
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular simulations of the interfacial properties in silk–hydroxyapatite composites†
Diego
López Barreiro‡
 a,
Zaira
Martín-Moldes‡
ab,
Adrián
Blanco Fernández
c,
Vincent
Fitzpatrick
b,
David L.
Kaplan
b and
Markus J.
Buehler
*ad
a,
Zaira
Martín-Moldes‡
ab,
Adrián
Blanco Fernández
c,
Vincent
Fitzpatrick
b,
David L.
Kaplan
b and
Markus J.
Buehler
*ad
aLaboratory for Atomistic and Molecular Mechanics (LAMM), 77 Massachusetts Avenue, 1-165, Cambridge, MA 02139, USA. E-mail: mbuehler@MIT.EDU; Fax: +1.617.452.2750; Tel: +1.617.452.2750
bDepartment of Biomedical Engineering, Tufts University, 4 Colby Street, Medford, MA 02155, USA
cInstituto de Cerámica de Galicia (ICG), Universidade de Santiago de Compostela, Avda. do Mestre Mateo, 25, 15706, Santiago de Compostela, A Coruña, Spain
dCenter for Computational Science and Engineering, Schwarzman College of Computing, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139, USA
First published on 11th July 2022
Abstract
Biomineralization is a common strategy used in Nature to improve the mechanical strength and toughness of biological materials. This strategy, applied in materials like bone or nacre, serves as inspiration for materials scientists and engineers to design new materials for applications in healthcare, soft robotics or the environment. In this regard, composites consisting of silk and hydroxyapatite have been extensively researched for bone regeneration applications, due to their reported cytocompatibility and osteoinduction capacity that supports bone formation in vivo. Thus, it becomes relevant to understand how silk and hydroxyapatite interact at their interface, and how this affects the overall mechanical properties of these composites. This theoretical–experimental work investigates the interfacial dynamic and structural properties of silk in contact with hydroxyapatite, combining molecular dynamics simulations with analytical characterization. Our data indicate that hydroxyapatite decreases the β-sheets in silk, which are a key load-bearing element of silk. The β-sheets content can usually be increased in silk biomaterials via post-processing methods, such as water vapor annealing. However, the presence of hydroxyapatite appears to reduce also for the formation of β-sheets via water vapor annealing. This work sheds light into the interfacial properties of silk–hydroxyapatite composite and their relevance for the design of composite biomaterials for bone regeneration.
1. Introduction
Bone is a biomineralized composite whose main components are collagen and carbonate-substituted hydroxyapatite minerals.1,2 Although bone has an innate capacity to self-regenerate, there is great interest in the tissue engineering field for developing robust and lightweight biomaterials that stimulate, control and accelerate the repair of large bone defects. Such biomaterials are usually three-dimensional scaffolds that combine polymers and inorganic osteoinductive materials. Synthetic polymers, such as polycaprolactone (PCL), polyglycolic acid (PGA), polylactic acid (PLA) or poly(lactic-co-glycolic acid) (PLGA) are commonly used.3,4 However, there is an increased shift towards the use of natural biopolymers, including silk, collagen, alginate or chitosan, due their biocompatibility and biodegradability, as well as their versatile aqueous processing into useful biomaterial formats.4–7 Moreover, recombinant DNA technology and developments in bioprocess engineering increasingly enable the use of fermentative processes for the biofabrication of rationally-designed protein biopolymers. This allows to rationally design novel protein biopolymers with fit-for-purpose physicochemical and biological properties8 while reducing our dependence on animal sources for their production.Silk proteins from Bombyx mori cocoons have outstanding mechanical properties that arise from a hierarchical structure.9,10 Silk is characterized by a high extensibility and toughness,11 and has been successfully applied as a soft matrix for the manufacturing of materials and composites with applications in biomedicine,12 flexible electronics13 and water purification,14 among many other areas of utility. The crystalline domain in silk (with repetitive motifs including GAGAGA, GAGAGS, GAGAGY, or GAGYGA) is composed of stacks of hydrophobic anti-parallel β-sheets held together by hydrogen bonds.15,16 The amorphous domain (with the amino acid sequence TGSSGFGPYVANGGYSGYEYAWSSESDFGTGS)17,18 constitutes the hydrophilic matrix in which the β-sheet crystals are embedded. The crystalline domains provide mechanical strength, whereas the amorphous domains provide flexibility and toughness.19 Last, the N-terminus has been suggested to play a key role in the self-assembly of silk into fibers.20
Although silk can be used for the fabrication of biomaterials that mimic the extracellular matrix, inherent osteoinductivity is absent.21 Thus, osteoinductive components need to be embedded in the silk matrix to enhance bone tissue regeneration. Several candidates are being investigated, including apatites,22,23 silica24,25 or bioactive glass.26,27 Particularly, hydroxyapatite (HAP) is an appealing choice4 because HAP has a chemical structure similar to that of the mineral components of natural bone, and silk can template the growth of HAP crystals along its fibers.28,29 Materials containing different forms of HAP (e.g., powder, nanoparticles) have shown osteogenic differentiation capacity in vitro, while supporting bone formation in vivo when blended with silk into composites.21,30–33 Moreover, the addition of silk has shown to improve the compressive strength and Young's modulus of HAP-based materials.34
Several approaches can be followed to prepare silk–HAP composites: mixing HAP and silk in solution;30,34 mineralizing preformed silk scaffolds in solutions containing precursor salts of HAP;35,36 or using recombinant chimeric silk-like polypeptides fused to biomineralization domains that nucleate the growth of HAP crystals.37 Regardless of the approach followed, the morphological, mechanical and structural properties of silk–HAP composites will impact their in vivo behavior and bone regeneration potential.23 These properties can be severely affected by the interfacial interactions between silk and HAP.38 Most of the research so far has focused on the biomineralization of the whole silk protein, with only a limited number of studies dissecting the interactions of each silk domain with HAP.28 This study addresses that gap by applying molecular dynamics (MD) simulations to unveil how the different domains in silk (amorphous, crystalline and N-terminus) interact with HAP. MD simulations can uncover structural and dynamic features of biomolecules with atomistic resolution. They have been successfully applied to study the interfacial properties of silk in contact with substrates like graphene39 and silica.40 MD literature pertaining silk–HAP composites mainly encompasses coarse grain simulations for applications like water filtration,29 or bioinspired self-folding materials.41 Here, we used fully atomistic MD simulations instead to investigate how the amorphous, crystalline and N-terminus silk domains interact with HAP (001) surfaces. These simulations revealed with atomistic resolution important interfacial features in silk–HAP composites, such as hydrogen bonding patterns, changes in secondary structure, or key interacting amino acids. The simulation results were complemented with experimental data obtained via FTIR. This allowed us to gain further insight into how the secondary structure of silk is impacted by the presence of HAP and the use a common consolidation method for silk biomaterials, water vapor annealing. Overall, our computational–experimental data suggest that HAP reduced the content of ordered, load-bearing β-sheet crystals in silk. This decrease was only partially overcome by the use of water vapor annealing. These results deepen our understanding of the interfacial phenomena occurring in silk–HAP composites while providing guidelines for controlling their mechanical properties.
2. Materials and methods
2.1. Computational modeling and simulations
The HAP (001) surface was used for these simulations.42–44 This surface is commonly used in computational simulations pertaining the interaction of HAP with biomacromolecules.45–47 The protonation of the inorganic–aqueous interface due to the presence of water is critical for MD simulations involving HAP and proteins, but this issue is oftentimes overlooked in computational studies. To that end, the Interface Force Field provides pH-resolved surface models of the HAP (001) surface at pH 5.44,48 Those surface models were used here to generate models at pH 7 by adjusting the protonation state, atomic environment and atomic charges on the hydroxyapatite surface (Fig. S1†). The software Materials Studio 4.449 was used to protonate several H2PO4− surface groups, until the concentration of surface H2PO4− and HPO42− groups became equal. Accordingly, the partial charges were modified for those surface-exposed dihydrogen phosphate groups deprotonated to monohydrogen phosphate. By increasing the pH from 5 to 7, one calcium ion was added for every two new monohydrogen phosphate groups to maintain charge neutrality.We used MD simulations to investigate the dynamic properties of the amorphous domain, β-sheet crystals and N-terminus of silk in contact with HAP at pH 7 (Fig. 1). Silk's crystalline structure and the N-terminus were obtained from the Protein Data Bank (PDB) files with the codes 2SLK and 3UA0, respectively. 2SLK is formed by a 3-layer stack of antiparallel β sheets. Each layer contains five antiparallel peptide chains with the sequence GAGAGS. 3UA0 is a homodimer forming an eight-stranded β-sheet.50 12 peptide chains with the sequence TGSSGFGPYVANGGYSGYEYAWSSESDFGTGS17,18 in a 3 by 4 arrangement were used to create the amorphous domain, following the procedure outlined elsewhere.13
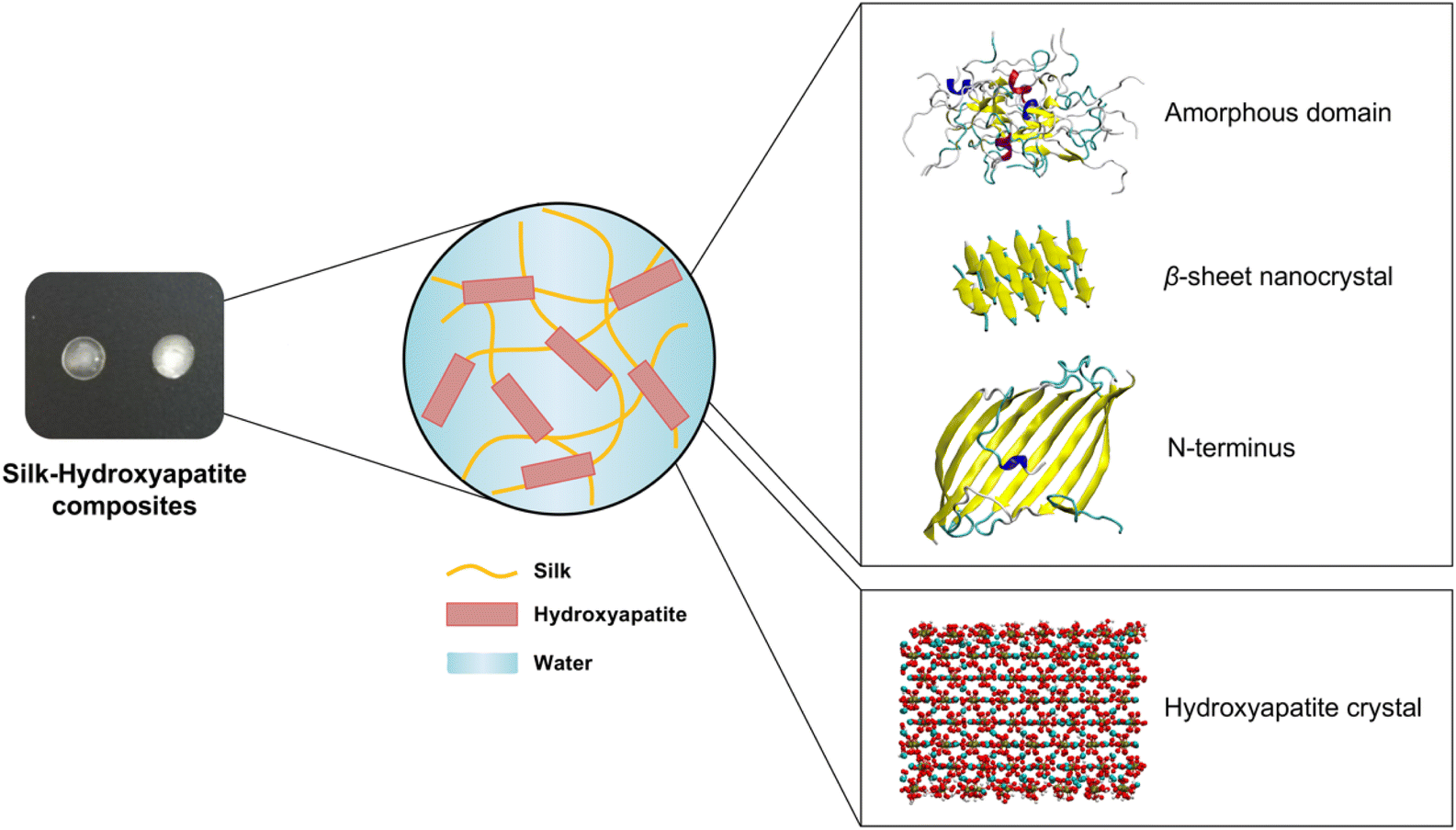 | ||
| Fig. 1 The objective of this work is to investigate the interfacial dynamic and structural properties of three different silk domains (amorphous, crystalline and N-terminus) in contact with HAP surfaces, and their implications for the bottom-up design of mineralized materials with controlled structural and mechanical properties. In this figure, a schematic representation of silk–HAP composites is shown, including snapshots of the initial configuration of the silk domains and the HAP unit cell used for MD simulations. | ||
MD simulations were performed in NAMD 2.13,51 using the Chemistry at HARvard Macromolecular Mechanics (CHARMM)52 force field for the solvated protein system and the Interface Force Field48 for HAP. The simulated systems intended to represent typical conditions used in the ex vivo preparation of silk–HAP scaffolds e.g., via 3D printing.34 The starting structures for the crystalline, amorphous and N-terminus domains were placed 5 Å above the HAP surface. The systems were solvated in a TIP3P water box with periodic boundary conditions and Na+ and Cl− ions were added as needed to neutralize the total charge of the system. The system dimensions were only allowed to change in the z direction, keeping the x–y plane dimensions fixed. The ShakeH algorithm53 was applied to all the bonds including hydrogen atoms. In all cases, 3D periodic boundary conditions were applied. The solvation box was made sufficiently large in the vertical direction to avoid spurious effects of self-interactions for the HAP–protein system.
The energy of the systems was minimized, followed by Langevin dynamics for 125 ns using the NPT ensemble at 300 K. Pressure (1 bar) was exerted through the Nosé–Hoover Langevin piston. The long-range electrostatic coulombic interactions were calculated using the particle mesh Ewald (PME) method with a grid spacing of 1 Å with periodic boundary conditions. A cutoff distance of 12 Å was applied for electrostatic and van der Waals interactions of the explicit solvent replicas, with a switch distance of 10 Å to avoid hard cuts. The timestep chosen was 2 fs. The position of the backbone of the peptide structures was restrained for the first 1 ns of simulation. The bulk of the hydroxyapatite atoms was kept fixed throughout the entire simulation, only allowing to move the most external atoms on the top and bottom surfaces. Three replicas were performed for each system, and simulation data was output every 0.2 ns.
2.2. Data analysis of MD simulations
Data produced during the simulations were analyzed using in-house TCL and Python scripts. Atomic structures were visualized using the Visual Molecular Dynamics (VMD) graphics software.54 The evolution of the root mean square deviation (RMSD) of the atomic positions during the simulation was analyzed for the protein backbone using the RMSD Trajectory Tool from VMD. The count of intraprotein and protein–HAP hydrogen bonds throughout the simulation trajectories was analyzed using the Hbonds plugin from VMD, following its default geometric criteria (distance cutoff of 3.0 Å and a D–H–A angle cutoff of 20°). Custom TCL scripts were developed to analyze in VMD the solvent accessible surface area (SASA) (using a standard water probe radius of 1.4 Å), the radius of gyration of the protein domains, and the evolution of the protein's secondary structure with the STRIDE algorithm.55 Close contacts between the protein domains and HAP were defined as contacts with a distance below 4 Å, excluding hydrogen atoms.2.3. Preparation of silk–HAP films
The silk solution used for film preparation was prepared as described previously.56 Briefly, pieces of Bombyx mori cocoons (5 g) were boiled for 30 minutes in 0.02 M aqueous Na2CO3, rinsed extensively in diH2O and dried overnight. Dry silk was then dissolved in 9.3 M aqueous LiBr solution at 60 °C for 4 hours. The silk/LiBr solution was dialyzed against distilled water for 2 days, with 10 water changes. The resulting solution was centrifuged at 9000 rpm at 4 °C twice for 20 min. The concentration of the silk solution was determined by calculating the weight ratio between the wet and dry solution. The extracted silk in solution was diluted to a concentration of 5% (w/v) in water. Commercial HAP (MP Biomedicals, Solon, OH, US, purity >99.99%) was dispersed in water by sonication and then mixed with the silk solution to obtain a suspension with HAP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :silk ratios of 1, 5 or 10 wt%.
:silk ratios of 1, 5 or 10 wt%.
For characterization purposes, multiple films of each sample were prepared by depositing 30 μL of each dissolved protein solutions onto different polydimethylsiloxane (PDMS) disks (R = 6 mm). Water vapor annealing experiments to induce β-sheet formation in silk films were performed using an isotemp vacuum oven for 24 h at room temperature, and then air dried.57 Scanning Electron Microscopy (SEM) of silk and silk–HAP films was performed on a Zeiss EVO-10MA microscope (Zeiss, Oberkochen, Germany) at 5 kV accelerating voltage. Prior to imaging, the samples were rapidly frozen with liquid nitrogen, cryofractured to expose the cross-sections, and coated with ∼10 nm gold using a SC7620 sputter coater (Quorum Technologies, UK).
2.4. Analysis of the secondary structure of silk–HAP films
Fourier Transform Infrared Spectroscopy (FTIR) of the films was carried out with an Attenuated Total Reflectance FTIR machine (Jasco, Oklahoma City, OK, USA). Films were measured using five independent replicas. 64 scans with a resolution of 4 cm−1 were recorded and averaged in the range of 400 to 4000 cm−1 for each sample. The protein secondary structure composition was determined by performing peak deconvolution over the amide I region (1595–1705 cm−1) with an in-house Python script using the lmfit package for curve fitting. This region is the most sensitive spectral region to protein secondary structure, which originates from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the amide group coupled with the in-phase bending of the N–H bond and stretching of the C–N bond.58,59 The baseline was corrected by fitting a straight line between the 1595–1705 cm−1 region. The deconvolution was carried out using five primary peaks assigned to different secondary structures: 1620 cm−1 (β-sheet), 1645, 1660 and 1670 cm−1 (random coil/helix), and 1700 cm−1 (turn).60 The peak positions were allowed to shift 4 cm−1 using the Levenberg–Marquardt algorithm, and a Gaussian model was selected for the band shape. A nonlinear least-squares method was used to obtain a reconstituted curve as close as possible to the original spectra. The statistical significance of the numerical values reported was assessed via a one-way analysis of variance (ANOVA) test.
O stretching vibration of the amide group coupled with the in-phase bending of the N–H bond and stretching of the C–N bond.58,59 The baseline was corrected by fitting a straight line between the 1595–1705 cm−1 region. The deconvolution was carried out using five primary peaks assigned to different secondary structures: 1620 cm−1 (β-sheet), 1645, 1660 and 1670 cm−1 (random coil/helix), and 1700 cm−1 (turn).60 The peak positions were allowed to shift 4 cm−1 using the Levenberg–Marquardt algorithm, and a Gaussian model was selected for the band shape. A nonlinear least-squares method was used to obtain a reconstituted curve as close as possible to the original spectra. The statistical significance of the numerical values reported was assessed via a one-way analysis of variance (ANOVA) test.
3. Results
3.1. Molecular dynamics simulations
The simulation results indicated that the three silk domains adsorbed on HAP (001) surfaces at pH 7 (Fig. 2), but the structural consequences of this adsorption were different for each domain. The following section reports these consequences in terms of hydrogen bonding, silk–HAP close contacting amino acids, radius of gyration, root mean square deviation (RMSD) of atomic positions, solvent accessible surface area (SASA) and secondary structure. The evolution of those parameters during the simulation is plotted in Fig. 3, and average values for the first and the last 5 ns of simulation are reported in Table 1.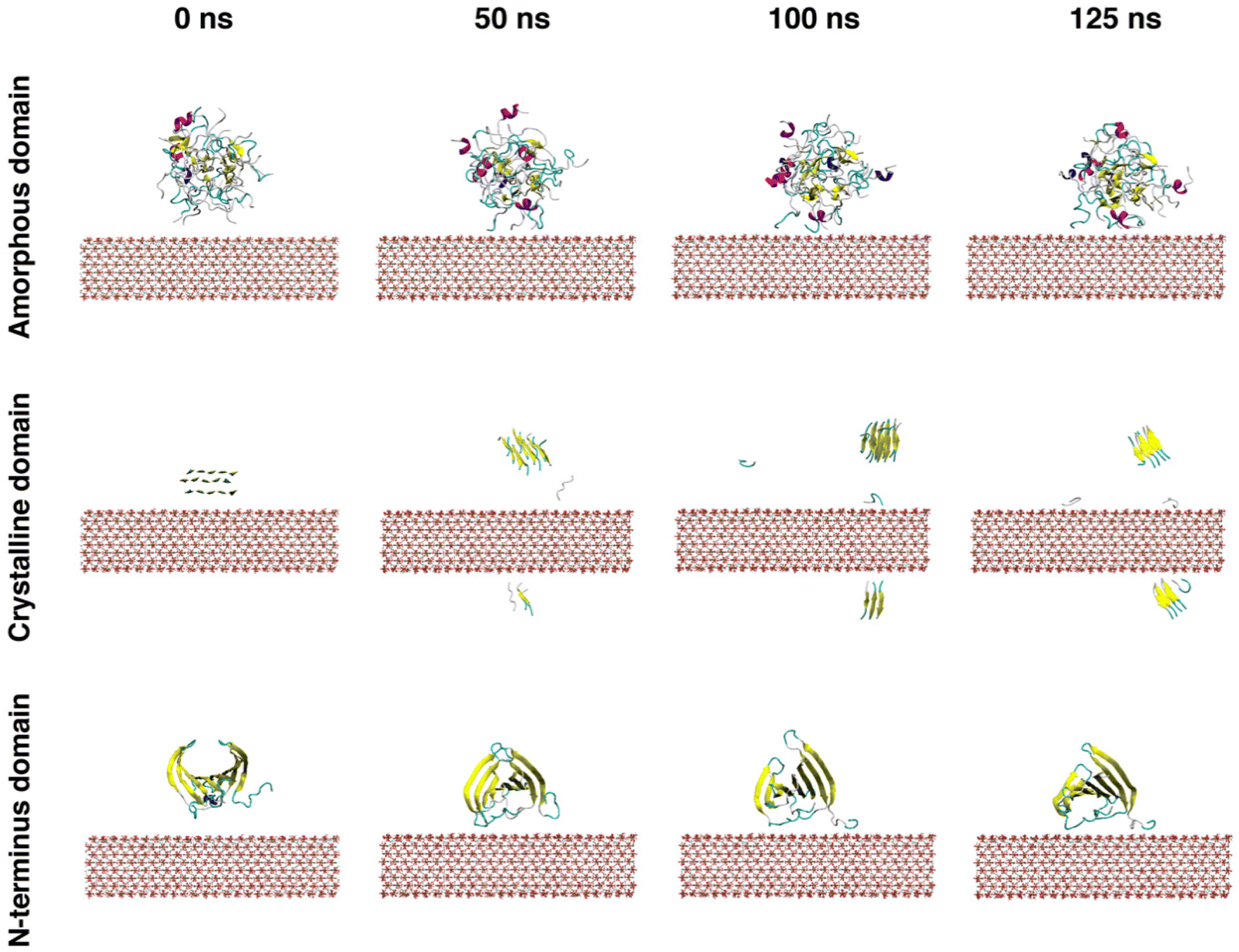 | ||
| Fig. 2 Depiction of MD snapshots of the interaction between the amorphous, crystalline and N-terminus domains of silk and HAP (001) surfaces resolved for pH 7. HAP atoms are plotted using the CPK representation, whereas the various silk domains are plotted using the cartoon representation. Water molecules and Na+ and Cl− ions are not shown for clarity. With regard to the secondary structures in silk domains, α-helices are colored in purple, 310 helices in blue, β-strands in yellow, turns in cyan and random coil regions white. | ||
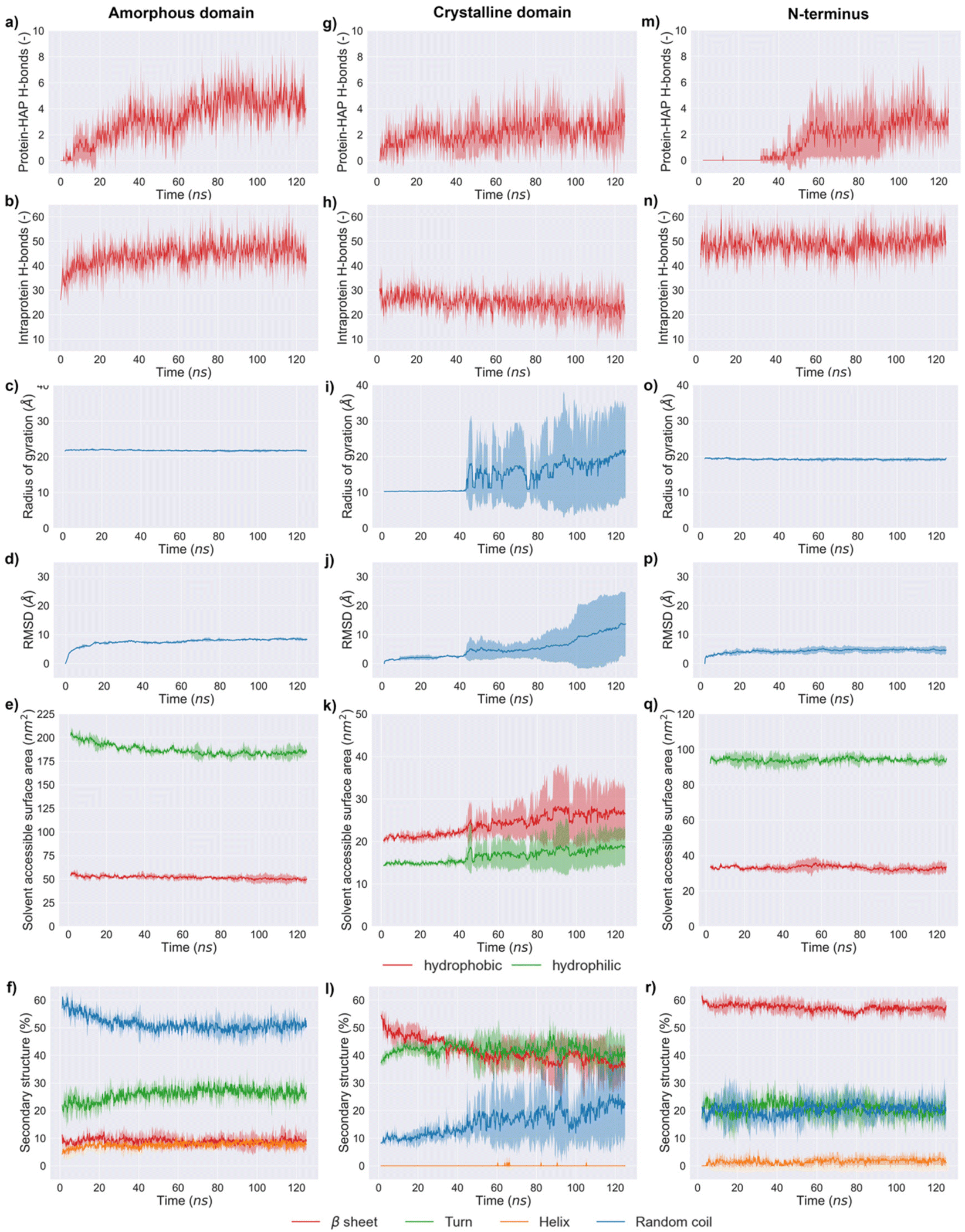 | ||
| Fig. 3 Temporal profile of key parameters during 125 ns of MD simulations: (a, g and m) protein–HAP hydrogen bonds for the different silk domains; (b, h and n) intraprotein hydrogen bonds for the different silk domains; (c, i and o) radius of gyration of silk domains; (d, j and p) root mean square displacement (RMSD) of silk domains; (e, k and q) solvent accessible surface area (SASA) of hydrophobic and hydrophilic amino acids in silk; (f, l and r) secondary structure of silk domains. | ||
| Amorphous domain | Secondary structure (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Protein-HAP H-bonds | Intraprotein H-bonds | SASAhydrophobic (nm2) | SASAhydrophilic (nm2) | R gyr (Å) | RMSD (Å) | β sheet | Turn | Random coil | Helix | |
| Initial 5 ns | 0 ± 0 | 35 ± 3 | 55.0 ± 1.5 | 201.1 ± 2.7 | 21.8 ± 0.1 | 3.9 ± 1.2 | 8.7 ± 1.1 | 21.7 ± 1.5 | 58.0 ± 1.6 | 5.3 ± 0.8 |
| Final 5 ns | 4 ± 1 | 45 ± 2 | 49.6 ± 0.8 | 185.2 ± 1.4 | 21.7 ± 0.1 | 8.4 ± 1.2 | 9.1 ± 0.6 | 26.2 ± 1.4 | 51.0 ± 1.0 | 8.0 ± 0.6 |
| Crystalline domain | Secondary structure (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Protein-HAP H-bonds | Intraprotein H-bonds | SASAhydrophobic (nm2) | SASAhydrophilic (nm2) | R gyr (Å) | RMSD (Å) | β Sheet | Turn | Random coil | Helix | |
| Initial 5 ns | 1 ± 0 | 27 ± 3 | 20.8 ± 0.5 | 14.7 ± 0.3 | 10.2 ± 0.0 | 1.1 ± 0.3 | 51.1 ± 2.3 | 39.0 ± 1.0 | 9.5 ± 1.0 | 0.0 ± 0.0 |
| Final 5 ns | 3 ± 1 | 23 ± 2 | 26.7 ± 0.3 | 18.7 ± 0.2 | 21.0 ± 0.5 | 13.1 ± 0.7 | 36.4 ± 0.8 | 40.0 ± 1.2 | 22.6 ± 1.5 | 0.0 ± 0.0 |
| N-terminus domain | Secondary structure (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Protein-HAP H-bonds | Intraprotein H-bonds | SASAhydrophobic (nm2) | SASAhydrophilic (nm2) | R gyr (Å) | RMSD (Å) | β sheet | Turn | Random coil | Helix | |
| Initial 5 ns | 0 ± 0 | 48 ± 4 | 33.1 ± 0.6 | 94.3 ± 0.9 | 19.5 ± 0.1 | 2.2 ± 0.7 | 59.6 ± 0.7 | 21.8 ± 1.9 | 18.5 ± 1.9 | 0.1 ± 0.3 |
| Final 5 ns | 3 ± 1 | 51 ± 3 | 33.1 ± 0.6 | 93.8 ± 0.7 | 19.2 ± 0.1 | 4.6 ± 0.1 | 57.1 ± 0.6 | 20.1 ± 1.3 | 20.6 ± 1.2 | 2.0 ± 0.7 |
3.2. Secondary structure of silk–HAP films
The secondary structure of silk films prepared with different HAP contents was analyzed by FTIR (Fig. 4a). The spectra showed the presence of a peak at 1040–960 cm−1 in all samples with 10 wt% HAP, characteristic of the presence of PO43− ions from HAP.29,34,61 Such a peak was not observed for the silk film control and silk films containing 1 wt% HAP (Fig. S2†), and it was only present in some of the replicas with 5 wt% HAP (Fig. S3†), indicating an inhomogeneous distribution of the HAP particles at such low concentrations. HAP samples without silk did not show any relevant changes in their FTIR spectra before and after water vapor annealing (Fig. S4†), hinting that any changes observed after water vapor annealing in the amide I region were caused by changes in the silk component of the films.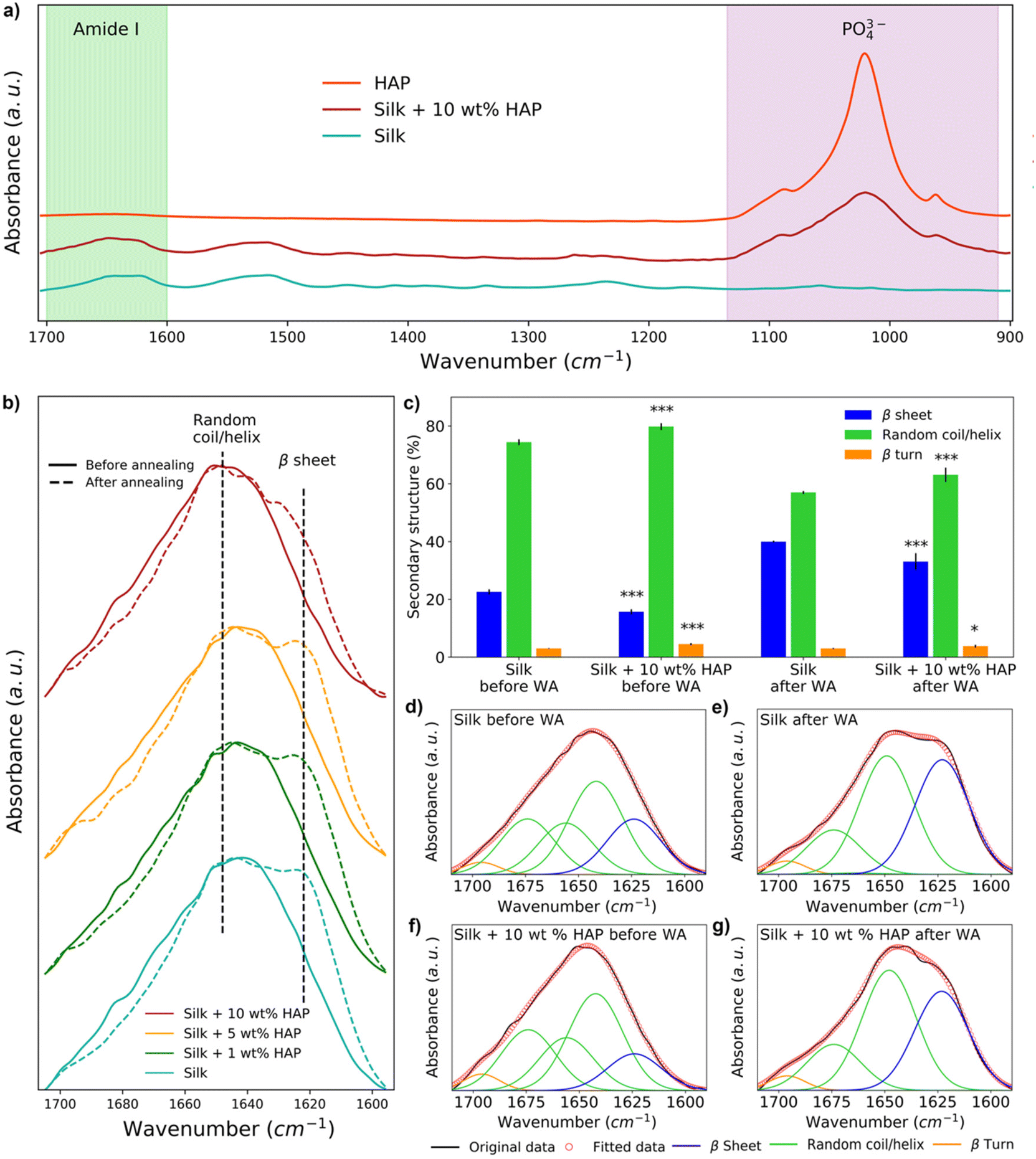 | ||
| Fig. 4 Analysis of the secondary structure of silk–HAP films with different HAP contents by means of Fourier-transform infrared spectroscopy (FTIR): (a) spectra of different silk–HAP films, highlighting peaks associated with the amide I region and the presence of phosphate groups from HAP; (b) amide I region for different silk–HAP regions, highlighting the position of peaks related to β sheets and random coil/helix secondary structures before and after water vapor annealing (WA); (c) quantitative analysis of the deconvolution of the amide I region (error bars represent standard deviation of the average of five individual samples); and (d–g) deconvoluted spectra of the amide I region of silk and silk–HAP 10 wt% films, highlighting the increase in β sheet structures after water vapor annealing (WA). | ||
The analysis of the spectra in the amide I region demonstrated that the water vapor annealing process caused an increase in the absorption band at 1620 cm−1 for all samples, which is associated with the presence of β-sheets (Fig. 4b). However, the intensity of the 1620 cm−1 band decreased as the concentration of HAP increased (Fig. 4b). This was also noticeable by deconvoluting the spectra of silk and silk and silk–HAP 10 wt% films (p = <0.001) (Fig. 4c–g). The deconvolution for films with 1 and 5 wt% HAP were not statistically different to the silk control films (Table S1†). Before water vapor annealing, silk films had a random coil/helix content of 74.4 ± 1.0% and a β-sheet content of 22.6 ± 0.9%, while for silk–HAP 10 wt% films the random coil/helix content was 79.8 ± 1.2% and the β-sheet content 15.7 ± 0.9%. After water vapor annealing the β-sheet content of the films increased to 40.0 ± 0.3%, while the random coil/helix content decreased to 57.0 ± 0.5%; the increase in β-sheets after water vapor annealing was lower for silk–HAP 10 wt% films (33.1 ± 2.9%), with a random coil/helix content of 63.1 ± 2.5%. Although we report numerical values for the secondary structure obtained by this means, we note that this methodology is associated with some level of uncertainty as discussed elsewhere,62 and therefore the deconvolution results should be interpreted as mere indicators of a trends, rather than as absolute values.
3.3. Surface characterization of silk–HAP films
SEM was used to assess the morphology of silk and silk–HAP films, as well as the distribution of HAP within the silk matrix. Given the differences identified for the secondary structure of silk and silk–HAP 10 wt% films, we analyzed their surface and cross-section. Silk control films were continuous and uniform (Fig. 5). In contrast, deposits of HAP crystals became visible on the surface of silk–HAP 10 wt% films, confirming a uniform distribution of HAP particles within the silk matrix. | ||
| Fig. 5 Mesostructure characterization: SEM images of the (a) surface and (b) cross-section area of silk films, and the (c) surface and (d) cross-section area of silk–HAP 10 wt% films. | ||
4. Discussion
The ability of silk to induce HAP mineralization has been demonstrated under varied experimental conditions.21,30–33 Here, we investigated the interfacial phenomena related to this process via fully atomistic MD simulations between three silk domains and HAP (001) surfaces. Our results showed that silk was richer in disordered structures (random coil and turns) when in contact with the HAP (001) surface, which agreed with earlier experimental data.21 The secondary structure of the crystalline domain of silk remained more stable than the amorphous and N-terminus domains during the first 40 ns of simulation. We attribute this to the stabilization effect of its highly concentrated network of hydrogen bonds, which required longer times to be disrupted by the HAP (001) surface. However, once the disruption was initiated, the crystal structure collapsed rapidly: random structures rose from 9.5 ± 1.0 to 22.6 ± 1.5% while β-sheets decreased from 51.1 ± 2.3 to 36.4 ± 0.8%. A similar effect was observed in MD simulations of silk β-sheet crystals in contact with silica surfaces.40 This weakening of β-sheet crystals when in contact with HAP is relevant from a mechanical point of view, as those crystals are key for the load-bearing properties of silk. The simulations also indicated that strands from the crystalline domain interacted with the HAP (001) surface only after losing their initial β-sheet conformation. This agreed with previous experimental data reporting the inability of β-sheet structures to nucleate the biomineralization of HAP.28The amorphous domain was very stable throughout the simulation, as shown by its radius of gyration (ca. 21.8 Å throughout the entire simulation). This domain only underwent a partial rearrangement of its secondary structure, with a reduction in random coil regions (from 58.0 ± 1.6% in the starting structure to 51.0 ± 1.0% during the last 5 ns of simulation) and a rise in turn structures. This was in line with its concomitant rise in intraprotein hydrogen bonds (35 ± 3 to 45 ± 2) and the reduction in SASA of the hydrophobic residues (55.0 ± 1.5 to 49.6 ± 0.8 nm2) (Fig. 3e). Close contacts between the amorphous domain and HAP took place mostly through glutamic acids. This agreed with previous experimental data suggesting that the deposition of HAP crystals on silk was templated by the high solvent exposure and the presence of carboxylic acid residues in the amorphous domain.28,63 The N-terminus was the least affected by the interaction with the HAP (001) surface, highlighting the stability of the cross-β structure of its homodimeric structure (Fig. 3r). This domain showed minimal variations in its radius of gyration (19.5 ± 0.1 to 19.2 ± 0.1 Å), the lowest RMSD of the three domains investigated (4.6 ± 0.1 Å), and almost negligible variation in SASA, both for hydrophilic (ca. 33 nm2) and hydrophobic (ca. 94 nm2) amino acids. The N-terminus is believed to play a key role in the oligomerization of silk into nanofibers,20 and therefore its high stability might indicate that silk retains its ability to form fibrillar structures in the presence of HAP (001) surfaces.
Overall, these MD simulations suggested that silk interacts with the HAP (001) surface preferentially through disordered regions and via charged amino acids. This has implications for the biofabrication of silk-like polypeptides. When designing recombinant silk-like polypeptides, focus is usually placed on utilizing motifs like GAGAGS64–66 due to their ability to form β-sheet crystals. This usually aims at enhancing the mechanical properties of materials derived from such polypeptides. However, our data highlight the important role of less ordered and/or more charged blocks in enhancing the interfacial interaction between silk and HAP.
FTIR data agreed with MD data, confirming that HAP reduced the β-sheets in silk (Fig. 4b). Given that a high content of β-sheets is connected to a larger strength in silk biomaterials,67 our results showed the need for postprocessing methods to enhance the mechanical properties of silk–HAP films. To that end, water vapor annealing is a post-synthesis method commonly used to enhance the formation of β-sheets in silk biomaterials to enhance their mechanical properties.62,68,69 Here, water vapor annealing increased the content of β-sheet structures in films, although this was more effective in the absence of HAP, as shown by FTIR data (Fig. 4c). Even though a reduction in β-sheet crystals would generally be regarded as disadvantageous for the mechanical properties of silk, we hypothesize that it could have two positive effects. First, less β-sheets could enhance the interfacial interaction between HAP and silk, since HAP crystals have shown a preferential nucleation on unstructured regions of silk.28 In fact, the present work showed that the crystalline domain was only able to interact with the HAP surface only after the β-sheet crystal was disrupted (Fig. 2). And second, silk biomaterials with a high content of β-sheets could limit the adhesion and spreading of cells, possibly due to the lack of contact sites for cellular attachment in β-sheets or the preference of cells to avoid the sharp edges of β-sheet crystals. To that end, recent work showed that the attachment of hMSCs cells and their alkaline phosphatase activity was 2.5 times larger in silk biomaterials with 25% β-sheets, compared to similar materials with 41% β-sheets.70 This suggested that lower crystallinity of the surface favored not only cell attachment but also viability and proliferation.67
5. Conclusion
This combined computational–experimental work described with atomistic resolution the interfacial interaction of the amorphous, crystalline and N-terminus domains of silk in contact with HAP (001) surfaces at pH 7 and the potential implications for the development of osteoinductive silk biomaterials. Overall, this work provided computational data with atomistic resolution to quantify dynamic and structural processes that occur during the formation of silk–HAP composites. HAP (001) surfaces weakened a key mechanical signature of silk (its β-sheet crystals). Conversely, the amorphous and N-terminus domains remained relatively unaltered during the MD simulations. The reduction in β-sheets was also confirmed experimentally by FTIR of silk–HAP films, and could be only partially overcome by water vapor annealing. Even though a reduction in β-sheet crystals would lower the mechanical strength of the silk component, it could facilitate the interaction between silk and HAP, as well as increase cell attachment.Author contributions
DLK and MJB acquired funding, conceptualized and supervised this research. DLB, ZMM, AFB and VF conducted the research, analyzed the data, and created the visuals. DLB and AFB developed the computational work and the scripts for analysis. ZMM and VF performed the experimental work. The manuscript was written by all the authors and they have given approval to its final version.Data availability
The authors declare that all the relevant data supporting the findings of this study are available within the paper or from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could influence the work reported in this.Acknowledgements
DLB, ZMM, VF, DLK and MJB acknowledge the support from the National Institutes of Health (U01 EB014976). MJB acknowledges the support from ONR (N000141612333 and N000141912375) and DLK from the AFOSR (FA9550-17-1-0333). All the authors acknowledge the support from MIT-Spain INDITEX Sustainability Seed Fund project “Multiscale modeling and production of silk-ceramic bone regeneration biomaterials”.References
- N. Reznikov, M. Bilton, L. Lari, M. Stevens and R. Kröger, Science, 2018, 360, eaao2189 CrossRef PubMed.
- S. Von Euw, Y. Wang, G. Laurent, C. Drouet, F. Babonneau, N. Nassif and T. Azaïs, Sci. Rep., 2019, 9, 1–11 CrossRef CAS PubMed.
- H. Qu, H. Fu, Z. Han and Y. Sun, RSC Adv., 2019, 9, 26252–26262 RSC.
- M. Milazzo, N. Contessi Negrini, S. Scialla, B. Marelli, S. Farè, S. Danti and M. J. Buehler, Adv. Funct. Mater., 2019, 29, 1903055 CrossRef.
- M. C. Catoira, L. Fusaro, D. Di Francesco, M. Ramella and F. Boccafoschi, J. Mater. Sci. Mater. Med., 2019, 30, 115 CrossRef PubMed.
- O. O. Ige, L. E. Umoru and S. Aribo, ISRN Mater. Sci., 2012, 2012, 1–20 CrossRef.
- S. Bhatia and S. Bhatia, in Natural Polymer Drug Delivery Systems, Springer International Publishing, 2016, pp. 95–118 Search PubMed.
- Y. J. Yang, A. L. Holmberg and B. D. Olsen, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 549–575 CrossRef CAS PubMed.
- S. Keten, Z. Xu, B. Ihle and M. J. Buehler, Nat. Mater., 2010, 9, 359 CrossRef CAS PubMed.
- D. Ebrahimi, O. Tokareva, N. G. Rim, J. Y. Wong, D. L. Kaplan and M. J. Buehler, ACS Biomater. Sci. Eng., 2015, 1, 864–876 CrossRef CAS PubMed.
- Y. Wang, J. Guo, L. Zhou, C. Ye, F. G. Omenetto, D. L. Kaplan and S. Ling, Adv. Funct. Mater., 2018, 28, 1–24 Search PubMed.
- X. Wang, Z. Ding, C. Wang, X. Chen, H. Xu, Q. Lu and D. L. Kaplan, J. Mater. Chem. B, 2018, 6, 2739–2746 RSC.
- D. López Barreiro, Z. Martín-Moldes, J. Yeo, S. Shen, M. J. Hawker, F. J. Martin-Martinez, D. L. Kaplan and M. J. Buehler, Adv. Mater., 2019, 31, 1904720 CrossRef PubMed.
- S. Ling, K. Jin, D. L. Kaplan and M. J. Buehler, Nano Lett., 2016, 16, 3795–3800 CrossRef CAS PubMed.
- C. Fu, Z. Shao and V. Fritz, Chem. Commun., 2009, 6515–6529 RSC.
- S.-W. Ha, H. S. Gracz, A. E. Tonelli and S. M. Hudson, Biomacromolecules, 2005, 6, 2563–2569 CrossRef CAS PubMed.
- C.-Z. Zhou, F. Confalonieri, M. Jacquet, R. Perasso, Z.-G. Li and J. Janin, Proteins: Struct., Funct., Bioinf., 2001, 44, 119–122 CrossRef CAS PubMed.
- G. Chen, N. Matsuhisa, Z. Liu, D. Qi, P. Cai, Y. Jiang, C. Wan, Y. Cui, W. R. Leow, Z. Liu, S. Gong, K. Zhang, Y. Cheng and X. Chen, Adv. Mater., 2018, 30, 1800129 CrossRef PubMed.
- W. Huang, S. Ling, C. Li, F. G. Omenetto and D. L. Kaplan, Chem. Soc. Rev., 2018, 47, 6486–6504 RSC.
- D. P. Tran, V. T. Lam, T. L. Tran, T. N. S. Nguyen and H. T. Thi Tran, Phys. Chem. Chem. Phys., 2018, 20, 19240–19249 RSC.
- Z. Ding, Z. Fan, X. Huang, Q. Lu, W. Xu and D. L. Kaplan, ACS Appl. Mater. Interfaces, 2016, 8, 24463–24470 CrossRef CAS PubMed.
- H. H. K. Xu, P. Wang, L. Wang, C. Bao, Q. Chen, M. D. Weir, L. C. Chow, L. Zhao, X. Zhou and M. A. Reynolds, Bone Res., 2017, 5, 17056 CrossRef CAS PubMed.
- M. Farokhi, F. Mottaghitalab, S. Samani, M. A. Shokrgozar, S. C. Kundu, R. L. Reis, Y. Fatahi and D. L. Kaplan, Biotechnol. Adv., 2018, 36, 68–91 CrossRef CAS PubMed.
- A. J. Mieszawska, N. Fourligas, I. Georgakoudi, N. M. Ouhib, D. J. Belton, C. C. Perry and D. L. Kaplan, Biomaterials, 2010, 31, 8902–8910 CrossRef CAS PubMed.
- Z. Martín-Moldes, D. Ebrahimi, R. Plowright, N. Dinjaski, C. C. Perry, M. J. Buehler and D. L. Kaplan, Adv. Funct. Mater., 2017, 28, 1702570 CrossRef PubMed.
- Q. Fu, E. Saiz, M. N. Rahaman and A. P. Tomsia, Mater. Sci. Eng., C, 2011, 31, 1245–1256 CrossRef CAS PubMed.
- X. Du, D. Wei, L. Huang, M. Zhu, Y. Zhang and Y. Zhu, Mater. Sci. Eng., C, 2019, 103, 109731 CrossRef CAS PubMed.
- B. Marelli, C. E. Ghezzi, A. Alessandrino, J. E. Barralet, G. Freddi and S. N. Nazhat, Biomaterials, 2012, 33, 102–108 CrossRef CAS PubMed.
- S. Ling, Z. Qin, W. Huang, S. Cao, D. L. Kaplan and M. J. Buehler, Sci. Adv., 2017, 3, e1601939 CrossRef PubMed.
- S. Bhumiratana, W. L. Grayson, A. Castaneda, D. N. Rockwood, E. S. Gil, D. L. Kaplan and G. Vunjak-Novakovic, Biomaterials, 2011, 32, 2812–2820 CrossRef CAS PubMed.
- F. A. Sheikh, H. W. Ju, B. M. Moon, H. J. Park, J. H. Kim, O. J. Lee and C. H. Park, Nanoscale Res. Lett., 2013, 8, 303 CrossRef PubMed.
- X. Huang, S. Bai, Q. Lu, X. Liu, S. Liu and H. Zhu, J. Biomed. Mater. Res., Part B, 2015, 103, 1402–1414 CrossRef CAS PubMed.
- Z. Ding, H. Han, Z. Fan, H. Lu, Y. Sang, Y. Yao, Q. Cheng, Q. Lu and D. L. Kaplan, ACS Appl. Mater. Interfaces, 2017, 9, 16913–16921 CrossRef CAS PubMed.
- V. Fitzpatrick, Z. Martín-Moldes, A. Deck, R. Torres-Sanchez, A. Valat, D. Cairns, C. Li and D. L. Kaplan, Biomaterials, 2021, 276, 120995 CrossRef CAS PubMed.
- Y. Jin, B. Kundu, Y. Cai, S. C. Kundu and J. Yao, Colloids Surf., B, 2015, 134, 339–345 CrossRef CAS PubMed.
- H. Liu, G. W. Xu, Y. F. Wang, H. S. Zhao, S. Xiong, Y. Wu, B. C. Heng, C. R. An, G. H. Zhu and D. H. Xie, Biomaterials, 2015, 49, 103–112 CrossRef CAS PubMed.
- N. Dinjaski, R. Plowright, S. Zhou, D. J. Belton, C. C. Perry and D. L. Kaplan, Acta Biomater., 2017, 49, 127–139 CrossRef CAS PubMed.
- F. Barthelat, Z. Yin and M. J. Buehler, Nat. Rev. Mater., 2016, 1, 16007 CrossRef CAS.
- Y. Cheng, L.-D. Koh, D. Li, B. Ji, Y. Zhang, J. Yeo, G. Guan, M.-Y. Han and Y.-W. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 21787–21796 CrossRef CAS PubMed.
- A. M. Grant, H. S. Kim, T. L. Dupnock, K. Hu, Y. G. Yingling and V. V. Tsukruk, Adv. Funct. Mater., 2016, 26, 6380–6392 CrossRef CAS.
- S. Ling, K. Jin, Z. Qin, C. Li, K. Zheng, Y. Zhao, Q. Wang, D. L. Kaplan and M. J. Buehler, Adv. Mater., 2018, 30, 1802306 CrossRef PubMed.
- S. J. Segvich, H. C. Smith and D. H. Kohn, Biomaterials, 2009, 30, 1287–1298 CrossRef CAS PubMed.
- S. Segvich, S. Biswas, U. Becker and D. H. Kohn, Cells Tissues Organs, 2009, 189, 245–251 CrossRef CAS PubMed.
- T.-J. Lin and H. Heinz, J. Phys. Chem. C, 2016, 120, 4975–4992 CrossRef CAS.
- A. Rimola, M. Corno, J. Garza and P. Ugliengo, Philos. Trans. R. Soc., A, 2012, 370, 1478–1498 CrossRef CAS PubMed.
- X. Dong, Q. Wang, T. Wu and H. Pan, Biophys. J., 2007, 93, 750–759 CrossRef CAS PubMed.
- J.-W. Shen, T. Wu, Q. Wang and H.-H. Pan, Biomaterials, 2008, 29, 513–532 CrossRef CAS PubMed.
- H. Heinz, T.-J. Lin, R. K. Mishra and F. S. Emami, Langmuir, 2013, 29, 1754–1765 CrossRef CAS PubMed.
- Materials Studio, https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio/, (accessed 31 January 2021).
- Y.-X. He, N.-N. Zhang, W.-F. Li, N. Jia, B.-Y. Chen, K. Zhou, J. Zhang, Y. Chen and C.-Z. Zhou, J. Mol. Biol., 2012, 418, 197–207 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell, Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- D. Frishman and P. Argos, Proteins: Struct., Funct., Bioinf., 1995, 23, 566–579 CrossRef CAS PubMed.
- D. N. Rockwood, R. C. Preda, T. Yücel, X. Wang, M. L. Lovett and D. L. Kaplan, Nat. Protoc., 2011, 6, 1612 CrossRef CAS PubMed.
- S. Zhou, W. Huang, D. J. Belton, L. O. Simmons, C. C. Perry, X. Wang and D. L. Kaplan, Acta Biomater., 2015, 15, 173–180 CrossRef CAS PubMed.
- J. Bandekar, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1992, 1120, 123–143 CrossRef CAS.
- H. Yang, S. Yang, J. Kong, A. Dong and S. Yu, Nat. Protoc., 2015, 10, 382–396 CrossRef CAS PubMed.
- S. Ling, N. Dinjaski, D. Ebrahimi, J. Y. Wong, D. L. Kaplan and M. J. Buehler, ACS Biomater. Sci. Eng., 2016, 2, 1298–1308 CrossRef CAS PubMed.
- J. Ming, Z. Liu, S. Bie, F. Zhang and B. Zuo, Mater. Sci. Eng., C, 2014, 37, 48–53 CrossRef CAS PubMed.
- X. Hu, D. Kaplan and P. Cebe, Macromolecules, 2006, 39, 6161–6170 CrossRef CAS.
- A. Takeuchi, C. Ohtsuki, T. Miyazaki, M. Kamitakahara, S. Ogata, M. Yamazaki, Y. Furutani, H. Kinoshita and M. Tanihara, J. R. Soc., Interface, 2005, 2, 373–378 CrossRef CAS PubMed.
- Q. Wang, X. Xia, W. Huang, Y. Lin, Q. Xu and D. L. Kaplan, Adv. Funct. Mater., 2014, 24, 4303–4310 CrossRef CAS PubMed.
- A. Ibáñez-Fonseca, D. Orbanic, F. J. Arias, M. Alonso, D. I. Zeugolis and J. C. Rodríguez-Cabello, Small, 2020, 16, 2001244 CrossRef PubMed.
- M. D. Golinska, M. K. Włodarczyk-Biegun, M. W. T. Werten, M. A. C. Stuart, F. A. de Wolf and R. de Vries, Biomacromolecules, 2014, 15, 699–706 CrossRef CAS PubMed.
- X. Hu, K. Shmelev, L. Sun, E. S. Gil, S. H. Park, P. Cebe and D. L. Kaplan, Biomacromolecules, 2011, 12, 1686–1696 CrossRef CAS PubMed.
- Q. Lu, X. Hu, X. Wang, J. A. Kluge, S. Lu, P. Cebe and D. L. Kaplan, Acta Biomater., 2010, 6, 1380–1387 CrossRef CAS PubMed.
- C. Malinowski, F. He, Y. Zhao, I. Chang, D. W. Hatchett, S. Zhai and H. Zhao, RSC Adv., 2019, 9, 40792–40799 RSC.
- Z. Wenhao, T. Zhang, J. Yan, Q. Li, P. Xiong, Y. Li, Y. Cheng and Y. Zheng, Acta Biomater., 2020, 116, 223–245 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation of hydroxyapatite surfaces resolved at pH 7, FTIR spectra of HAP and silk–HAP, deconvolution of the amide I peak for different silk–HAP materials. See DOI: https://doi.org/10.1039/d2nr01989b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |