
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Template synthesis of dual-functional porous MoS2 nanoparticles with photothermal conversion and catalytic properties†
Xuefeng
Pan
ab,
Radwan M.
Sarhan
a,
Zdravko
Kochovski
a,
Guosong
Chen
 c,
Andreas
Taubert
b,
Shilin
Mei
*a and
Yan
Lu
*ab
c,
Andreas
Taubert
b,
Shilin
Mei
*a and
Yan
Lu
*ab
aDepartment for Electrochemical Energy Storage, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, Berlin 14109, Germany. E-mail: yan.lu@helmholtz-berlin.de; shilin.mei@helmholtz-berlin.de
bInstitute of Chemistry, University of Potsdam, Potsdam 14476, Germany
cThe State Key Laboratory of Molecular Engineering of Polymers and Department of Macromolecular Science, Fudan University, Shanghai 200433, China
First published on 5th April 2022
Abstract
Advanced catalysis triggered by photothermal conversion effects has aroused increasing interest due to its huge potential in environmental purification. In this work, we developed a novel approach to the fast degradation of 4-nitrophenol (4-Nip) using porous MoS2 nanoparticles as catalysts, which integrate the intrinsic catalytic property of MoS2 with its photothermal conversion capability. Using assembled polystyrene-b-poly(2-vinylpyridine) block copolymers as soft templates, various MoS2 particles were prepared, which exhibited tailored morphologies (e.g., pomegranate-like, hollow, and open porous structures). The photothermal conversion performance of these featured particles was compared under near-infrared (NIR) light irradiation. Intriguingly, when these porous MoS2 particles were further employed as catalysts for the reduction of 4-Nip, the reaction rate constant was increased by a factor of 1.5 under NIR illumination. We attribute this catalytic enhancement to the open porous architecture and light-to-heat conversion performance of the MoS2 particles. This contribution offers new opportunities for efficient photothermal-assisted catalysis.
Introduction
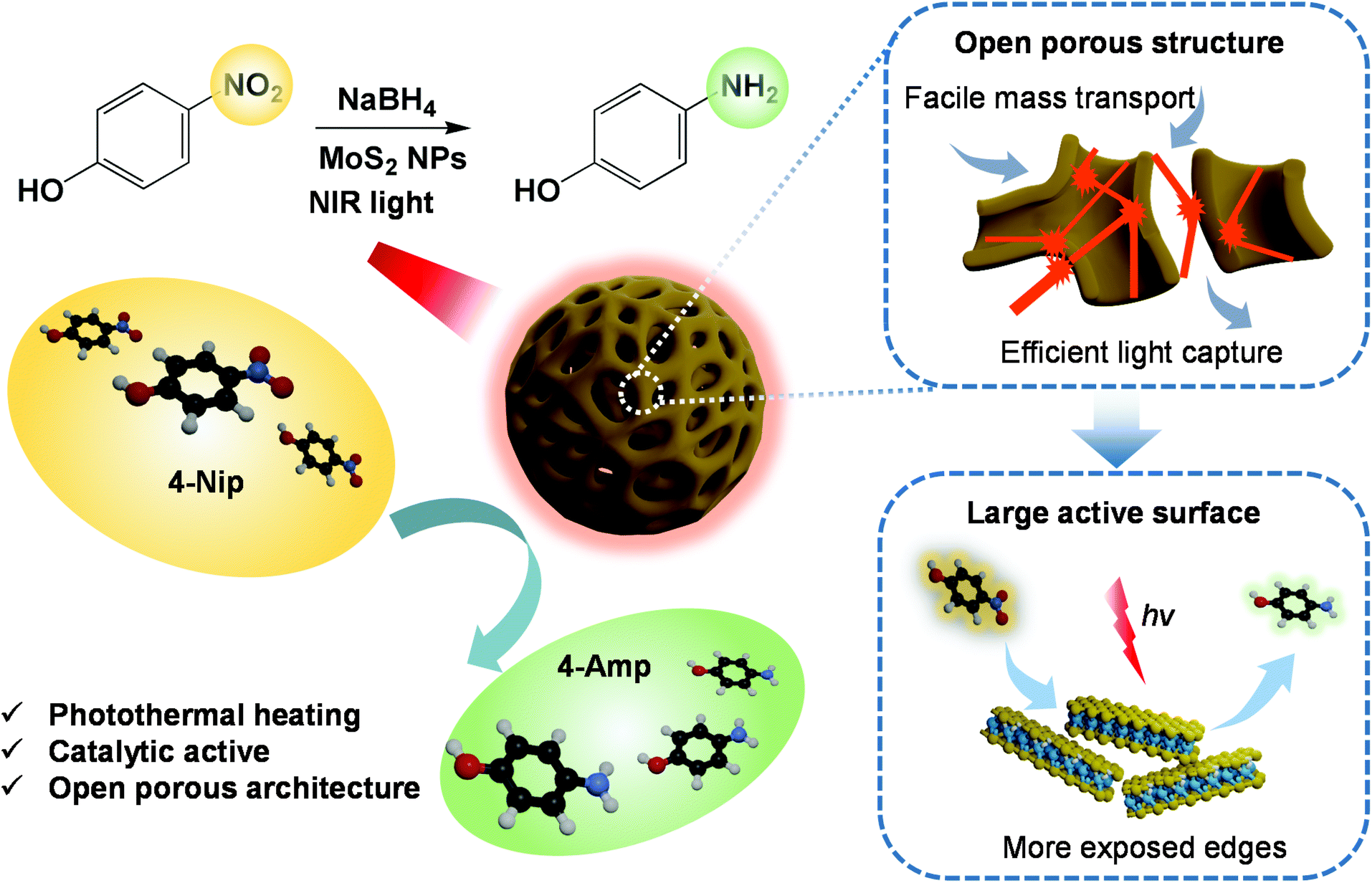
Over the past decades, solar-driven photothermal conversion has attracted intensive attention, which enables broad applications such as environmental remediation, water desalination and photothermal-assisted catalysis under mild conditions with high efficiency.1–4 Therefore, it is essential to develop photothermal conversion catalysts that can strongly harvest solar energy ranging from UV to the visible light, and even to the near-infrared light (NIR) to accelerate the degradation of water contaminants by the converted heat.5–7 In principle, the rational design of efficient photothermal conversion-assisted catalysts includes, but is not limited to, active composition as well as structural optimization. Nowadays, various solar absorbers including plasmonic noble metals (e.g., gold and silver),8,9 narrow bandgap semiconductors (e.g., copper sulfide and tungsten oxide),10–12 carbon-based nanostructures (e.g., carbon nanotubes and graphenes),13,14 and organic compounds (e.g., polyaniline (PANI), polypyrrole and polydopamine (PDA)) with outstanding photothermal conversion performance are successfully fabricated.15–17 For instance, a bismuth vanadate photocatalyst modified with PANI was applied in the water oxidation reaction under NIR light irradiation. The water oxidation photocurrent was increased by more than 300% than that of pristine catalysts with the assistance of photothermal heating of PANI.17 Recently, our group has successfully developed a gold@PDA nanoreactor for the fast reduction of 4-nitrophenol (4-Nip). The photoheating caused by PDA under NIR irradiation greatly enhanced the reduction reaction catalyzed by the gold nanoparticles embedded inside the nanoreactor.18 Generally, the catalyst and the photothermal converter are often two different species according to the above-mentioned reports. A hybridization process is usually required to combine the unique function of each material, which often involves complicated synthetic routes and is undesirable from the economic point of view. Furthermore, the activity of the catalytic species is possibly being impaired due to the modification by the photothermal conversion components, which may reduce the active surface or induce diffusion-controlled sluggish kinetics. Therefore, developing a dual-functional catalyst utilizing its intrinsic photothermal conversion and catalytic properties is in great demand.Among various photothermal conversion materials, molybdenum disulfide (MoS2) has shown great potential in the fields of hydrogen generation, pollutant degradation and photothermal therapy due to its tunable chemical and physical properties.19–24 The photothermal effect of MoS2 is mainly explained by the generation of electron-hole pairs upon light excitation and their relaxation via a non-radiative decay mechanism.25–27 In addition, MoS2 exhibits a good catalytic performance in the reduction of 4-Nip.28 The surface area and crystallinity of the catalytic surface are additional important parameters that determine their performances. Large surface area with a plenty of exposed edges can provide abundant active sites for the catalytic reaction.29 In addition, metallic 1T-phase MoS2 has been reported to exhibit a stronger NIR absorption and a higher photothermal conversion efficiency than those of the 2H-phase.22 However, their instability and tedious synthesis protocol are inevitable. For the more stable 2H phase of MoS2, the unsaturated sulfur (S) atoms at the edge sides of MoS2 nanosheets determine the catalytic activity, while the other S atoms in bulk MoS2 contribute little to the overall activity.30,31 Thus, fabrication of MoS2 with abundant exposed active edges is an effective method to improve the catalytic performance. Furthermore, several approaches including bandgap engineering, defect design, and structural heterojunction via hybridization with other nanomaterials such as titanium oxide and nickel oxide have been developed to simultaneously optimize their catalytic and photothermal conversion performance.26,32,33 Inevitably, these modification approaches also involve complicated synthetic procedures, which lead to additional production costs. Thus, facile fabrication of MoS2 catalysts with efficient photothermal conversion effects and rich active sites on the catalytic surface remains a challenge.
Recently, it has been revealed that the rational design of nanostructured MoS2 (e.g., hollow structure) could enable enhanced light scattering and capturing, reduced distance for charge migration, and abundant surface reactive area.34–36 However, less efforts have been made to explore the more complicated surface/internal nanostructure of MoS2 due to the limitation of commonly used synthetic methods (e.g., hard template, hydrothermal method, and physical deposition). In contrast, soft templates such as surfactant micelles, organic molecules, and polymer assemblies are good alternatives to prepare novel nanostructures.37,38 Unlike the harsh conditions needed to remove the hard templates, soft polymer templates can be readily eliminated by solvent dissolution or direct calcination. In particular, block copolymers open up new possibilities for the construction of advanced architectures due to their tunable microphase-separation when serving as soft templates.
In this work, we developed a series of dual-functional MoS2 particles with well-designed nanostructures, combining the photothermal conversion effect and catalytic property using a soft-template strategy (Scheme 1). Polystyrene-b-poly(2-vinylpyridine) (PS-b-P2VP) nanoparticles with tunable swollen structures were first fabricated and then used as soft templates.39 Different volume fractions (determined by the molecular weight) of the P2VP moiety and swelling conditions were applied to tailor the microphase separation of the PS-b-P2VP particles, inducing a series of soft templates with significantly different nanostructures.40 The template can be directly complexed with the ammonium tetrathiomolybdate ((NH4)2MoS4) as the MoS2 precursor. After calcination, MoS2 nanoparticles with pomegranate-like, hollow, and open porous structures were successfully constructed. The dependence of the photothermal conversion effect as well as the catalytic performance on their morphologies was systematically investigated, demonstrating the importance of structure–property relationship in the photothermal conversion-assisted catalysis.
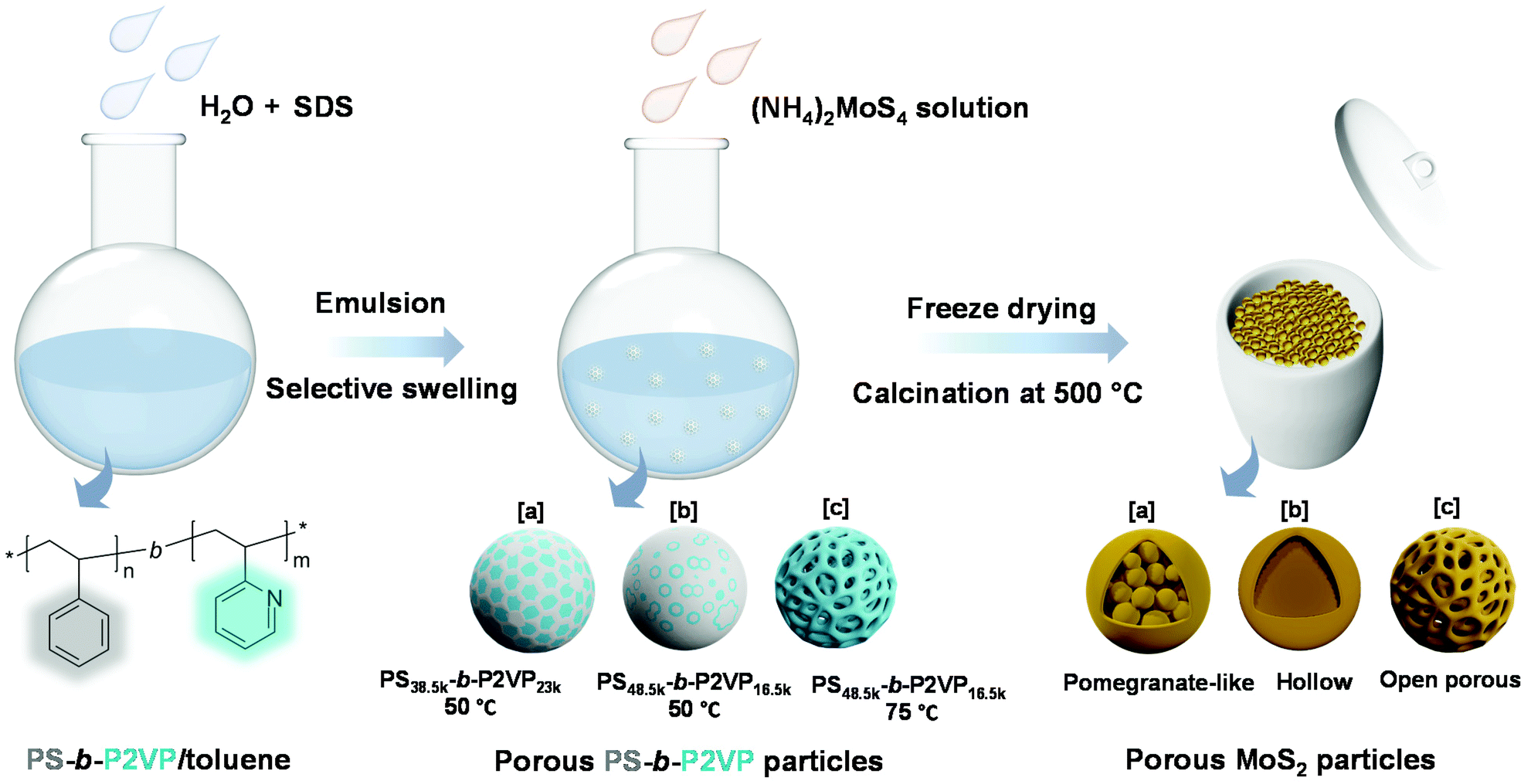 | ||
| Scheme 1 Synthesis strategy of MoS2 particles with various morphologies using PS-b-P2VP as a sacrificial soft template. | ||
Results and discussion
Material synthesis and characterization
Block copolymers composed of two (or more) covalently bonded homopolymer chains show rich functional groups and microphase separation structure, promoting their increasing application in the formation of ordered hybrid structures and porous functional materials.41–47 A simple pore-generating method termed “selective swelling” has shown its great potential in extending the micropattern diagram as well as the porous structure of block copolymers.48,49 The methodology is based on the difference in polarity (or interaction) of the constituent blocks and the swelling solvent. The pores/voids are typically derived from deformed domains of the minor component dispersed in the continuous phase of the major component. In this contribution, PS-b-P2VP particles with two different molecular weight fractions of the P2VP moiety were generated by a delicate emulsion-swelling method (Fig. S1a and S1b†). Ethanol, which has strong interaction with the P2VP block but poor interaction with the PS block, was applied as the selective swelling solvent. For PS38.5k-b-P2VP23k and PS48.5k-b-P2VP16.5k, the template particles tend to form solid nanostructures upon swelling in ethanol at 50 °C (Fig. S1c and S1d†). It is also found that dense and spherical P2VP nanodomains (∼20 nm) were uniformly distributed inside the polymer particles in PS38.5k-b-P2VP23k. For PS48.5k-b-P2VP16.5k, an irregular hollow space was also generated inside. In this scenario, swelling of both polymer particles results in only slight deformation, and therefore, no porous structures were obtained under this mild condition (Fig. 1a and b). This is mainly due to the slight swelling degree of the P2VP chains at this temperature (50 °C). Moreover, the osmotic pressure of the slightly swollen P2VP domains is too weak to drive the plastic deformation of the rigid PS domains. Therefore, solid or non-porous complex nanostructures of MoS2 can be expected using these featured polymer particles as templates for the subsequent step of MoS2 generation. Notably, when the temperature increased from 50 to 75 °C, an open porous nanostructure with interconnected micellar fibers was fabricated (Fig. 1c). At this temperature, a strong chain movement is triggered, which gives rise to the structural reorganization of the polymer chains into network-like spheres. Accordingly, an open porous structure of MoS2 particles can be obtained by applying these porous particles as synthetic templates. Note that a lower temperature (30 °C) led to a slight swelling of the polymer chains, which is similar to that performed at 50 °C (Fig. S1e†). In contrast, a higher temperature (90 °C) led to the deformation of the polymer particles (Fig. S1f†).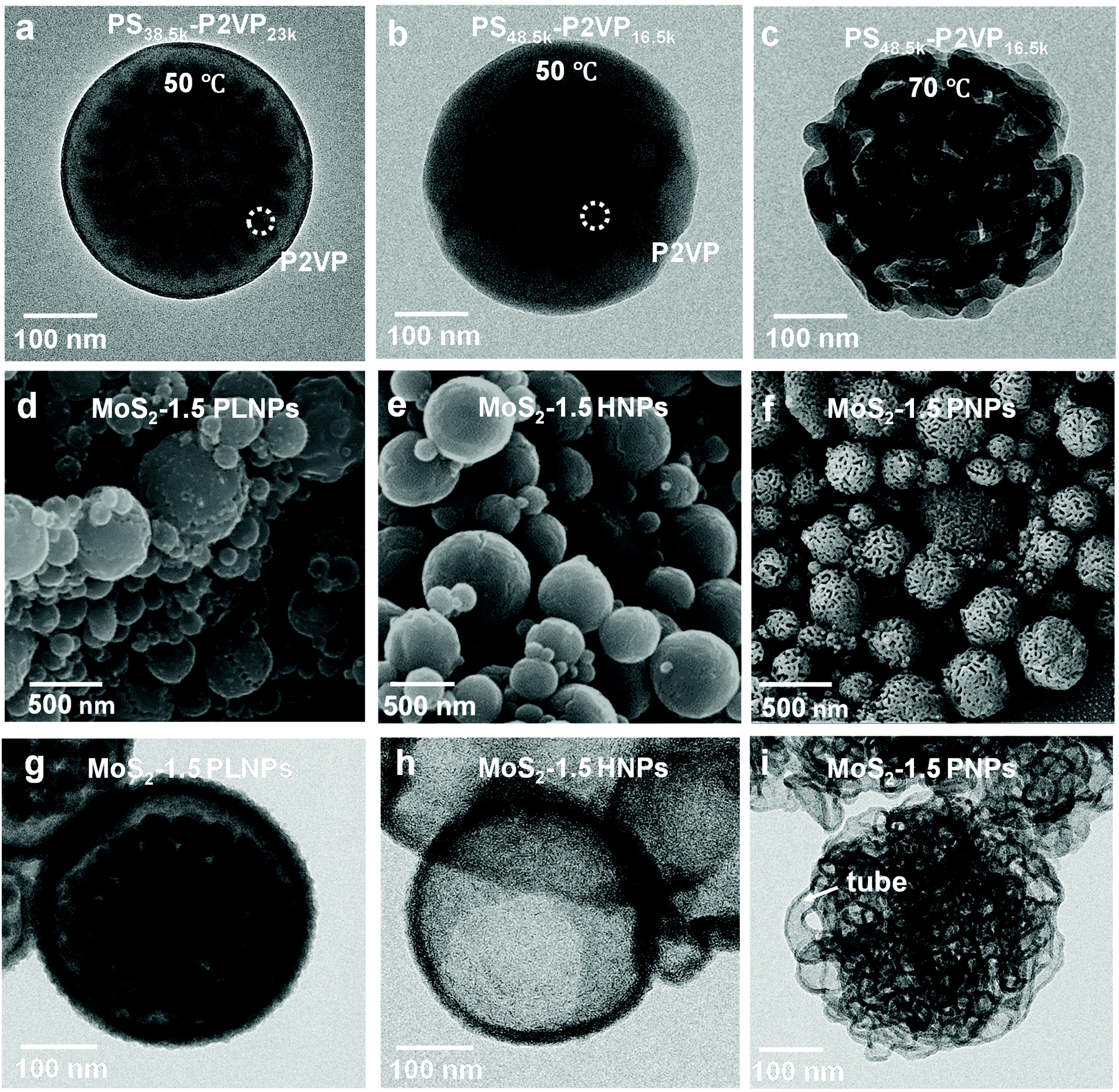 | ||
| Fig. 1 TEM images of (a) PS38.5k-b-P2VP23k and (b) PS48.5k-b-P2VP16.5k particles obtained by swelling at 50 °C, and (c) PS48.5k-b-P2VP16.5k particles obtained by swelling at 75 °C. The polymer particles were stained with I2 vapor for 20 min. The dark contrast shows the distribution of P2VP domains. (d) SEM and (g) TEM images of the MoS2-1.5 PLNPs. (e) SEM and (h) TEM images of the MoS2-1.5 HNPs. (f) SEM and (i) TEM images of the MoS2-1.5 PNPs. | ||
Block copolymers have been reported to serve as templates for the synthesis of nanoparticles via coordination with inorganic precursors.50,51 Benefiting from the good affinity of the P2VP block to the inorganic precursors, anionic MoS42− was simply complexed with the PS-b-P2VP template with selective loading onto the P2VP domain.52,53 After the thorough incorporation of a molybdenum precursor into the PS-b-P2VP particle, the introduction of a trace amount of H+ and 3-MPA into the solution leads to the decomposition of MoS42− into MoSx (x = 2–3). In this experiment, 3-MPA can serve as a stabilizer and prevent the aggregation of the composite particles. Finally, MoS2 particles with various internal nanostructures were obtained after calcination in an argon atmosphere. During the calcination step, MoS42− was pyrolyzed into MoS2 as the backbone, while much of the polymer template was calcined, leaving a small amount of carbon residues and hollow channels. For PS38.5k-b-P2VP23k particles swollen at 50 °C, MoS42− cations diffused inside the polymer particle and coordinated with small spherical P2VP domains due to the relatively large volume fraction of the P2VP block (37.4%), compared to PS48.5k-b-P2VP16.5k with a fraction of 25.4%. This results in the formation of pomegranate-like nanoparticles (PLNPs) that contain a solid outer layer (∼25 nm thick) with numerous dense nanospheres (∼30 nm) inside, as shown in Fig. 1d and g. A limited free space was also observed in PLNPs. In contrast, MoS2 hollow nanoparticles (HNPs) were obtained using PS48.5k-b-P2VP16.5k particles swollen at 50 °C as templates (Fig. 1e). The HNP particles possess a thin layer of MoS2 shell (∼9 nm), without any visible internal MoS2 spheres (Fig. 1h). The observed small particles also exhibit the same internal structure as the big ones (Fig. S2a† and 2b). This might be ascribed to the fact that few MoS42− cations can diffuse into the internal part of PS48.5k-b-P2VP16.5k particles primarily limited by their thicker blocking layer (rigid PS layers) compared with PS38.5k-b-P2VP23k. When it comes to PS48.5k-b-P2VP16.5k particles swollen at 75 °C, the MoS2 porous nanoparticles (PNPs) preserve the original loose structure of the polymer template (Fig. 1f). Due to the free space between interlocked fibers, the MoS42− cations can easily diffuse and adsorb onto the internal part of the porous polymer template. After calcination, the polymer was burnt out leading to the formation of the MoS2 particles with multi-interconnected hollow channels (Fig. 1i). The open porous MoS2 particles with an interconnected hollow channel structure show several advantages when compared with particles of other morphologies including high porosity, light weight, and large accessible surface area. These are of special relevance to the practical applications including drug delivery, water purification, and surface catalysis. To further optimize the particle structure, we successfully tuned the thickness of the deposited MoS2 layers by varying the concentration of the inorganic precursor during the synthesis. Fig. 2a–c show the SEM images of a series of MoS2 particles prepared with different MoS42− concentrations. The MoS2 PNPs prepared with different concentrations of (NH4)2MoS4 (1, 1.5, and 3 mg mL−1) are denoted as MoS2-1, MoS2-1.5 and MoS2-3 PNPs, respectively. These three samples show a broad size distribution with a similar trend that the diameter of the majority concentrates between 200 and 500 nm (Fig. S2c–e†). By increasing the precursor concentration, more MoS42− were coordinated onto the PS-b-P2VP particles and the distance between the separated micellar fibers became narrower. The layer thickness of the hollow channels in the particles increased from ∼7 to 12 nm on average, when the concentration of precursor increased from 1 to 3 mg mL−1. The presence of tiny MoS2 flakes with generally single-layered structures was identified from the HRTEM images (Fig. 2d), suggesting the abundance of exposed active edges. The outer and inner diffraction rings as observed from the selected-area electron diffraction (SAED) pattern correspond to the (110) and (100) reflection of MoS2, respectively (Fig. 2e). It is worth noting that the isolated MoS2 nanoflakes (short sheets of length ∼5 nm) of the MoS2-1.5 PNPs might be favorable for the utilization of active edges. Carbon generated from the polymer template during calcination also prevents the aggregation of the MoS2 nanoflakes. On the contrary, the nanoflakes of bulk MoS2 (Fig. 2f) fabricated without any polymer template show a huge tendency to fuse together, resulting in the formation of a few layers of stacked multilayers (long sheets of length ∼9 nm). Besides, an irregular bulk phase without any defined structure was found from the inset SEM image.
 | ||
| Fig. 2 TEM images of MoS2 PNPs prepared at precursor concentrations of (a) 1, (b) 1.5, and (c) 3 mg mL−1. The insets are the corresponding SEM images with a scale bar of 500 nm. (d) HRTEM image and (e) SAED pattern of MoS2-1.5 PNPs. (f) HRTEM image of bulk MoS2 prepared without any polymer template; the inset is the corresponding SEM image with a scale bar of 500 nm. | ||
Taking MoS2-1.5 PNPs, for example, the X-ray diffraction (XRD) profile verifies that MoS2 in all the PNPs exhibits a typical 2H phase (Fig. 3a, see the others in Fig. S3†).54 The main diffraction peaks at 32° and 56.8° correspond to the (100) and (110) reflections, respectively. Moreover, the weak (002) reflection at 14.4° indicates the relatively limited layers of MoS2. The chemical composition of the obtained MoS2-1.5 PNPs was further confirmed by Raman spectroscopy, as shown in Fig. 3b. The two Raman peaks at 1345 and 1587 cm−1 represent the D and G bands of sp2 carbon, respectively, while the Raman peaks at around 377 and 403 cm−1, correspond to the E12g and out-of-plane A1g vibration modes of the MoS2.55 The amount of carbon and MoS2 was calculated based on the molybdenum oxide residue from the TGA measurement in air (Fig. 3c and S4†). The MoS2-1.5 PNPs show ∼38.9 wt% of carbon species, corresponding to 61.1 wt% of MoS2 in the particles. In contrast, ∼69.2 wt% of MoS2 was left in the MoS2-3 PNPs on account of higher amounts of MoS42− precursors during coordination, as summarized in Table S1.† From the N2 adsorption/desorption isotherm curves in Fig. 3d, a distinct hysteresis loop can be observed with a typical type-IV isotherm, referring to a mesoporous structure. The pore size distribution curve displays a narrow range of mesopores centered at ∼5 nm and a broad range (10–20 nm) centered at ∼15 nm. The specific surface area (SBET) of these MoS2 particles calculated according to the BET equation is summarized in Fig. S5 and Table S2.† There is a clear decreasing tendency of SBET for the MoS2-1, MoS2-1.5 and MoS2-3 PNPs. The SBET of MoS2-1.5 PNPs was calculated to be 106.6 m2 g−1, which is around 1.3-fold of that of the MoS2-3 PNPs. Meanwhile, the bulk MoS2 possesses the lowest SBET of 36.3 m2 g−1. The SBET apparently decreased with the increase in the layer thickness, mainly due to the gradually diminished gap between the thick MoS2 frameworks of the particles. Consequently, a reduced porosity as well as a lower specific surface area was obtained, as confirmed from the above-mentioned SEM and TEM images.
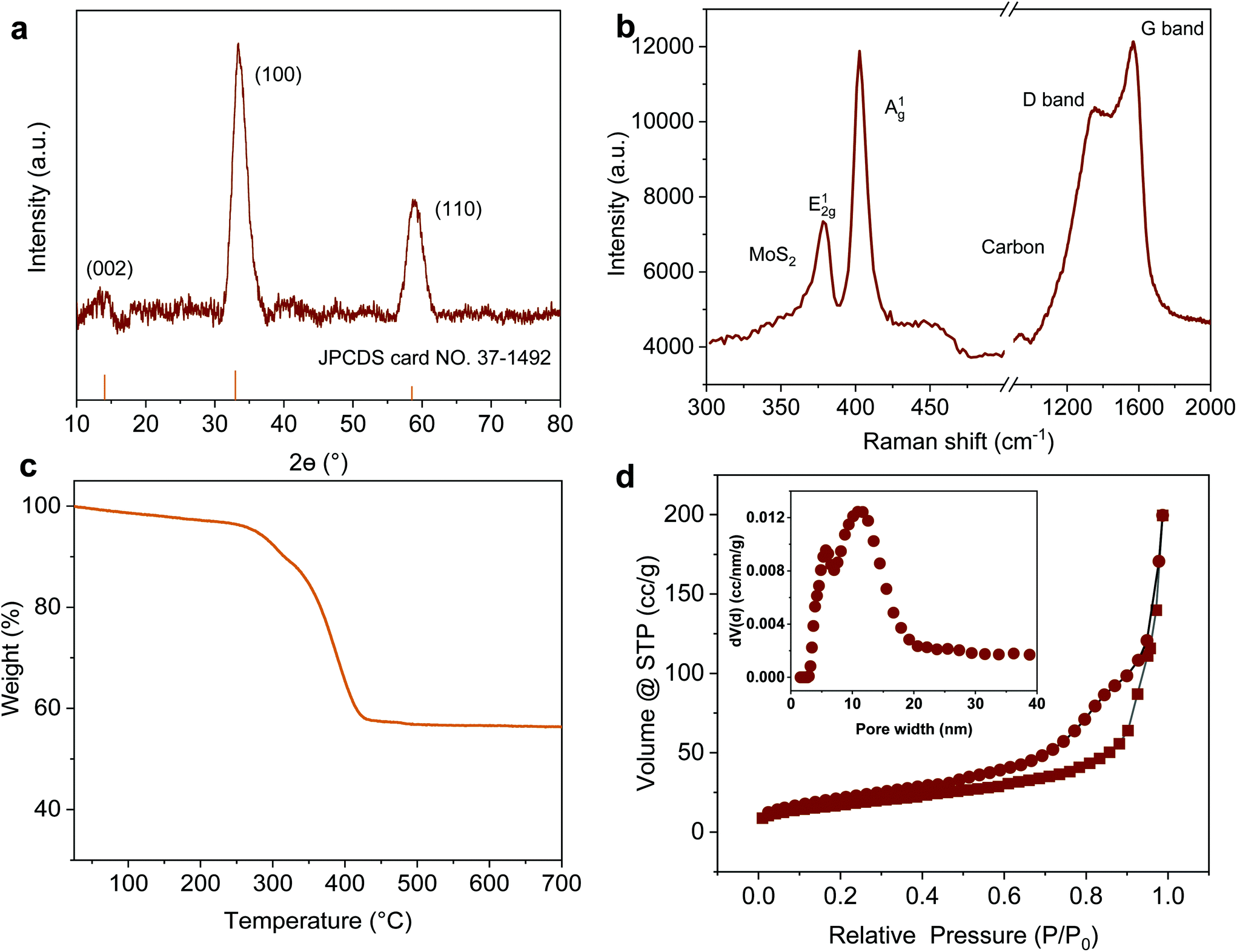 | ||
| Fig. 3 (a) XRD pattern, (b) Raman spectrum, (c) TGA in air and (d) N2 adsorption/desorption isotherm curves of MoS2-1.5 PNPs; the inset is the curve of pore size distribution. | ||
Photothermal conversion performance
To assess the photothermal conversion performance of the MoS2 particles, we monitored the temperature change of the MoS2 dispersion upon light irradiation as illustrated in Fig. 4a. A NIR-laser (808 nm, 3 W cm−2) was adopted to irradiate the dispersions of the MoS2 particles placed in a quartz cuvette, while the temperature was simultaneously recorded using a thermocouple connected to a digital thermometer. The thermal evolution of the MoS2-1.5 PNP dispersion with time was first visualized using an IR camera (Fig. 4b). The temperature of MoS2 particle dispersions clearly increased with the irradiation time. To precisely track the temperature variation of the MoS2 dispersion during NIR irradiation, quantitative results were further collected from the digital thermometer. Fig. 4c displays the photothermal heating curves of MoS2 particles fabricated with different polymer templates. The temperature of the MoS2 PNP dispersion increased by ∼20 °C, while the dispersion containing the same concentration of MoS2-1.5 HNPs and MoS2-1.5 PLNPs exhibited a temperature increase of ∼16 °C. The bulk MoS2 particle synthesized without any polymer template showed a much lower temperature increment of ∼11 °C. As a reference, the pure water displayed a temperature change of ∼5 °C under the same light intensity. Moreover, when the laser is off, the temperature profile of the MoS2 particle dispersions decreased slowly compared to that of the bulk MoS2, showing that their defined hollow and porous structures may lead to a better thermal insulating effect (Fig. S6†). These results indicate the dependence of the photothermal conversion performance on the surface/internal structure of the MoS2 particles. It has been reported that hollow nanostructures improve the light harvesting via multiple scattering of the photons.56,57 This, in turn, will enhance the light-to-heat conversion efficiency. The hollow architectures facilitate localization and harvesting of the light, resulting in a relatively high particle temperature. Subsequently, the solution temperature increases as the heat dissipates to the solution. Therefore, our nanostructured MoS2 particles (MoS2-1.5 PLNPs, MoS2-1.5 PNPs and MoS2-1.5 HNPs) show better photothermal conversion performance than that of bulk MoS2. For MoS2-1.5 PLNPs, however, the conversion efficiency could be limited by the insufficient free space. In addition, the MoS2-1.5 PNP particles possess more complicated porous construction than that of the MoS2-1.5 HNPs, which is likely to lead to efficient light scattering and capture. The complex open architecture of the MoS2-1.5 PNPs efficiently accelerates the mass and heat transport, resulting in a higher overall temperature of the dispersion. Hence, the photothermal conversion performance of the MoS2-1.5 PNPs stands out among these three featured MoS2 particles.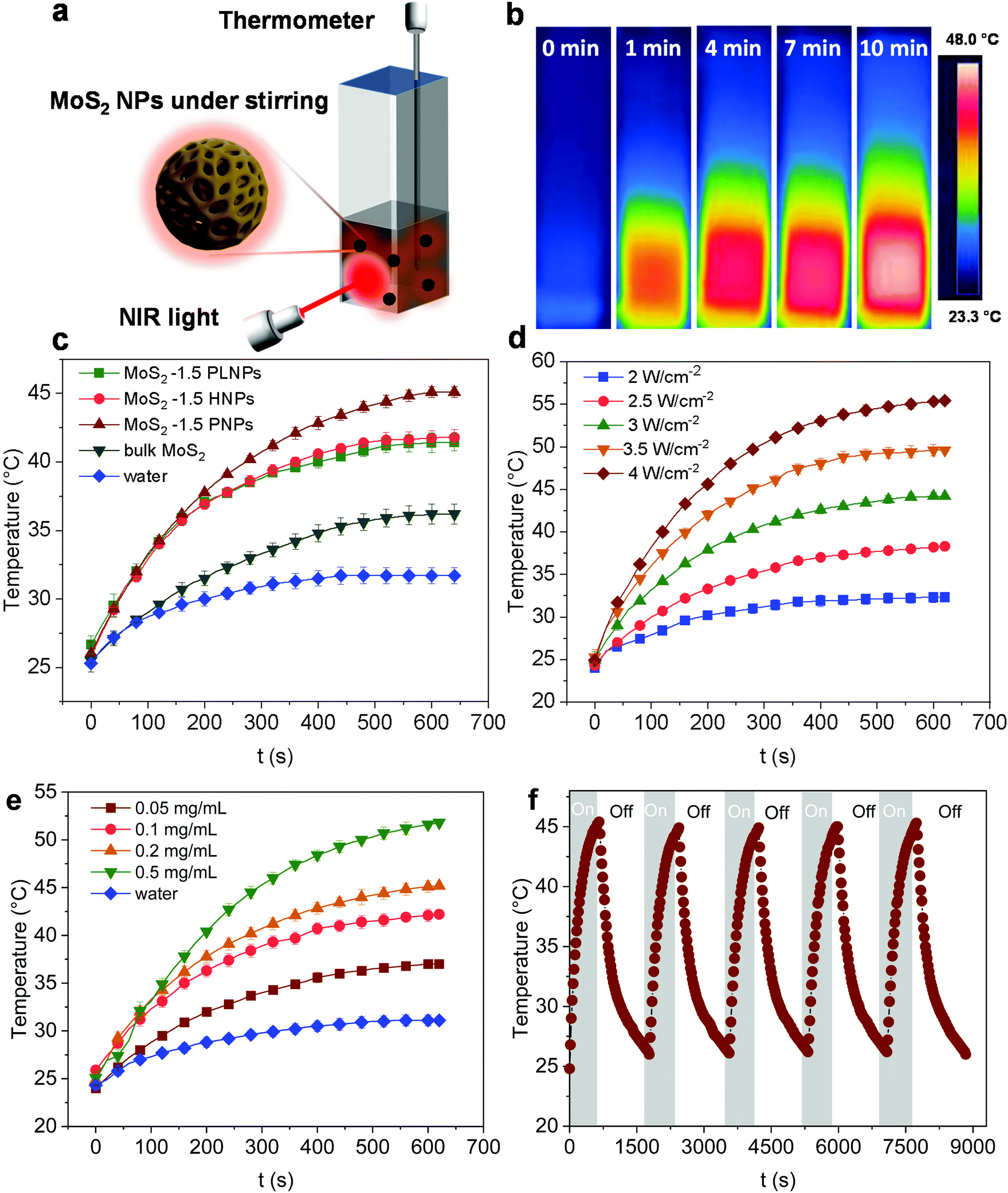 | ||
| Fig. 4 (a) Schematic of the photothermal conversion setup. (b) Temperature variation of the MoS2-1.5 PNP dispersion at 0.1 mg mL−1 visualized using an IR camera. (c) Photothermal curves of the MoS2 particles with different morphologies at 0.1 mg mL−1 and 3 W cm−2. (d) Photothermal curves of the MoS2-1.5 PNP dispersion at 0.1 mg mL−1 in response to different intensities of NIR light. (e) Photothermal curves of the MoS2-1.5 PNP dispersion in response to different concentrations at 3 W cm−2. (f) Stability test of the MoS2-1.5 PNP dispersion at 0.1 mg mL−1 and 3 W cm−2. | ||
We further investigated the dependence of the photothermal effect on the irradiation power and the particle concentration. As shown in Fig. 4d, when the laser intensity increased from 2 to 4 W cm−2, a temperature increment from ∼5 to ∼32 °C was observed for the MoS2-1.5 PNP dispersion. Additionally, when the concentration of the MoS2 PNP dispersion was elevated from 0.05 to 0.5 mg mL−1, the temperature increment of the dispersions increased from ∼14 to ∼29 °C under a constant laser power of 3 W cm−2 (Fig. 4e). This strongly implies the tunable capability of the photothermal behavior of our MoS2 particles, which is of great importance for many applications including photothermal-assisted catalysis and photothermal cancer therapy. In addition to the enhanced photothermal performance, MoS2 PNPs showed excellent stability over a long irradiation period. The temperature increment of the particles displayed a stable reversible behavior using successive ON/OFF irradiation cycles (Fig. 4f). No obvious structural change was observed from the TEM image (Fig. S7†). It is noteworthy that the dispersions of the MoS2 PNPs with different wall thicknesses exhibited similar photothermal conversion performances and a temperature ∼10 °C higher than that of the bulk MoS2 dispersion under the same irradiation conditions (Fig. S8†).
Catalytic reduction of 4-Nip
As a classic model reaction, the reduction of 4-Nip into 4-aminophenol (4-Amp) can be conducted under ambient conditions by NaBH4 in the presence of a catalyst. In addition, this reduction reaction can be facilely monitored by UV-vis absorption spectroscopy and commonly used as a benchmark to compare the catalytic performance of various catalysts.58,59 In this work, this model reaction was employed to unfold the catalytic efficiency of our MoS2 particles with different morphologies. Importantly, we also evaluated the catalytic performance under NIR irradiation by taking advantage of the high photothermal efficiency of the MoS2 particles. Unlike conventional heating over a large macroscopic area, nanoscale heating by laser irradiation highly localizes the heat around the particles compared to the bulk solution and, therefore, might accelerate the rate of surface catalytic reactions.60,61 It is noteworthy that the distinctive structure of the porous surface as well as the interconnected hollow channels in the MoS2 PNPs increases the exposed active MoS2 edges and accessible surface area for the catalytic surface reactions. In addition, these open porous particles also exhibit excellent photothermal performance. On the basis of these considerations, the catalytic performance of MoS2 PNPs was thereafter explored in the presence/absence of NIR light.Fig. 5a displays the typical time-dependent UV-vis absorption spectra of 4-Nip after introducing MoS2 PNPs particles as catalysts. As shown, at time zero, a distinct peak of the absorption spectrum is located at 400 nm, assigned to the 4-nitrophenolate ions.62 As the reaction proceeds, a new peak appears at 290 nm and increases in absorption intensity, confirming the gradual reduction of 4-Nip into 4-Amp. After 15 minutes, the absorption peak of the 4-Nip at 400 nm was diminished, revealing that 4-Nip molecules were completely converted into 4-Amp. First, the reaction was performed in the darkness to compare the catalytic activity of the three MoS2 PNPs with different wall thicknesses by comparing the reaction rates, as shown in Fig. 5b. The pseudo-first-order reaction was ensured by an excess amount of NaBH4 and the apparent reduction rate (kapp) of 4-Nip was calculated by monitoring the absorbance intensity at 400 nm over time according to eqn (1):28
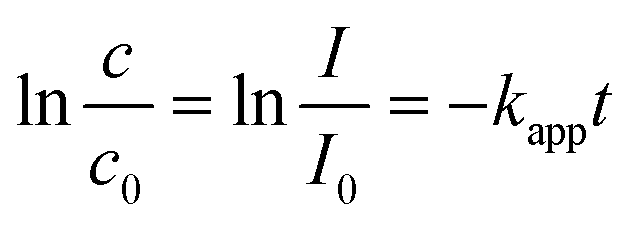 | (1) |
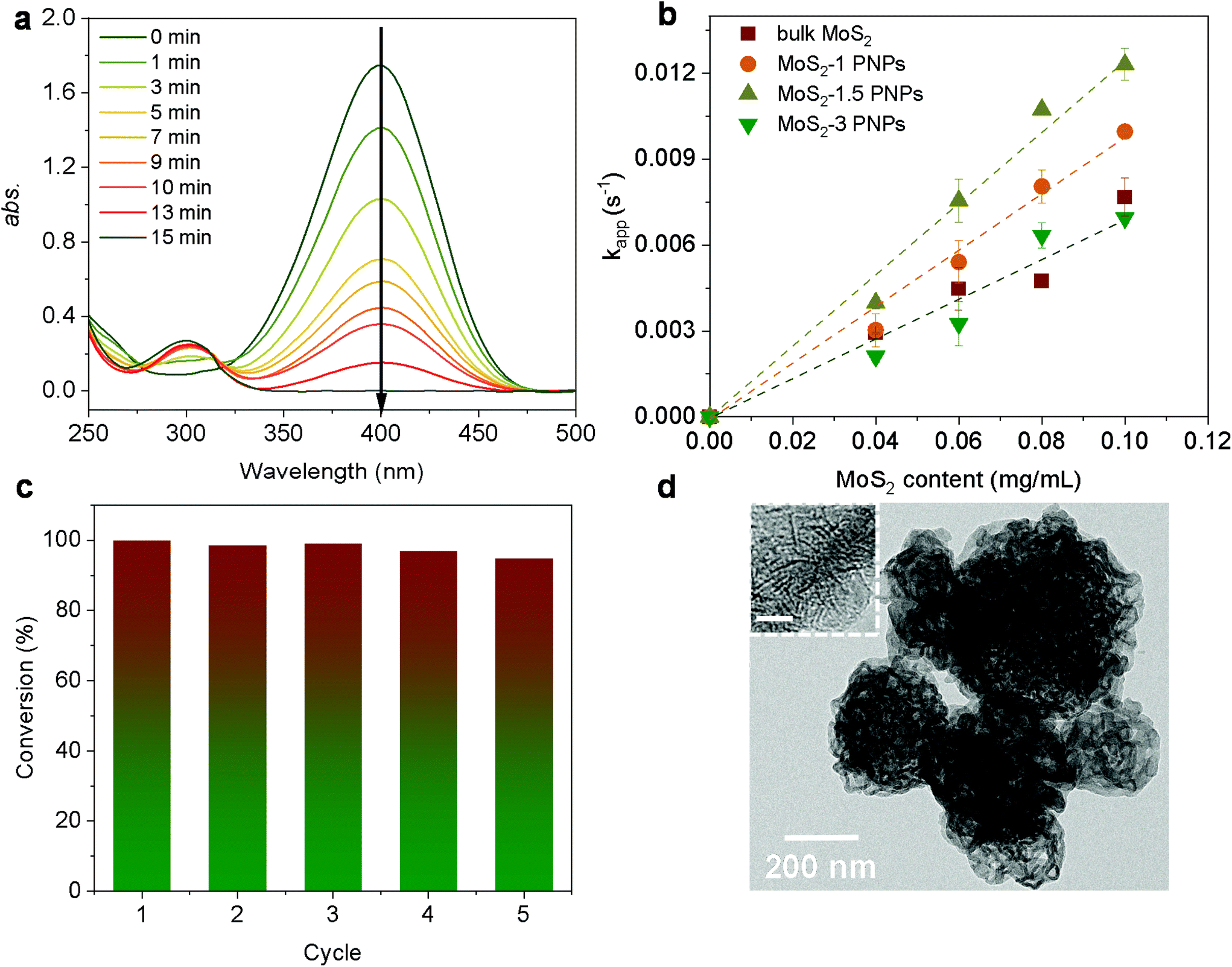 | ||
| Fig. 5 (a) UV-vis absorption spectra of the reduction of 4-Nip with the concentration of the MoS2-1.5 PNPs at 0.05 mg mL−1, 4-Nip at 0.1 mM, and NaBH4 at 20 mM at room temperature. (b) Rate constants, kapp, of different MoS2 PNPs as a function of catalytic concentration. (c) Catalytic recycling test of the MoS2-1.5 PNPs at 0.05 mg mL−1. (d) TEM image of the MoS2-1.5 PNPs after the recycling tests; the inset is the corresponding HRTEM image with a scale bar of 5 nm. | ||
Furthermore, the effect of the photoheating on the catalytic performance of the particles under light irradiation was explored. The MoS2-1.5 PNPs were utilized for the catalytic measurements with light due to the best catalytic performance in the darkness at room temperature. The MoS2-1.5 PNP dispersion was first continuously stirred and irradiated with NIR light for 15 minutes to ensure a stable temperature (here 37 °C; Fig. S11a†). The catalytic reduction of 4-Nip was then triggered after the introduction of NaBH4 under continuous light irradiation, and the reaction was simultaneously monitored using UV-vis spectroscopy (Fig. S11b†). It is worth mentioning that the absorption of 4-Nip showed no changes in the absence of NaBH4, indicating that the light solely cannot initiate the reaction (Fig. S11c and 11d†). As shown in Fig. 6a and S12,† light irradiation has led to a higher reduction rate of 0.012 s−1, which obviously surpassed the rate of the reaction performed in the darkness (0.003 s−1). The temperature of the reaction solution also increased from 20 to 37 °C by light irradiation. The enhanced reaction rate could therefore be ascribed to the high temperature induced by the photothermal conversion performance of the MoS2 particles. To elucidate the role of the photoheating effect, the reaction was performed in the darkness with external heating at 37 °C in a water bath, as observed in the irradiation experiment. As a result, the reaction rate increased from 0.003 to 0.008 s−1, suggesting the strong influence of the operating temperature. However, the reaction rate is still much lower than that of the photo-irradiated samples. It is also worth mentioning that photo-generated charges could possibly contribute to the reaction and enhance the reduction rate.63 If this is true, the activation energy (Ea) of the reaction should be reduced significantly.64 To ease this concern, we compared the Ea of the reaction in the presence/absence of light. The reactions were conducted at different operating temperatures with/without light irradiation and the corresponding reaction rates were further determined from the linear slope of ln(c/c0) vs. t, as shown in Fig. 6b and c. These reaction rates were determined in the form of a typical Arrhenius plot in Fig. 6d (see summary of kapp in Table S4†) to calculate the Ea from the linear slopes according to the following eqn (2):65
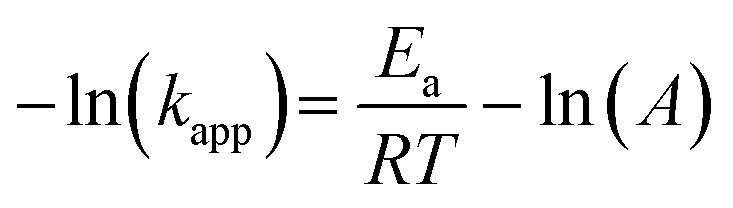 | (2) |
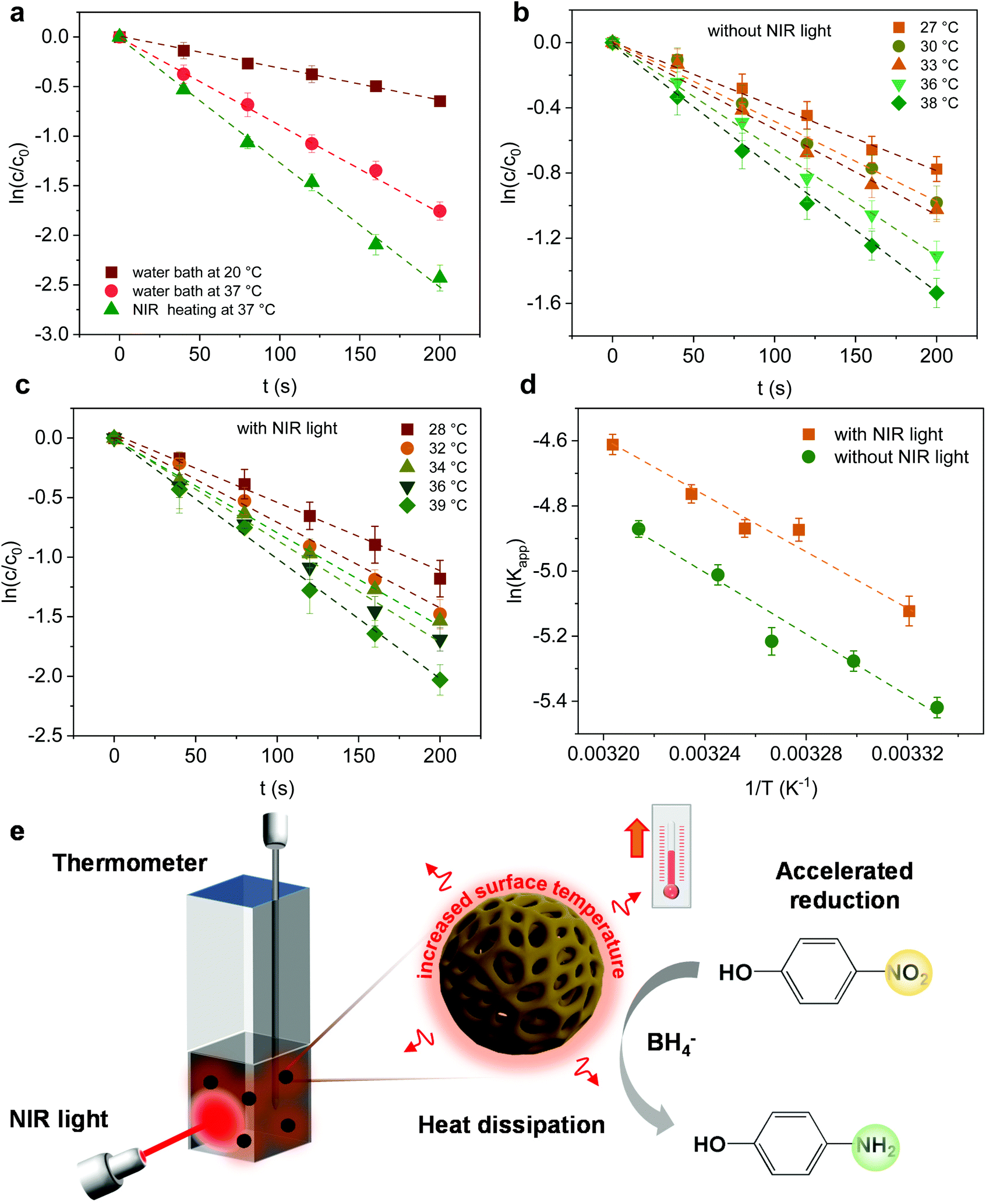 | ||
| Fig. 6 (a) Plots of c/c0versus reaction time with the concentration of MoS2-1.5 PNPs at 0.05 mg mL−1, 4-Nip at 1 mM, and NaBH4 at 0.2 M under different heating conditions. (b) Plots of I/I0versus reaction time in the darkness. (c) Plots of I/I0versus reaction time in the presence of light. (d) Reaction rates as a function of temperature in the presence (yellow line) and absence (green line) of 808 nm laser irradiation. The concentrations of MoS2-1.5 PNPs, 4-Nip and NaBH4 are 0.03 mg mL−1, 1 mM and 0.2 M, respectively. (e) Scheme of the reduction reaction of 4-Nip under NIR irradiation. | ||
 | ||
| Scheme 2 Schematic of 4-Nip reduction with porous MoS2 under NIR light illumination. | ||
Conclusions
In summary, we have designed a series of dual-functional MoS2 particles including pomegranate-like, hollow, and open porous particles using a soft PS-b-P2VP template. These nanoparticles with different well-defined internal structures show an efficient photoheating effect. The porous MoS2 nanoparticles displayed the highest photothermal conversion performance, benefitting from the localization of the light in their internal network-like structures. Moreover, this high photothermal conversion behavior has been demonstrated to accelerate the reduction reaction of 4-Nip under NIR light irradiation, confirming the synergistic properties of these MoS2 particles. Thus, the open porous MoS2 particles with interconnecting nanochannels were shown to serve as efficient nanocatalysts, while their abundant active sites, fast reactant diffusion pathway and local surface heating play a key role in enhancing the overall catalytic performance.Experimental section/methods
Assembled block copolymer nanospheres
For PS48.5k-b-P2VP16.5k, 40 mg of the polymer powder was dissolved in 4 mL of toluene and introduced to 80 mL of deionized water with 0.12 mL of the surfactant solution (SDS, 0.5 wt%). The mixture was left to emulsify under sonication for 30 min. The solution was then evaporated using a rotary evaporator at 40 °C in a water bath, resulting in solidified polymer nanospheres dispersed in water. To completely remove toluene, the solution was left at 40 °C overnight. The dispersion was centrifuged at 9000 rpm for 2 h, and the obtained particles were redispersed in 80 mL of ethanol. The particle dispersion was further heated at 50 and 75 °C for 1 h to induce the porous structure, respectively. After cooling down, the polymer dispersion was then evaporated to remove ethanol. For PS38.5k-b-P2VP23k, the particles were obtained using the same protocol, while the swelling of the polymer was only performed in ethanol at 50 °C.Nanostructured MoS2 particles
First, 150 mL of (NH4)2MoS4 solution was added to the as-prepared polymer dispersion (30 mg mL−1, 15 mL) with final concentrations of (NH4)2MoS4 maintained at 1, 1.5 and 3 mg mL−1, respectively. After stirring at 250 rpm for 30 min, HCl (0.16 mL, 1 M) was added dropwise into the solution, resulting in the decomposition of a slight amount of the precursor into molybdenum sulfide (MoSx, x = 2–3). To improve the hydrophilicity of MoS2 in the aqueous solution, 100 μL of 3-MPA was added into the solution under stirring. After stirring for 30 min, the brown solution was separated by centrifugation at 8000 rpm for 40 min and washed three times with fresh water. The obtained dark brown particles were freeze-dried overnight. The final product of MoS2 particles was obtained by calcination at 500 °C in an argon atmosphere for 2 h at a rate of 3 °C min−1. In comparison to MoS2 particles prepared with different polymer templates, bulk MoS2 was fabricated by following the same process without any polymer template.Photothermal conversion performance
The photothermal conversion performance of the MoS2 particles was measured using an aqueous dispersion of the particles with different concentrations (0.05–0.5 mg mL−1). First, 1 mL of the dispersion was placed in a quartz cuvette under 300 rpm of stirring and directly irradiated under 808 nm NIR laser (PhotonTec Berlin, turn-key 808 nm diode laser system) with different intensities (2.0–4.0 W cm−2) for approximately 10 min. To avoid direct heating by light, the thermometer was kept far away from the laser spot, while the distance between the laser spot and the thermometer was fixed at ca. 6 cm. The temperature changes of the dispersion were recorded using a digital thermometer (P 300 Thermometer, Dostmann electronics) every 20 seconds. The IR camera (FLIR-E8 XT) was employed to visualize the temperature changes of the dispersion every 3 min.Catalytic reduction of 4-nitrophenol
Materials characterization
X-ray diffraction analysis (XRD, Bruker D8) measurements were conducted using a monochromatic X-ray beam with CuKa radiation at a scan rate of 0.05° min−1. N2 adsorption/desorption isotherms were obtained using a Quantachrome Autosorb-1 system at 77 K. The specific surface area was calculated using the Brunauer-Emmett-Teller (BET) method based on a multipoint analysis. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were performed using a JEOL JEM-2100 instrument operating at an accelerating voltage of 200 kV. Scanning electron microscopy (SEM) measurement was carried out using a LEO Gemini 1530 microscope operating at 5 kV. Thermogravimetric analysis (TGA) was performed using a Mettler Toledo TGA 1 Stare thermal instrument in the temperature range of 25–900 °C at a heating rate of 10 °C min−1 in air. The Raman spectra were recorded using an InVia 2R9X81 Raman spectrometer equipped with a detector of Renishaw Centrus 2MYA08 and a HeNe laser (a wavelength of 532 nm).Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors acknowledge Mr. J. Yang at Department of Chemistry/Functional Materials, Technische Universität Berlin for the TGA measurement. The authors also thank Mr. Z. Zhang at Department of Chemistry, Humboldt-Universität zu Berlin for the Raman measurement. Y. Lu thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 410871749 for financial support.References
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 RSC.
- N. Meng, J. Ren, Y. Liu, Y. Huang, T. Petit and B. Zhang, Energy Environ. Sci., 2018, 11, 566–571 RSC.
- M. Gao, L. Zhu, C. K. Peh and G. W. Ho, Energy Environ. Sci., 2019, 12, 841–864 RSC.
- M. Tan, J. Wang, W. Song, J. Fang and X. Zhang, J. Mater. Chem. A, 2019, 7, 1244–1251 RSC.
- T. Zhang, F. Meng, M. Gao, W. L. Ong, K.-G. Haw, T. Ding, G. W. Ho and S. Kawi, EcoMat, 2021, 3, e12152 CAS.
- Y. Cao, M. Derakhshani, Y. Fang, G. Huang and C. Cao, Adv. Funct. Mater., 2021, 31, 2106231 CrossRef CAS.
- H. Liu, H.-G. Ye, M. Gao, Q. Li, Z. Liu, A.-Q. Xie, L. Zhu, G. W. Ho and S. Chen, Adv. Sci., 2021, 8, 2101232 CrossRef CAS PubMed.
- K. Xu, J. Wu, C. F. Tan, G. W. Ho, A. Wei and M. Hong, Nanoscale, 2017, 9, 11574–11583 RSC.
- S. Hu, B.-J. Liu, J.-M. Feng, C. Zong, K.-Q. Lin, X. Wang, D.-Y. Wu and B. Ren, J. Am. Chem. Soc., 2018, 140, 13680–13686 CrossRef CAS PubMed.
- D. Mateo, J. Albero and H. García, Energy Environ. Sci., 2017, 10, 2392–2400 RSC.
- Z. Zhang, X. Jiang, B. Liu, L. Guo, N. Lu, L. Wang, J. Huang, K. Liu and B. Dong, Adv. Mater., 2018, 30, 1705221 CrossRef PubMed.
- L. Jiang, X. Yuan, G. Zeng, J. Liang, X. Chen, H. Yu, H. Wang, Z. Wu, J. Zhang and T. Xiong, Appl. Catal., B, 2018, 227, 376–385 CrossRef CAS.
- Y. Yang, R. Zhao, T. Zhang, K. Zhao, P. Xiao, Y. Ma, P. M. Ajayan, G. Shi and Y. Chen, ACS Nano, 2018, 12, 829–835 CrossRef CAS PubMed.
- H. Yuan, F. Liu, G. Xue, H. Liu, Y. Wang, Y. Zhao, X. Liu, X. Zhang, L. Zhao, Z. Liu, H. Liu and W. Zhou, Appl. Catal., B, 2021, 283, 119647 CrossRef CAS.
- Y. Chai, H. Ma, X. Ma, X. Zhang, Y. He, Y. Wang, Q. Jiang, X. Wang, J. Ji and M. Xue, J. Mater. Chem. A, 2020, 8, 10891–10897 RSC.
- S. Mei, X. Xu, R. D. Priestley and Y. Lu, Chem. Sci., 2020, 11, 12269–12281 RSC.
- M. Zhao, T. Chen, B. He, X. Hu, J. Huang, P. Yi, Y. Wang, Y. Chen, Z. Li and X. Liu, J. Mater. Chem. A, 2020, 8, 15976–15983 RSC.
- S. Mei, Z. Kochovski, R. Roa, S. Gu, X. Xu, H. Yu, J. Dzubiella, M. Ballauff and Y. Lu, Nano-Micro Lett., 2019, 11, 83 CrossRef CAS PubMed.
- Y.-C. Chen, A.-Y. Lu, P. Lu, X. Yang, C.-M. Jiang, M. Mariano, B. Kaehr, O. Lin, A. Taylor, I. D. Sharp, L.-J. Li, S. S. Chou and V. Tung, Adv. Mater., 2017, 29, 1703863 CrossRef PubMed.
- J. Ke, J. Liu, H. Sun, H. Zhang, X. Duan, P. Liang, X. Li, M. O. Tade, S. Liu and S. Wang, Appl. Catal., B, 2017, 200, 47–55 CrossRef CAS.
- D. A. Reddy, H. Park, S. Hong, D. P. Kumar and T. K. Kim, J. Mater. Chem. A, 2017, 5, 6981–6991 RSC.
- Z. Zhou, B. Li, C. Shen, D. Wu, H. Fan, J. Zhao, H. Li, Z. Zeng, Z. Luo, L. Ma and C. Tan, Small, 2020, 16, 2004173 CrossRef CAS PubMed.
- M.-H. Wu, L. Li, N. Liu, D.-J. Wang, Y.-C. Xue and L. Tang, Process Saf. Environ. Prot., 2018, 118, 40–58 CrossRef CAS.
- X. Li, L. Kong, W. Hu, C. Zhang, A. Pich, X. Shi, X. Wang and L. Xing, J. Adv. Res., 2022, 37, 255–266 CrossRef CAS.
- D. Xu, Z. Li, L. Li and J. Wang, Adv. Funct. Mater., 2020, 30, 2000712 CrossRef CAS.
- J. Wang, Y. Li, L. Deng, N. Wei, Y. Weng, S. Dong, D. Qi, J. Qiu, X. Chen and T. Wu, Adv. Mater., 2017, 29, 1603730 CrossRef PubMed.
- X. Yang, Y. Yang, L. Fu, M. Zou, Z. Li, A. Cao and Q. Yuan, Adv. Funct. Mater., 2018, 28, 1704505 CrossRef.
- S. García-Dalí, J. I. Paredes, J. M. Munuera, S. Villar-Rodil, A. Adawy, A. Martínez-Alonso and J. M. D. Tascón, ACS Appl. Mater. Interfaces, 2019, 11, 36991–37003 CrossRef PubMed.
- X. Hai, W. Zhou, K. Chang, H. Pang, H. Liu, L. Shi, F. Ichihara and J. Ye, J. Mater. Chem. A, 2017, 5, 8591–8598 RSC.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- W. Bao, N. J. Borys, C. Ko, J. Suh, W. Fan, A. Thron, Y. Zhang, A. Buyanin, J. Zhang and S. Cabrini, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- M. Guo, Z. Xing, T. Zhao, Z. Li, S. Yang and W. Zhou, Appl. Catal., B, 2019, 257, 117913 CrossRef CAS.
- X. Yu, R. Du, B. Li, Y. Zhang, H. Liu, J. Qu and X. An, Appl. Catal., B, 2016, 182, 504–512 CrossRef CAS.
- T. Quan, N. Goubard-Bretesché, E. Härk, Z. Kochovski, S. Mei, N. Pinna, M. Ballauff and Y. Lu, Chem. – Eur. J., 2019, 25, 4757–4766 CrossRef CAS PubMed.
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H.-M. Cheng and L. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef PubMed.
- R. R. Poolakkandy and M. M. Menamparambath, Nanoscale Adv., 2020, 2, 5015–5045 RSC.
- Y. Cui, T. Zhu, A. Li, B. Liu, Z. Cui, Y. Qiao, Y. Tian and D. Qiu, ACS Appl. Mater. Interfaces, 2018, 10, 6956–6964 CrossRef CAS PubMed.
- S. Mei, L. Wang, X. Feng and Z. Jin, Langmuir, 2013, 29, 4640–4646 CrossRef CAS PubMed.
- S. Mei and Z. Jin, Small, 2013, 9, 322–329 CrossRef CAS PubMed.
- H. Fan and Z. Jin, Macromolecules, 2014, 47, 2674–2681 CrossRef CAS.
- W. Han, P. Hou, S. Sadaf, H. Schäfer, L. Walder and M. Steinhart, ACS Appl. Mater. Interfaces, 2018, 10, 7451–7458 CrossRef CAS PubMed.
- L. Guo, Y. Wang and M. Steinhart, Chem. Soc. Rev., 2021, 50, 6333–6348 RSC.
- N. Hampu, J. R. Werber, W. Y. Chan, E. C. Feinberg and M. A. Hillmyer, ACS Nano, 2020, 14, 16446–16471 CrossRef CAS PubMed.
- H. Yang, X. Shi, S. Chu, Z. Shao and Y. Wang, Adv. Sci., 2021, 8, 2003096 CrossRef CAS PubMed.
- D. Liu, Y. Liao, E. J. Cornel, M. Lv, T. Wu, X. Zhang, L. Fan, M. Sun, Y. Zhu, Z. Fan and J. Du, Chem. Mater., 2021, 33, 7972–7985 CrossRef CAS.
- E. Kang, B. Graczykowski, U. Jonas, D. Christie, L. A. G. Gray, D. Cangialosi, R. D. Priestley and G. Fytas, Macromolecules, 2019, 52, 5399–5406 CrossRef CAS PubMed.
- Y. Wang, U. Gösele and M. Steinhart, Chem. Mater., 2008, 20, 379–381 CrossRef CAS.
- J. Zhou and Y. Wang, Macromolecules, 2020, 53, 5–17 CrossRef CAS.
- Y. Liu, J. Wang, M. Zhang, H. Li and Z. Lin, ACS Nano, 2020, 14, 12491–12521 CrossRef PubMed.
- X. Li, J. Iocozzia, Y. Chen, S. Zhao, X. Cui, W. Wang, H. Yu, S. Lin and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 2046–2070 CrossRef CAS PubMed.
- S. Mei, J. Cao and Y. Lu, J. Mater. Chem. A, 2015, 3, 3382–3389 RSC.
- T. Quan, Y. Xu, M. Tovar, N. Goubard-Bretesché, Z. Li, Z. Kochovski, H. Kirmse, K. Skrodczky, S. Mei, H. Yu, D. Abou-Ras, M. Wagemaker and Y. Lu, Batter. Supercaps, 2020, 3, 747–756 CrossRef CAS.
- D. Yang and R. F. Frindt, J. Appl. Phys., 1996, 79, 2376–2385 CrossRef CAS.
- X. Zheng, J. Xu, K. Yan, H. Wang, Z. Wang and S. Yang, Chem. Mater., 2014, 26, 2344–2353 CrossRef CAS.
- S. H. Hwang, J. Yun and J. Jang, Adv. Funct. Mater., 2014, 24, 7619–7626 CrossRef CAS.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 3, 1139 CrossRef.
- S. Wunder, Y. Lu, M. Albrecht and M. Ballauff, ACS Catal., 2011, 1, 908–916 CrossRef CAS.
- P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- W. Feng, L. Chen, M. Qin, X. Zhou, Q. Zhang, Y. Miao, K. Qiu, Y. Zhang and C. He, Sci. Rep., 2015, 5, 17422 CrossRef CAS PubMed.
- N. R. S. Sibuyi, K. L. Moabelo, M. Meyer, M. O. Onani, A. Dube and A. M. Madiehe, J. Nanobiotechnol., 2019, 17, 122 CrossRef PubMed.
- R. D. Neal, R. A. Hughes, P. Sapkota, S. Ptasinska and S. Neretina, ACS Catal., 2020, 10, 10040–10050 CrossRef CAS.
- Q. Xiao, S. Sarina, A. Bo, J. Jia, H. Liu, D. P. Arnold, Y. Huang, H. Wu and H. Zhu, ACS Catal., 2014, 4, 1725–1734 CrossRef CAS.
- N. Li, Z. Liu, M. Liu, C. Xue, Q. Chang, H. Wang, Y. Li, Z. Song and S. Hu, Inorg. Chem., 2019, 58, 5746–5752 CrossRef CAS PubMed.
- Z. A. Piskulich, O. O. Mesele and W. H. Thompson, J. Phys. Chem. A, 2019, 123, 7185–7194 CrossRef CAS PubMed.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CrossRef CAS.
- J. Lü, Y. Fu, Y. Song, D. Wang and C. Lü, RSC Adv., 2016, 6, 14247–14252 RSC.
- L. Forni, Catal. Today, 1999, 52, 147–152 CrossRef CAS.
- Y. Zhou, Q. Ji, H. Liu and J. Qu, Environ. Sci. Technol., 2018, 52, 7477–7485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr01040b |
| This journal is © The Royal Society of Chemistry 2022 |