulating the binding of phytic acid, which serves as the main storage form of P in plant seeds, within the assembled hollow spheres of diamine-modified C60. The intercalation process and subsequent pyrolysis lead to the formation of N and P co-doped hollow carbon spheres (NP@CHSs) with different P/N species (Fig. 1). When used as an electrocatalyst for the ORR, NP@CHSs exhibit excellent catalytic activity and stability. The performance is superior in contrast to both fullerene carbons with single N doping and the commercial Pt/C catalyst. By comparing the structural and molecular features of NP@CHSs, we also analyze the relationship between the state of the N–C–P network and the electrocatalytic performance of fullerene-derived carbon spheres.
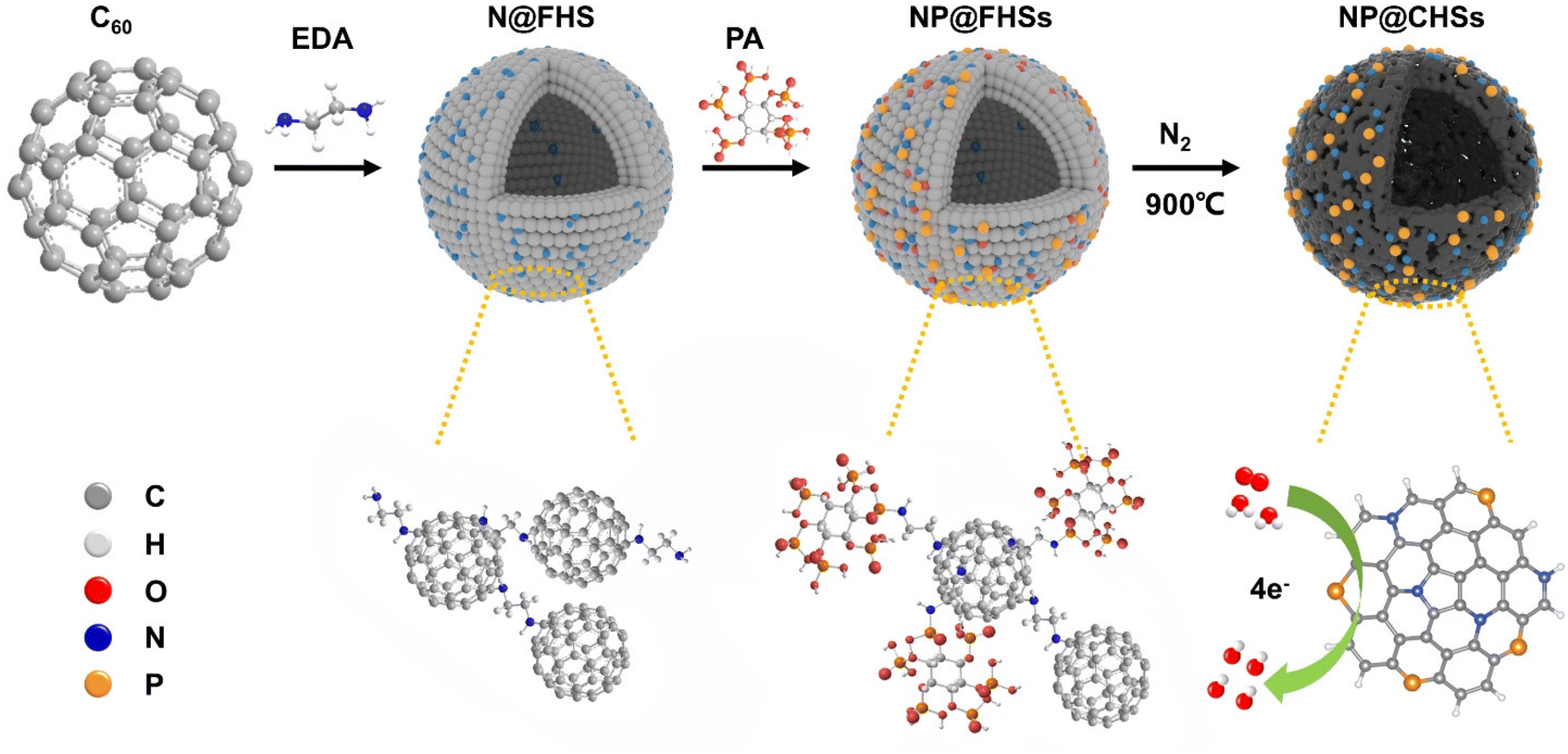 | ||
| Fig. 1 The scheme for the fabrication of N/P co-doped fullerene-derived carbon spheres for ORR catalysis. | ||
2. Experimental
2.1. Materials
Pristine fullerene (C60) powder (purity 99.9%) was purchased from Henan Fullerene Nano New Materials Technology Co. Ltd. Toluene, ethanol, ethylenediamine (EDA), and hydrogen peroxide (H2O2, 30 wt%) were purchased from Sinopharm Chemical Reagent Co. Ltd. Tetrabutylammonium hydroxide (TBAH, 40 wt%) was purchased from J&K Scientific Co. Ltd. Phytic acid solution (PA, 70 wt%) was purchased from Shanghai Macklin Biochemical Co. Ltd. Nafion solution (5 wt%) and the Pt/C catalyst were purchased from Suzhou Sinero Technology Co. Ltd. All chemicals were used as received.2.2. Formation of EDA and PA incorporated fullerene hollow spheres
100 mg of C60 was dissolved in 50 ml of toluene and sonicated for 30 minutes. 600 μl of EDA was dropped into the solution and sonicated for 25 minutes. Then, 150 μl of TBAH and 2 ml of H2O2 were dropped into the mixture solution and stirred at 60 °C for 2 hours. The precipitate was collected by centrifugation and washed three times with ethanol. The sample was finally dried under vacuum and named N@FHS.Based on the above process, various volumes of PA solution (2.4 ml, 4.8 mL, 7.2 mL, and 9.6 mL) were added into the ethanol dispersions of N@FHS and stirred vigorously at room temperature for 12 hours. The precipitate was collected by centrifugation and washed two times with ethanol. The samples were finally dried under vacuum and remarked NP1@FHS, NP2@FHS, NP3@FHS, and NP4@FHS (Table 1).
| Assembled samples | C60s | N@FHS | NP1@FHS | NP2@FHS | NP3@FHS | NP4@FHS |
|---|---|---|---|---|---|---|
| Carbonized samples | C60CS | N@CHS | NP1@CHS | NP2@CHS | NP3@CHS | NP4@CHS |
C60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EDA:PA ratio :EDA:PA ratio |
100:0:0 |
1:60:0 |
1:60:30 |
1:60:60 |
1:60:80 |
1:60:100 |
| Morphology | Irregular particles | Hollow sphere | ||||
2.3. Formation of N and P co-doped carbon hollow spheres
The above fullerene hollow spheres of N@FHS and NP@FHSs were heated to 900 °C at a heating speed of 3 °C min−1 and kept for 2 hours in a tubular furnace under a constant flow of nitrogen gas. The resultant products were marked N@CHS and NP@CHSs (NP1@CHS, NP2@CHS, NP3@CHS, and NP4@CHS), respectively (Table 1).2.4. Electrochemical measurement
Electrochemical measurements were performed on a CS2350H dual unit electrochemical workstation using a standard three-electrode cell in an alkaline medium (0.1 M KOH). Hg/HgCl, graphite, and a modified glassy carbon electrode (GCE) were used as the reference, counter, and working electrodes, respectively.Catalyst inks were made by the following steps: (1) mixing 5 mg of samples in 960 μl of ethanol and 40 μl of Nafion solution (5 wt%) and (2) ultrasonication of the mixture for 1 hour. Then 20 μl of the ink was dropped onto a glassy carbon (GC) disk to form a uniform film at a catalyst loading of 0.5 mg cm−2.
Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) techniques were employed to determine the electrochemical behavior and ORR kinetics of the catalysts. CV plots were recorded in an alkaline medium (0.1 m KOH) under an N2/O2 environment and in the potential range of 0–1.2 V. LSV plots were recorded at a scan rate of 10 mV s−1 with different rotating speeds (400–2500 rpm) in an O2 saturated 0.1 M KOH aqueous solution. The potentials measured in this work were calibrated regarding the reversible hydrogen electrode (RHE) through ERHE = ESCE + 0.059pH + 0.241.
The kinetic parameters involved in the typical ORR process, including the electron transfer number (n) and kinetic current density (Jk), were analyzed based on the Koutecky–Levich (K–L) equation:
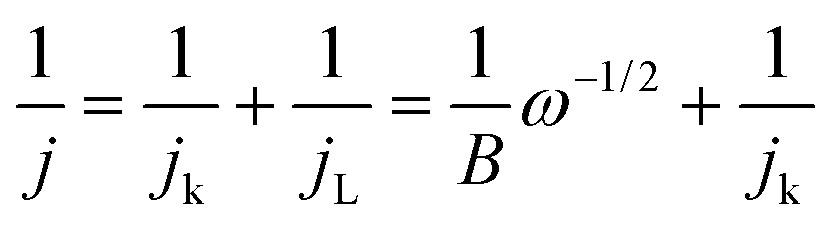
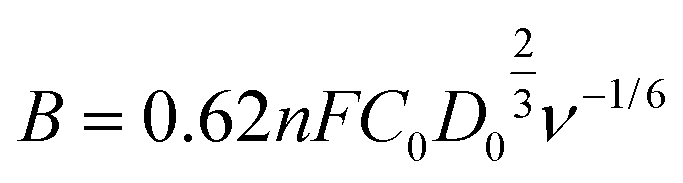
In which, J, JL, and Jk are the measured current density, the limiting current density, and the kinetic current density, respectively, ω is the rotation speed in rpm, F is the Faraday constant (96485 C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3), D0 is the diffusion coefficient of O2 in 0.1 M KOH (1.90 × 10−5 cm2 s−1), and ν is the kinetic viscosity (0.01 cm2 s−1).
The stability of the catalysts was tested by chronoamperometric current–time (i–t) measurements at a constant potential of 0.5 V vs. reversible hydrogen electrode (RHE) for 10 hours, and an accelerated durability test at a scan rate of 100 mV s−1 in an O2-saturated 0.1 M KOH solution for 5000 cycles. Methanol tolerance of i–t responses was measured with/without injecting 1 M methanol into the electrolyte at 0.55 V vs. RHE in an O2-saturated 0.1 M KOH solution.
The performance of the homemade setup Zn–air battery was tested using NP3@CHS (2 mg cm−2) on carbon paper as the air cathode, a polished zinc foil as the anode and an aqueous solution of 6 M KOH as the electrolyte. Current–voltage (I–V) polarization curves were recorded using the LSV technique at room temperature. All tests were conducted in static air without an additional O2 supply.
3. Results and discussion
3.1. Control assembly for fullerene hollow spheres mixed with EDA and PA
Fullerenes have a strong tendency to aggregate into superstructures with different morphologies in polar solvents, which may cause a significant change in their chemical/physical properties.22,23 Furthermore, the incorporation of functional groups by co-assembly or chemical modification may further tune the properties of fullerene superstructures. As an electron-deficient molecule, fullerenes may readily undergo nucleophilic addition reactions. When mixed with EDA, the amine group may bind to C60 fullerene and induce the assembly of amine-C60 into spherical aggregates in toluene.24,25 In this work, the mixing ratio of C60 and EDA is about 1:60. The addition of TBAH and H2O2 in the reaction system may further promote the reaction of amines with fullerenes.26 This mixture system results in the formation of hollow fullerene spheres (N@FHS). As shown in Fig. 2 and S2,† N@FHS has a diameter of about 900 nm and a wall thickness of 100 nm. Different from the assembly structures of bare C60s, the diamine may bridge C60s and form cross-linked C60 assemblies.27,28 C60s are prone to pack densely due to π–π interaction, while bridging by the diamine may enlarge the spacing of C60s in the superstructures. It thus might facilitate the binding of other molecules in the structure. Taking advantage of amine-functionalized fullerene hollow spheres, we control the binding of the P component by mixing different amounts of phytic acid (PA), which possesses a high density of negatively charged phosphate groups. The SEM observation indicates that there are no obvious morphological changes for the EDA:PA molar ratios of 2:1 (NP1@FHS), 1:1 (NP2@FHS), 3:4 (NP3@FHS), and 3:5 (NP4@FHS) in the fullerene hollow spheres (Fig. S1†).
 | ||
| Fig. 2 (a) SEM image of N@FHS; (b) SEM image and (c) TEM image of NP3@CHS; and (d) high-angel annular dark-field (HAADF)-STEM image of NP3@CHS and the corresponding elemental mappings of (e) C, (f) N and (g) P. | ||
The FTIR spectrum of N@FHS (Fig. S3†) shows the vibration and bending adsorption of the N–H bond around 3400 cm−1 and 1650 cm−1, the vibration and deformation absorptions of CH2 at 2970 and 1460 cm−1, and the vibration absorption of the C–N bond adjacent to the carbon cage at 1250 cm−1. After the adsorption of PA, the adsorption peaks for the C–N bond become strengthened and wider for NP@FHSs. The absorption of the N–H bond also shifts to a lower wavenumber. These results indicate the interaction between the amine group on C60 and PA in NP@FHSs.
3.2. Fullerene-derived hollow carbon spheres with N and P components
To stabilize the N and P doping in the carbon matrix for the ORR, heat treatment for N@FHS and NP@FHSs was applied at 900 °C under an N2 atmosphere to obtain fullerene-derived hollow carbon spheres with various N and P hybrid states. The transformed carbon spheres show a similar spherical morphology for N@CHS (without P), NP1@CHS, NP2@CHS, and NP3@CHS (Fig. 2 and Fig. S4–S6†), while NP4@CHS, which formed from a high mixing ratio of EDA:PA, changes to broken spherical structures (Fig. S4†). We suggest that it is caused by pyrolysis of a large amount of PA in the structure. As the O atoms within PA can deplete partial C atoms in C60s during the heat treatment, it thus may induce structural defects in the carbon matrix. But when too many carbon defects exist, the spherical structure might destruct easily.
The structural changes of NP@CHSs from heat treatment are also analyzed by the X-ray diffraction (XRD) technique (Fig. 3a and b). The structures of N@FHS and NP@FHSs show broad diffraction peaks in the XRD patterns (Fig. 3a). The peak centered at 2θ = 6.8° (d = 1.30 nm) should correspond to the diamine-C60 adduct, as the diameter of C60 is about 1 nm and the molecular length of EDA is 0.38 nm.29 In comparison to N@FHS, the peak around 6.8° becomes broader for NP@FHSs. It suggests the binding of PA with diamine-C60 in the superstructure. After the heat treatment, the diffraction peaks at <10° disappear (Fig. 3b). The broad peaks centered at 21° and 44° should arise from the polymorph carbon structure.30 These results indicate the successful transformation of fullerene-derived carbons after the heat treatment at 900 °C. Compared to other NP@CHSs, NP4@CHS shows broader diffraction around 20°. The peak is also shifted to a higher degree (23°). It implies the formation of more dense packing of carbons in NP4@CHS, which might be due to the seriously broken carbon network during the heat treatment.
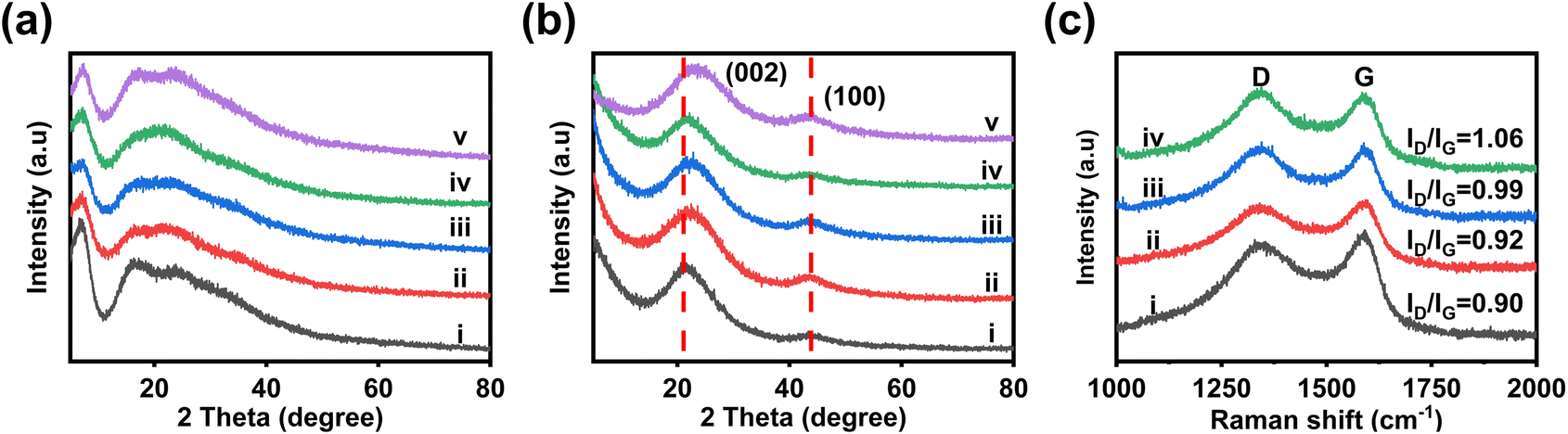 | ||
| Fig. 3 (a) XRD patterns of (i) N@FHS, (ii) NP1@FHS, (iii) NP2@FHS, (iv) NP3@FHS, and (v) NP4@FHS. (b) XRD patterns of (i) N@CHS, (ii) NP1@CHS, (iii) NP2@CHS, (iv) NP3@CHS, and (v) NP4@CHS. (c) Raman spectra of (i) N@CHS, (ii) NP1@CHS, (iii) NP2@CHS, (iv) NP3@CHS, and (v) NP4@CHS. | ||
Raman spectroscopy is employed to study the doping effect from N and P for the carbon features of various NP@CHSs. The spectra of NP@CHSs exhibit two strong bands around 1340 and 1590 cm−1, corresponding to the D band from the defected or disordered graphitic carbon and the G band from in-plane vibration of sp2 carbons (Fig. 3c).31 The typical peaks from C60 (two Ag bands and six Hg bands)32 or Raman peaks of N@FHS and NP@FHS disappeared for NP@CHSs (Fig. S7†). It confirms the successful structural transformation of packed C60s to carbon networks after the heat treatment. The intensity ratios of the D band and G band (ID/IG) for NP1@CHS, NP2@CHS, NP3@CHS, and NP4@CHS are 0.90, 0.92, 0.99 and 1.06. This result suggests that the presence of more PA in the fullerene superstructures might induce the formation of more defects in the carbon structures.
The elemental mapping analysis of NP@CHSs proves the homogenous distribution of N, P and C elements across the carbon structure (Fig. 2). The chemical states of doped elements in N@CHS and NP@CHSs are analyzed by X-ray photoelectron spectroscopy (XPS). The survey wide-range spectra recorded in the range of 0–900 eV reveal the presence of N, O, and C or N, P, O, and C elements in N@CHS and NP@CHSs (Fig. 4a and Fig. S8–S11†). The C 1s spectra of NP@CHSs show the main peak of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.7 eV) with small peaks of the C–O/CN/C–P bond (285.6 eV), the CO/C–N bond (286.7 eV), and the O–CO bond (289.2 eV) (Fig. 4b).33 The N 1s spectra of N@CHS and NP@CHSs can be deconvoluted into four peaks (Fig. 4c and Fig. S8–S11†), which can be assigned to primary pyridinic N (398.5 eV), pyrrolic N (400.0 eV), graphitic N (401.1 eV), and oxidized N (404.4 eV).33 For the P 2p spectra of NP@CHSs, the peak in the region of 128–140 eV can be fitted into two states, which are P–C at 132.6 eV and P–O at 133.9 eV (Fig. 4d).34 These data indicate the existence of a P–C–N network in NP@CHSs. According to the XPS results, we estimate the content ratios of N and P in the spheres (Table S2†). The N content of N@CHS is about 8.64 atom% and that of NP@CHSs is in the range of 3.44–4.64%. The P content of NP@CHSs is varied in the range of 2.63–3.46 atom% according to the addition amount of PA in NP@FHSs. Although the addition ratios of EDA:PA are different for NP@FHSs, the N:P ratios in NP@CHSs are quite similar. We suggest that N in the structure should play the main role in the binding of P. It thus causes a similar ratio of N and P in NP@CHSs for different mixing amounts of PA. Comparing the proportions of N states in NP@CHSs, we can find that the ratio of graphitic N increases with the P amount in the carbon spheres except for the case of NP4@CHS, whose carbon network is largely broken during the heat treatment (Table S1†). The total proportion of graphitic N and pyridinic N of NP@CHSs is shown in the order of NP3@CHS(71.82%) > NP2@CHS(62.57%) > NP1@CHS(59.52%) > NP4@CHS(56.73%) (Fig. 4e and Table S2†). It implies that the presence of P may influence the formation of C–N configuration, especially more active graphitic N and pyridinic N in the structure. The difference in the proportions of P–O and P–C in NP@CHSs (Fig. 4f) also reveals that a more active P–C bond with less oxidized P is inclined to form in the carbon framework under a higher mixing amount of PA.35
C (284.7 eV) with small peaks of the C–O/CN/C–P bond (285.6 eV), the CO/C–N bond (286.7 eV), and the O–CO bond (289.2 eV) (Fig. 4b).33 The N 1s spectra of N@CHS and NP@CHSs can be deconvoluted into four peaks (Fig. 4c and Fig. S8–S11†), which can be assigned to primary pyridinic N (398.5 eV), pyrrolic N (400.0 eV), graphitic N (401.1 eV), and oxidized N (404.4 eV).33 For the P 2p spectra of NP@CHSs, the peak in the region of 128–140 eV can be fitted into two states, which are P–C at 132.6 eV and P–O at 133.9 eV (Fig. 4d).34 These data indicate the existence of a P–C–N network in NP@CHSs. According to the XPS results, we estimate the content ratios of N and P in the spheres (Table S2†). The N content of N@CHS is about 8.64 atom% and that of NP@CHSs is in the range of 3.44–4.64%. The P content of NP@CHSs is varied in the range of 2.63–3.46 atom% according to the addition amount of PA in NP@FHSs. Although the addition ratios of EDA:PA are different for NP@FHSs, the N:P ratios in NP@CHSs are quite similar. We suggest that N in the structure should play the main role in the binding of P. It thus causes a similar ratio of N and P in NP@CHSs for different mixing amounts of PA. Comparing the proportions of N states in NP@CHSs, we can find that the ratio of graphitic N increases with the P amount in the carbon spheres except for the case of NP4@CHS, whose carbon network is largely broken during the heat treatment (Table S1†). The total proportion of graphitic N and pyridinic N of NP@CHSs is shown in the order of NP3@CHS(71.82%) > NP2@CHS(62.57%) > NP1@CHS(59.52%) > NP4@CHS(56.73%) (Fig. 4e and Table S2†). It implies that the presence of P may influence the formation of C–N configuration, especially more active graphitic N and pyridinic N in the structure. The difference in the proportions of P–O and P–C in NP@CHSs (Fig. 4f) also reveals that a more active P–C bond with less oxidized P is inclined to form in the carbon framework under a higher mixing amount of PA.35
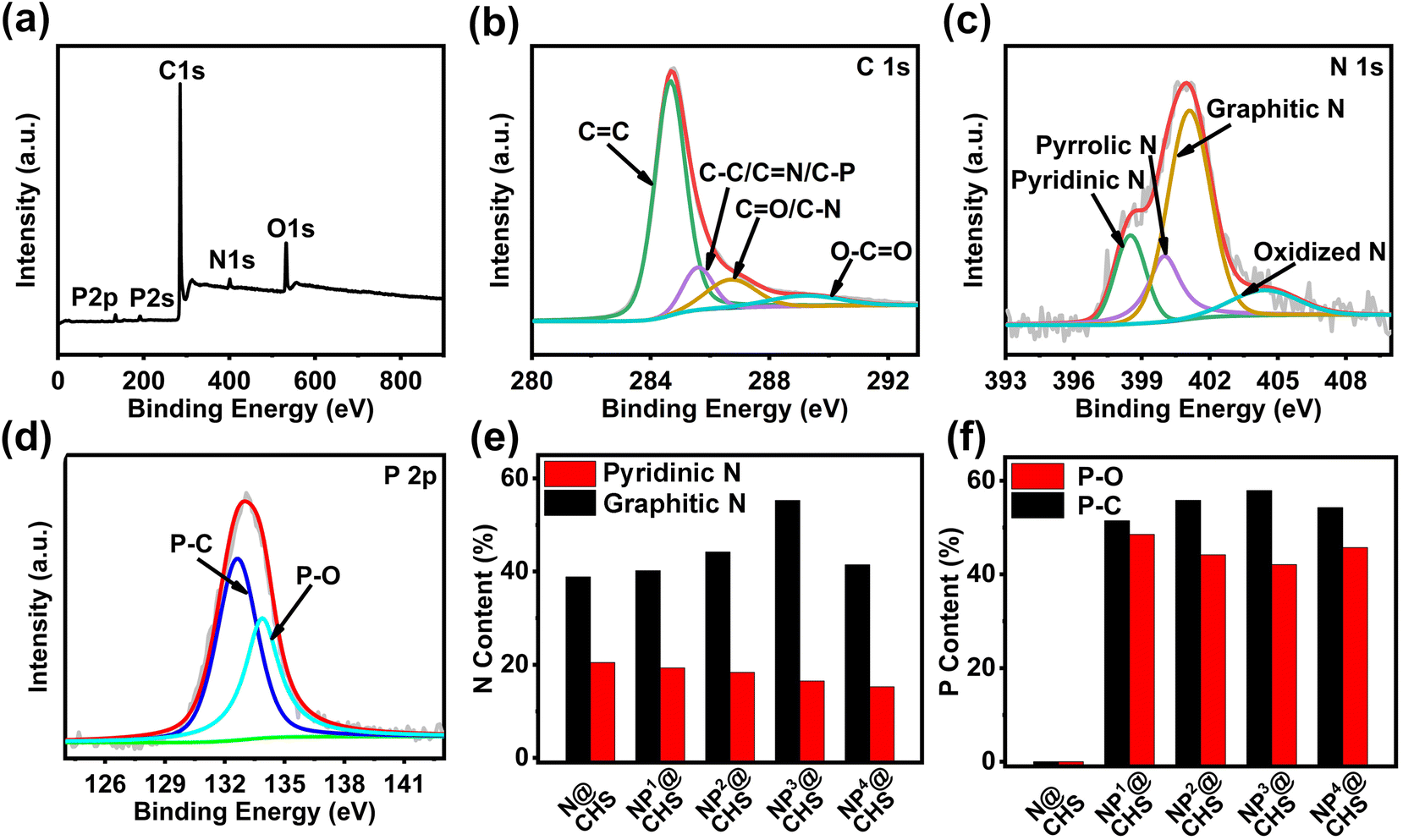 | ||
| Fig. 4 (a) XPS survey spectrum of NP3@CHS; (b) the high-resolution C 1s spectrum of NP3@CHS; (c) the high-resolution N 1s spectrum of NP3@CHS; (d) the high-resolution P 2p spectrum of NP3@CHS; (e) the relative contents of graphitic N and pyridinic N of NP@CHS and NP@CHSs based on the N 1s spectra; and (f) the relative contents of P–C and P–O of NP@CHS and NP@CHSs based on the P 2p spectra. | ||
HRTEM observation indicates the presence of a porous structure in the carbon spheres (Fig. S6†). Carbonization may cause the unfolding of C60s, which can lead to the increase of porous spaces in the structure. The isotherms of NP@CHSs show type IV characteristics with a H3 hysteresis loop for mesoporous materials with a small number of micropores (Fig. S12†). The influence of N/P doping on the porous structures of NP@CHSs is investigated according to the comparison of nitrogen sorption. The surface areas of NP1@CHS (398 m2 g−1), NP2@CHS (401 m2 g−1), and NP3@CHS (418 m2 g−1) show increasing inclination with the P mixing amount. But NP4@CHS possesses a much smaller surface area of 233 m2 g−1 (Fig. S12†). This result suggests that the addition of P in the C60 superstructure might promote the formation of structural defects and produce more pores during the heat treatment, while for NP4@CHS from the large addition of PA, it may cause the collapse of the carbon network and lead to a smaller surface area.
3.3. NP@CHSs as electrocatalysts for the oxygen reduction reaction (ORR)
The electrocatalytic activity of NP@CHSs for the ORR is measured by CV in an alkaline KOH electrolyte (Fig. 5a). Negligible reduction peaks can be observed within the potential range of 0–1.2 V in an N2-saturated solution for all the carbon spheres while showing an obvious cathodic peak in an O2-saturated KOH solution. This indicates the ORR process successfully occurred at the electrode surface.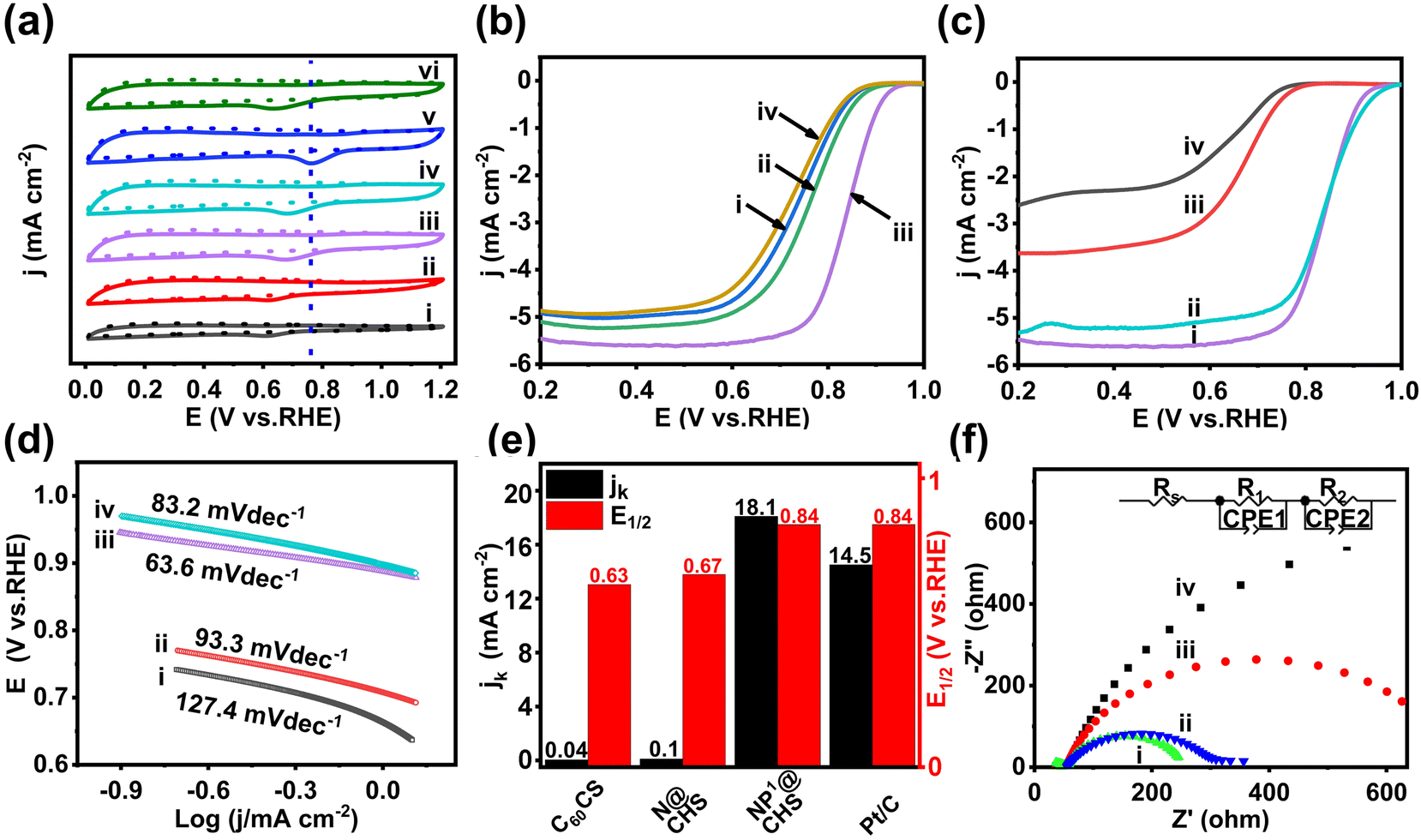 | ||
| Fig. 5 (a) CV curves of (i) C60CS, (ii)N@CHS, (iii) NP1@CHS, (iv) NP2@CHS, (v) NP3@CHS and (vi) NP4@CHS in N2 (dotted lines) and O2 (solid lines) saturated 0.1 M KOH solution at a scan rate of 50 mV s−1 and (b) LSV curves of (i) NP1@CHS, (ii) NP2@CHS, (iii) NP3@CHS and (iv) NP4@CHS at a scan rate of 10mV s−1 at 1600 rpm. (c) LSV curves of (i) C60CS, (ii) N@CHS, (iii) NP3@CHS and (iv) Pt/C at a scan rate of 10 mV s−1 at 1600 rpm. (d) Tafel plots of (i) C60CS, (ii) N@CHS, (iii) NP3@CHS and (iv) Pt/C; (e) the comparison of jK at 0.8 V and E1/2 of C60CS, N@CHS, NP3CHS and Pt/C; and (f) Nyquist plots of (i) C60CS, (ii) N@CHS, (iii) NP3@CHS and (iv) Pt/C. | ||
The reduction peaks of NP@CHSs appear to be more positive in contrast to those of N@CHS and C60CS. The peak potential of NP@CHSs is inclined to increase with the additional amount of P and also higher than N@CHS and C60CS. NP3@FHS shows the maximum value of peak potential (0.77 V) and the largest peak current density of 6.61 mA cm−2. This illustrates that the co-doping of P and N may further enhance the ORR catalytic activity of N-doped fullerene-derived carbons. The dramatic shift of the reduction peak to less cathodic potentials for NP4@FHS should be due to the broken carbon structure with a small surface area and less active sites.
To better understand the doping effect of P on the activity of NP@CHSs, LSV measurements were performed on a rotating disk electrode (RDE) in an O2-saturated 0.1 M KOH at a rotation rate of 1600 rpm min−1. The ORR polarization curves are shown in Fig. 5b. The limiting current density (JL), onset (E0), and half-wave potential (E1/2) are 5.468 mA cm−2, 0.954 V, and 0.841 V for NP3@CHS (Table S2†). Compared with the values of N@CHS and C60CS, NP3@NCS possesses much superior ORR activity and is also comparable to the commercial Pt/C catalyst (JL 5.306 mA cm−2, E0 0.976 V, E1/2 0.840 V) in alkaline solution (Fig. 5c and Table S2†). The ORR activity of NP3@CHS is also shown to exceed most of the other reported fullerene-derived electrocatalysts (Table S3†).
The kinetic parameters from the Tafel plots, Koutecky–Levich (K–L) plots, and Nyquist plots confirm the enhanced charge transfer in NP@CHS. The Tafel slope is reversely proportional to the charge transfer coefficient, reflecting the nature of electron transfer in the elementary reaction. NP3@CHS displays a Tafel slope of 63.6 mV dec−1, which is much lower than those of N@CHS (93.3 mV dec−1) and C60CS (127.4 mV dec−1), and even lower in contrast to the commercial Pt/C catalyst (83.2 mV dec−1) (Fig. 5d and Fig. S13†). The kinetic current density (jk) calculated from the K–L equation also indicates that NP3@CHS with a value of 18.1 mA cm−2 has a higher density than those of N@CHS (0.1 mA cm−2) and Pt/C (14.5 mA cm−2) (Fig. 5e). The Nyquist plots obtained by electrochemical impedance spectroscopy (EIS) can identify the electrode conductivity or the charge transfer resistance (Fig. 5f). NP3@CHS exhibits a semicircle in the high-frequency area, whose radius is smaller than those of N@CHS, C60CS, and Pt/C. The smaller radius means a lower charge transfer resistance and easier charge transfer of NP3@CHS at the electrode–electrolyte interface.
The voltammetric profiles of NP3@CHS show the enhancement of current density with increasing rotation rates (400–2500 rpm min−1) (Fig. 6a and Fig. S14 and S15†). It is caused by the decrease of electrolyte-electrode interface concentration polarization with the rotation rate. Fig. 6b and Fig. S14 and S15† show the K–L plots at different potentials. The parallel linear plots suggest first-order reaction kinetics toward the concentration of dissolved O2 and a similar electron transfer number (n) for the ORR at a different potential. According to the linear slope, the estimated n of NP3@CHS is 3.88, close to Pt/C (3.94) (Fig. 6c). It implies the dominance of the 4e− process for NP3@CHS in the range of 0.2–1.2 V. In comparison, the other carbon spheres without P show much lower n, of which N@CHS is 2.87 and C60CS is 2.13 (Fig. 6c). These results indicate that the co-doping of N and P in carbon spheres may facilitate the 4e− ORR process rather than the 2e− ORR process of carbon-based catalysts. This also suggests that the synergistic effect from N/P co-doping may change the ORR kinetics of carbon-based catalysts.
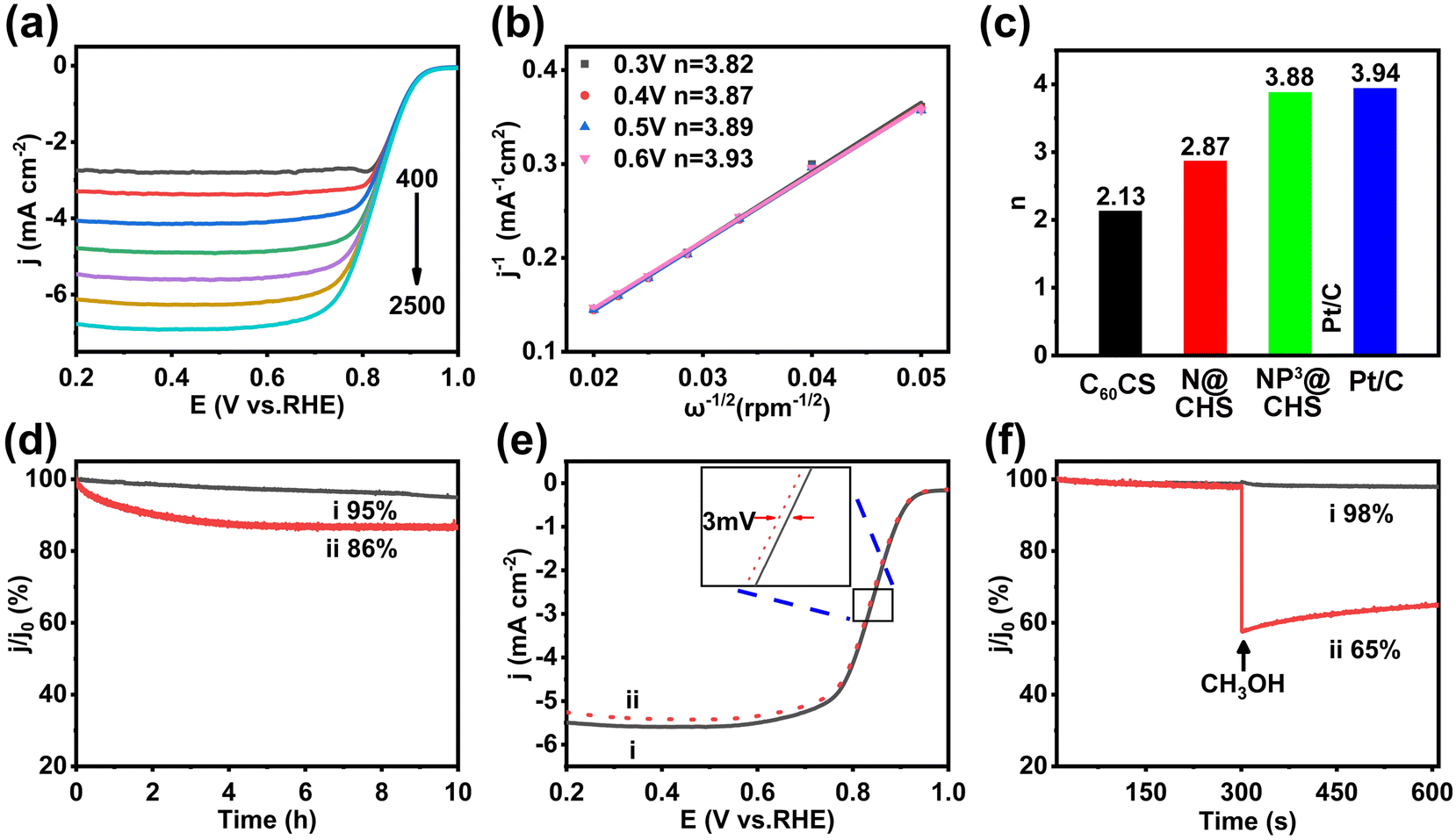 | ||
| Fig. 6 (a) LSV curves of NP3@CHS at a series of rotation speeds from 400 rpm to 2500 rpm; (b) the fitted K–L plots at different potentials; (c) the calculated electron-transfer numbers of the ORR for C60CS, N@CHS, NP3CHS and Pt/C; (d) the i–t response curves of (i) NP3@CHS and (ii) Pt/C; (e) LSV curves of NP3@CHS before and after 5000 CV cycles; and (f) the i–t response curves for the methanol immunity experiments of (i) NP3@CHS and (ii) Pt/C. | ||
Besides activity, long-term stability is also an essential aspect of carbon-based electrocatalysts. We evaluated the stability of NP3@CHS by current–time (i–t) chronoamperometry and the accelerated durability test (ADT). The i–t response of NP3@CHS reveals small attenuation of 5% after 10 hours, while Pt/C can only maintain 86% (Fig. 6d). The ADT durability test of NP3@CHS also shows good maintenance of the catalytic activity, with only 3 mV loss in the half-wave potential after 5000 CV cycles (Fig. 6e). In addition, NP3@CHS also exhibits a remarkably high tolerance to methanol with negligible loss (maintenance of 98% current density) compared to that of Pt/C (maintenance of 65% current density) (Fig. 6f). All these results demonstrate that the ORR activity and stability of fullerene-based carbon catalysts can be greatly improved through the co-doping of N and P, which may be even better than that of the commercial Pt/C catalyst.
Considering the high ORR activity of NP3@CHS and its application potential for Zn–air batteries, we also use it to build a typical Zn–air battery. NP3@CHS loaded on a conductive carbon paper is acted as the cathode. Its battery performance was measured and compared with Pt/C. A high and stable open-circuit voltage of 1.48 V with a peak-power density of 150.8 mW cm−2 is achieved for the Zn–air battery with NP3@CHS, which is superior to that of Pt/C (1.47 V, 140.3 mW cm−2) (Fig. 7a and b). Fig. 7c shows the successful charging of an LED light powered by the assembled Zn–air batteries with the NP3@CHS cathode. These results reveal the possibility of the NP3@CHS electrocatalyst used for Zn–air batteries.
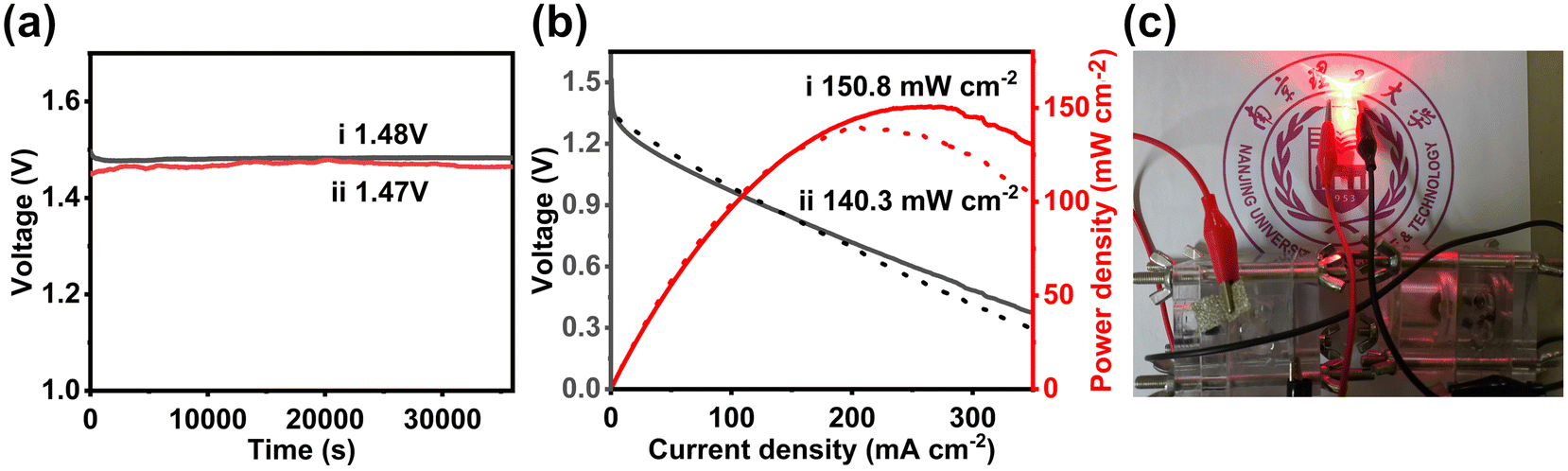 | ||
| Fig. 7 The performance of Zn–air batteries using (i) NP3@CHS and (ii) Pt/C as air cathodes. (a) Open circuit voltages and (b) polarization and power density curves. (c) Photograph of a red LED lamp powered by the assembled Zn–air batteries using NP3@CHS. | ||
3.4. ORR catalytic mechanism of NP@CHSs by simulations
Extensive studies have clarified that the ORR activity of metal-free carbon materials originates from the doping-induced charge transfer from adjacent C atoms.36,37 The introduction of P and N in carbon frameworks may change the charge density and geometric distribution of adjacent C atoms due to their higher electronegativity.38 We suggest that the excellent ORR activity of NP3@CHS should arise from the changes of C–N and P–C by doping N and P in the carbon skeleton from C60s.To understand the catalytic activity over N- and P-doping sites in carbon frameworks, density functional theory (DFT) calculations were performed. Different types of N and P coordination and configurations were constructed in the fullerene carbon models (Fig. 8), which are an undoped carbon ring (CR), a pyridinic N-doped pentagon carbon ring (PN-PR), a graphitic N-doped pentagon carbon ring (GN-PR), a pyridinic N/P-doped pentagon carbon ring (PN-PR-P), a graphitic N/P-doped pentagon carbon ring (GN-PR-P) and graphitic N/P-doped hexagon carbon rings (GN-HR-P).
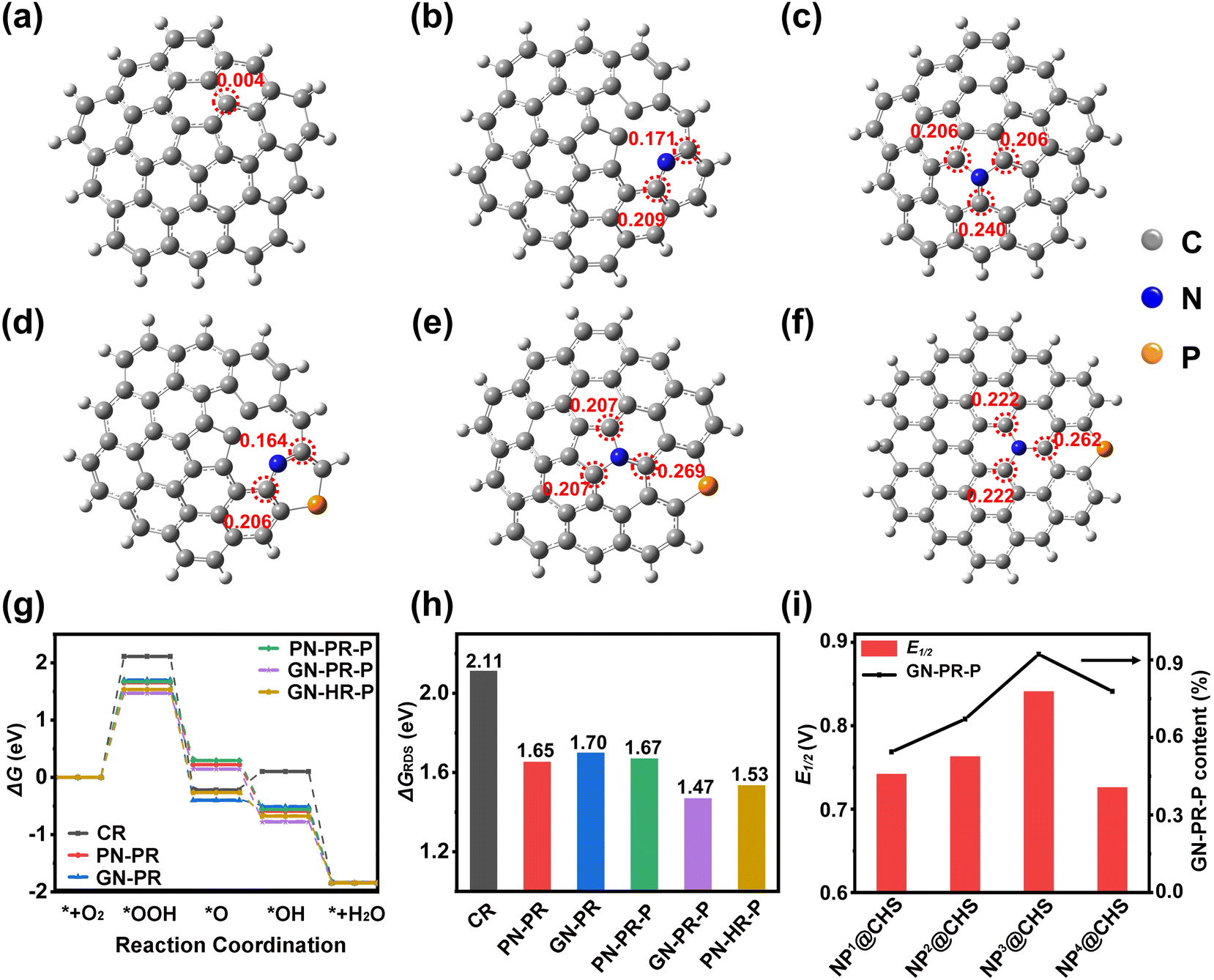 | ||
| Fig. 8 The models and atomic charge of (a) a CR, (b) a PN-PR, (c) a GN-PR, (d) PN-PR-P, (e) GN-PR-P, and (f) GN-HR-P. (g) ΔG values of all models for the ORR. (h) The comparison of ΔGRDS of different models. (i) The correlation between E1/2 and the content of GN-PR-P in various NP@CHSs. | ||
Based on the calculation results, the undoped CR exhibits a low charge (Fig. 8a). The doping of N (PN and GN) can increase the atomic charge of C adjacent to N. The GN model may induce a higher positive charge of the C atom (Fig. 8b and c). The additional doped P with a larger atomic radius could further promote charge delocalization in the carbon rings.39,40 However, the P influence on carbons of PN and GN is different. The atomic charge of adjacent C in PN-PR-P decreases from 0.209 (PN-PR) to 0.206, while that of C in GN-PR-P increases from 0.240 (PN-PR) to 0.269 (Fig. 8d and e). This indicates that P doping may influence the GN carbons larger than PN carbons. The comparison of GN-PR-P and GN-HR-P reveals that N/P co-doping may lead to a higher positive charge of C in the PR than in the HR and generate asymmetric charge distribution with greater polarization (Fig. 8e and f).41 It may enhance the oxygen adsorption and charge transfer in the ORR process.
The electron reduction of O2 generally consists of four elementary steps, involving five activated states of *+O2, *OOH, *O, *OH, and *+OH−.42 Among these, the rate-determining step (RDS) with the largest ΔG cis the key to the catalytic activity. As shown in Fig. 8g, the RDS of the ORR is the formation step of *OOH. The GN-PR and PN-PR show lower ΔGRDS than CR without doping (Fig. 8h). But PN-PR-P with N/P doping shows a little increase of ΔGRDS in contrast to the PN-PR. It means that P doping in PN carbon might suppress the catalytic activity, while P doping in GN carbons causes an obvious decrease in ΔGRDS and GN-PR-P possesses the lowest ΔGRDS. This means that the PR model with GN and P coordination gives more rational ΔG and higher activity than the N single doping models or N/P co-doped HR and PN-PR models. Based on the XPS results, we calculated the GN-P content of NP@CHSs. NP3@CHS has the highest ratio of GN-P (Fig. 8i), which is consistent with the order of ORR catalytic properties and the calculation result. These results confirm that the benefit of the fullerene structure with pentagon carbon may induce a more significant synergistic effect of N and P coordination on ORR catalysis.
4. Conclusions
In summary, fullerene-derived carbon catalysts with N/P co-doping are fabricated by controlling the intercalation of the P source into assembled diamine-fullerene hollow spheres. The enriched N moieties in the fullerene superstructure facilitate the binding of the P component to the carbon matrix, while the regulation of P intercalation could induce the formation of more graphitic N in the carbon frameworks. The resultant N/P co-doping carbon spheres possess high surface areas and homogenous distribution of N and P. They also exhibit superior ORR performance to the fullerene-derived carbons without doping or single N doping, and the commercial Pt/C catalyst. The excellent ORR activity is proved to arise from the fullerene structure and the significant synergistic effect of N and P coordinated C sites. This work presents a facile fabrication strategy for a new type of fullerene-derived electrocatalyst with controllable doping of heteroatoms. It may also promote their potential applications in other electrochemical processes or devices.Author contributions
Synthesis, F. M., B. J. and L. J.; investigation and data analysis, F. M., B. J. and S. W.; draft writing, B. J., S. W. and Q. J.; and project supervision, Q. J. and W. J. All authors approved this version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Instrument & Equipment Open Funding of Nanjing University of Science and Technology, the National Natural Science Foundation of China No. 21875108 and the Fundamental Research Funds for the Central Universities (No. 30921013106).References
- Y. Zhou, G. Chen and J. Zhang, J. Mater. Chem. A, 2020, 8, 20849–20869 RSC.
- C. Hu, Q. Dai and L. Dai, Cell Rep. Phys. Sci., 2021, 2, 100328 CrossRef CAS.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- D.-W. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576–591 RSC.
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
- R. Zhao, Y. Chen and S. Huang, Fundam. Res., 2021, 1, 807–823 CrossRef.
- Y. Rangraz and M. M. Heravi, RSC Adv., 2021, 11, 23725–23778 RSC.
- Y. Gao, Q. Wang, G. Ji, A. Li and J. Niu, RSC Adv., 2021, 11, 5361–5383 RSC.
- N. M. Sanchez-Ballester, G. Rydzek, A. Pakdel, A. Oruganti, K. Hasegawa, M. Mitome, D. Golberg, J. P. Hill, H. Abe and K. Ariga, J. Mater. Chem. A, 2016, 4, 9850–9857 RSC.
- T. Xu, W. Shen, W. Huang and X. Lu, Mater. Today Nano, 2020, 11, 100081 CrossRef.
- Z. Peng, Q. Jiang, P. Peng and F. Li, Eng. Sci., 2021, 14, 27–38 CAS.
- A. V. Baskar, M. R. Benzigar, S. N. Talapaneni, G. Singh, A. S. Karakoti, J. Yi, A.H Al-Muhtaseb, K. Ariga, P.M. Ajayan and A. Vinu, Adv. Funct. Mater., 2021, 32, 2106924 CrossRef.
- K. Ariga and L. K. Shrestha, Adv. Mater., 2021, 2, 582–597 RSC.
- K. Mao, W. Zhang, J. Dai and X. C. Zeng, Nanoscale, 2019, 11, 19422–19428 RSC.
- X. Chen, J. Chang and Q. Ke, Carbon, 2018, 126, 53–57 CrossRef CAS.
- Y. Wang, M. Jiao, W. Song and Z. Wu, Carbon, 2017, 114, 393–401 CrossRef CAS.
- J. Zhu, Y. Huang, W. Mei, C. Zhao, C. Zhang, J. Zhang, I. S. Amiinu and S. Mu, Angew. Chem., Int. Ed., 2019, 58, 3859–3864 CrossRef CAS PubMed.
- S. H. Noh, C. Kwon, J. Hwang, T. Ohsaka, B.-J. Kim, T.-Y. Kim, Y.-G. Yoon, Z. Chen, M. H. Seo and B. Han, Nanoscale, 2017, 9, 7373–7379 RSC.
- X. Xue, H. Yang, T. Yang, P. Yuan, Q. Li, S. Mu, X. Zheng, L. Chi, J. Zhu, Y. Li, J. Zhang and Q. Xu, J. Mater. Chem. A, 2019, 7, 15271–15277 RSC.
- Z. He, P. Wei, N. Chen, J. Han and X. Lu, Chem. – Eur. J., 2021, 27, 1423–1429 CrossRef CAS PubMed.
- Z. He, P. Wei, T. Xu, J. Han, X. Gao and X. Lu, Mater. Chem. Front., 2021, 5, 7873–7882 RSC.
- S. S. Babu, H. Möhwald and T. Nakanishi, Chem. Soc. Rev., 2010, 39, 4021–4035 RSC.
- Q. Tang, G. Zhang, B. Jiang, D. Ji, H. Kong, K. Riehemann, Q. Ji and H. Fuchs, SmartMat, 2021, 2, 109–118 CrossRef.
- K. Matsuoka, S. Matsumura, T. Akiyama and S. Yamada, Chem. Lett., 2008, 37, 932–933 CrossRef CAS.
- T. Akiyama, Bull. Chem. Soc. Jpn., 2019, 92, 1181–1199 CrossRef CAS.
- K. Matsubayashi, K. Kokubo, H. Tategaki, S. Kawahama and T. Oshima, Fullerenes, Nanotubes, Carbon Nanostruct., 2009, 17, 440–456 CrossRef CAS.
- Y. Sun, C. Cao, C. Liu, J. Liu, Y. Zhu, X. Wang and W. Song, Carbon, 2017, 125, 139–145 CrossRef CAS.
- F. F. Contreras-Torres, E. V. Basiuk, V. A. Basiuk, V. Meza-Laguna and T. Yu. Gromovoy, J. Phys. Chem. A, 2012, 116, 1663–1676 CrossRef CAS PubMed.
- T. Akiyama, Y. Ono, H. Miyamura, J. Saito, K. Kimura, S. Higashida and T. Oku, J. Nanopart. Res., 2018, 20, 252 CrossRef.
- B. Jiang, Q. Tang, W. Zhao, J. Sun, R. An, T. Niu, H. Fuchs and Q. Ji, CrystEngComm, 2020, 22, 6287–6294 RSC.
- R. Yadav and C. K. Dixit, J. Sci.: Adv. Mater. Devices, 2017, 2, 141–149 Search PubMed.
- M. Sathish and K. Miyazawa, Molecules, 2012, 17, 3858–3865 CrossRef CAS PubMed.
- C. Zhang, L. Hou, C. Cheng, Z. Zhuang, F. Zheng and W. Chen, ChemElectroChem, 2018, 5, 1891–1898 CrossRef CAS.
- R. Li, Z. Wei and X. Gou, ACS Catal., 2015, 5, 4133–4142 CrossRef CAS.
- Y. Qian, S. Jiang, Y. Li, Z. Yi, J. Zhou, T. Li, Y. Han, Y. Wang, J. Tian, N. Lin and Y. Qian, Adv. Energy Mater., 2019, 9, 1901676 CrossRef.
- C. Li, Z. Chen, A. Kong, Y. Ni, F. Kong and Y. Shan, J. Mater. Chem. A, 2018, 6, 4145–4151 RSC.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- Z. Liu, M. Wang, X. Luo, S. Li, S. Li, Q. Zhou, W. Xu and R. Wu, Appl. Surf. Sci., 2021, 544, 148912 CrossRef CAS.
- L. Ge, D. Wang, P. Yang, H. Xu, L. Xiao, G. X. Zhang, X. Lu, Z. Duan, F. Meng, J. Zhang and M. An, Nanoscale, 2019, 11, 17010–17017 RSC.
- J. Zhu, M. Xiao, P. Song, J. Fu, Z. Jin, L. Ma, J. Ge, C. Liu, Z. Chen and W. Xing, Nano Energy, 2018, 49, 23–30 CrossRef CAS.
- T. Najam, S. S. A. Shah, W. Ding, J. Jiang, L. Jia, W. Yao, L. Li and Z. Wei, Angew. Chem., Int. Ed., 2018, 57, 15101–15106 CrossRef CAS PubMed.
- J. Ma, L. Gong, Y. Shen, D. Sun, B. Liu, J. Zhang, D. Liu, L. Zhang and Z. Xia, Front. Mater., 2019, 6, 294 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental information, TEM and SEM images of C60-derived carbon spheres, FTIR, Raman and XPS spectra of C60-derived carbon spheres, and electrochemical performance of C60-derived carbon spheres. See DOI: https://doi.org/10.1039/d2nr02358j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |