
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNarcissistic self-sorting of n-acene nano-ribbons yielding energy-transfer and electroluminescence at p–n junctions†
Philip
Schäfer
a,
Christiaan
de Vet
 a,
Leire
Gartzia-Rivero
ab,
Guillaume
Raffy
a,
Min-Tzu
Kao
a,
Christian
Schäfer
a,
Laura J.
Romasanta
c,
Bertrand
Pavageau
c,
Yu-Tang
Tsai
ad,
Lionel
Hirsch
d,
Dario M.
Bassani
a and
André
Del Guerzo
*a
a,
Leire
Gartzia-Rivero
ab,
Guillaume
Raffy
a,
Min-Tzu
Kao
a,
Christian
Schäfer
a,
Laura J.
Romasanta
c,
Bertrand
Pavageau
c,
Yu-Tang
Tsai
ad,
Lionel
Hirsch
d,
Dario M.
Bassani
a and
André
Del Guerzo
*a
aUniv. Bordeaux, CNRS, Bordeaux INP, Institut des Sciences Moléculaires UMR 5255, 351 Cours de la Libération, F-33400 Talence, France. E-mail: andre.del-guerzo@u-bordeaux.fr
bDepartment of Physical Chemistry, University of the Basque Country (UPV/EHU), Apartado 644, 48080 Bilbao, Spain
cCNRS, Solvay, Univ. Bordeaux, LOF, UMR 5258, 178 Avenue du Dr Albert Schweitzer, F-33600 Pessac, France
dUniv. Bordeaux, CNRS, Bordeaux INP, ENSCPB, IMS, CNRS UMR 5218, F-33600 Pessac, France
First published on 9th May 2022
Abstract
The 2,3-didecyloxy derivative of an n-type anthracene (n-BG) and a p-type tetracene (p-R) have been synthesized and their self-assembly into nano-ribbons studied. Hyperspectral fluorescence imaging revealed their narcissistic self-sorting, leading to separated nanoribbons emitting with very different colors (blue or green for n-BG, depending on the growth solvent, and red for p-R). It is unique that the usual origins of self-sorting, such as specific H-bonding, different growth kinetics, or incompatible steric hindrance can be ruled out. Hence, the narcissistic behaviour is herein proposed to originate from a so-far unconsidered cause: the discrepancy between the quadrupolar character of n-BG and dipolar character of p-R. At the p–n junctions of these nanoribbons, inter-ribbon FRET and electro-luminescence switch-on were observed by fluorescence/luminescence microscopy.
Introduction
Orthogonally self-assembled systems,1 sometimes self-sorted, are essential to generate complexity and function in biological systems, and have therefore been transposed to biomimetics,2–5 tissue-engineering,6–8 and optoelectronics.9–14 van Esch defined orthogonal self-assembly as the independent formation of two different supramolecular structures, each with their own characteristics that coexist within a single system;15 Whitesides initially described it as an individual but simultaneous self-assembly.16 Orthogonal self-assembly is very important to preserve integrity of multiple components and achieve synergistic property enhancement or introduction of new behaviour, whether macroscopically or at interfaces. Self-sorting is certainly one of the most appealing mechanisms to achieve this, as it also infers ‘one-pot’ processing. It can occur as a narcissistic self-sorting, in which identical molecules assemble separately from other components present, or a social self-sorting, which involves specific blending of components.17 The use of orthogonal binding motives has been considered of prime importance to achieve narcissistic self-sorting.18 In many cases, multiple H-bonding sites are introduced and tuned to differentiate the binding patterns19,20 of the different components, or very different constituents such as lipids and peptides are used.21–23 Some of these ‘molecular codes’, as classified by Würthner,24 can be readily distinguished in the molecular structures. However, in the less evident cases, for example a rare case of narcissistic self-sorting without H-bonds of perylene derivatives, only the contribution of steric hindrance could be proposed as origin of the sorting.25The focus on self-sorted heterojunctions is also quite rare, whether localized or co-axial,26 and to the best of our knowledge always involve H-bonding systems. Shinkai and coworkers demonstrated the formation of a dual network of p- and n-type semiconductor-based nanofibers due to differentiated growth kinetics, leading to small amounts of hetero-junctions and resulting photo-current in a device.27 From the few comparable studies, some common features appear: (i) the dual network formation is based on differentiated assembly kinetics28 (due to different thermodynamic stabilities or pKa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29); (ii) H-bonds are of prime importance in the self-recognition; (iii) the functional polyaromatic unit is surrounded by medium-to-large bulky groups (cholesteric units,27 amido-trioctyloxyphenyl units,19 peptides30). The latter substituents contribute to reduce the density of functional polyaromatic core within the material.
29); (ii) H-bonds are of prime importance in the self-recognition; (iii) the functional polyaromatic unit is surrounded by medium-to-large bulky groups (cholesteric units,27 amido-trioctyloxyphenyl units,19 peptides30). The latter substituents contribute to reduce the density of functional polyaromatic core within the material.
In this work, compact polyaromatic, H-bond-free fluorescent n-acene derivatives are used to form narcissistic self-sorted p- and n-type self-assemblies with generation of electroluminescence at interfaces. Such a system would be expected to be very sensitive to incomplete self-sorting, and necessitates favourable interfacial interactions. On a fundamental level, this work contributes unravelling mechanisms that lead to a remarkable narcissistic self-sorting of relatively similar polyaromatic molecules having the same side chains, without forming H-bonds. In addition, in this work hyper-spectral confocal fluorescence microscopy (HS-CFM), rarely used in similar contexts,31 is revealed as an efficient tool to image multi-component materials and interfaces therein, whereas these are commonly considered difficult to characterize.32
Results and discussion
Self-sorting
Previous work on n-acenes has shown that 2,3-dialkoxy substitution promotes formation of anisotropic self-assemblies such as nanofibers of 2,3-didecyloxyanthracene (DDOA)33 or nanoribbons of 9,10-diphenyl-2,3-dihexadecyloxyanthracene34 in alcohols and DMSO. If compared, ribbons are wider than fibres with a higher width/height aspect ratio, which in contrast tend to form twisted bundles, branching and gels. Ribbons are nevertheless thinner than typical crystals. The alkoxy chains have been shown to alter the solubility, and also promote a sheet-like packing of the molecules alternating aromatic cores and alkyl chains in nanoribbons.34 Therefore, 9,10-dicyano-2,3-didecyloxyanthracene (herein called n-BG ‘n-type-blue/green’) was prepared as an electro-deficient n-type self-assembling derivative, whereas 2,3-didecyloxy-6,11-diphenyl-5,12-diphenylethynyltetracene (p-R ‘p-type-red’) can be expected to act as a p-type derivative owing to the tetracene core (Scheme 1 and Fig. 1a). The synthesis of n-BG is based on the 9,10-bromination of previously described DDOA35 and subsequent substitution with cyano groups. p-R is obtained from 2,3-didecyloxy-6,11-diphenyltetracene-5,12-dione similar to previously described procedures36,37 (Scheme 1 and ESI†). | ||
| Scheme 1 Final steps of the syntheses of n-BG and p-R. | ||
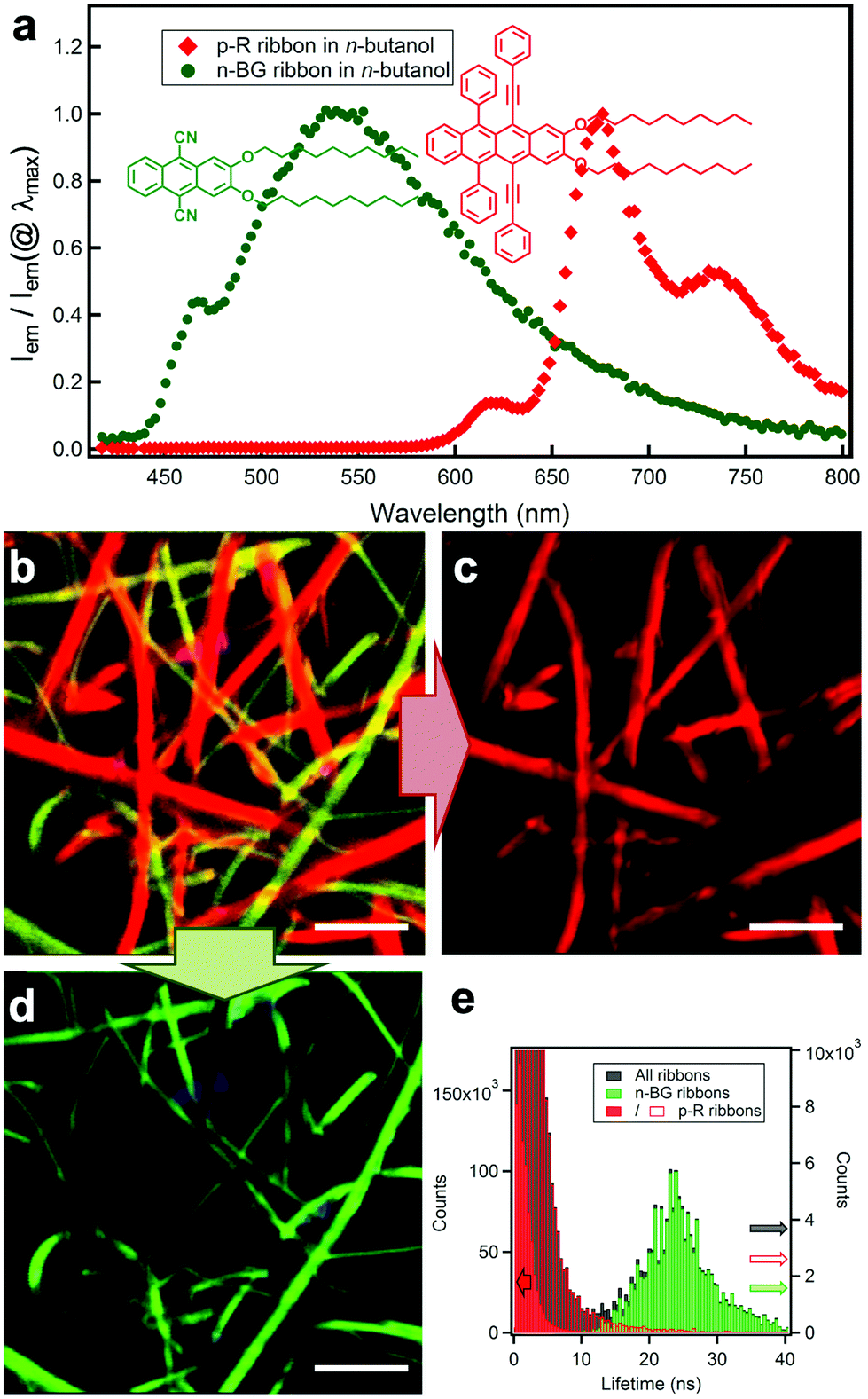 | ||
| Fig. 1 (a) Molecular structures of n-BG (green) and p-R (red) and relative emission intensity spectra Iem/Iem(@λmax) of the two different nanoribbons in n-butanol. (b) Hyperspectral image: the emission spectrum of each pixel is converted into the corresponding RGB-color, λex = 375 nm. The spectra in (a) are obtained from individual ribbons from this map. (c) Digital bandpass filter (650 nm–780 nm) applied on (b) highlighting the ribbons attributed to p-R. (d) Digital bandpass filter (405 nm–550 nm) applied on (b) showing the ribbons attributed to n-BG (scale bar: 5 μm). Note: some blue emissive spots can be attributed to photo-oxidized p-R (see ESI†). (e) Fluorescence lifetime distribution of the same area (black histogram): the longer lifetimes are correlated to the green-emissive (green histogram), and the shorter to the red-emissive (red histogram) ribbons. Red histogram is displayed in two different scales (full, left scale; empty: right scale). | ||
Spectroscopic studies in solution show that the anthracene n-BG emits blue light from a π–π* transition in apolar cyclohexane solvent (Table 1; Tables S1, S2 and Fig. S3 in ESI†). In the polar solvent acetone the emission becomes unstructured and shifted to cyan, and is attributed to an intramolecular charge-transfer ICT state with an excited state dipole of 14 D, as determined by a Lippert–Mataga analysis (Fig. S1†).38 The tetracene p-R emits very efficiently in the red spectral region, showing weak solvatochromism (Tables 1, S1 and Fig. S2†). As compared to the tetraphenyl (rubrene) analogue,36 the extension of the conjugation through the aryls results in red-shifted absorption and emission by ∼50 nm, with a 2.5× larger extinction coefficient and a 1.7× larger radiative constant kr, both related to the oscillator strength (kr calculated using the Strickler-Berg relation39 matches the experimental value, also implying a full solubilization of p-R at 10−5 M in toluene; Table S1†).
| λ em/nm | Φ em | τ/ns | k r/107 s−1 | k nr/107 s−1 | |
|---|---|---|---|---|---|
| a Cyclohexane. b Acetone (kr calculated for longest τ, a second lifetime of 2.5 ns contributes for 8% of the emission). c λ em and τ in mixed ribbons in n-butanol, Φem of n-BG NR in DCM/butanol (λem = 480 nm), kr and knr estimated by combining all data. d Toluene. e Could not be determined with accuracy (see Experimental). | |||||
| n-BG /sol | 435 | 0.32 | 6.0 | 5.3 | 11.4 |
| n-BG /sol | 462 | 0.26 | 6.2 | 3.8b | 12.3b |
| n-BG /NR | 545 | ∼0.26 | 23.6 | ∼1 | ∼3 |
| p-R /sol | 6030–0/6500–1 | 0.73 | 8.3 | 8.8 | 3.2 |
| p-R /NR | 6200–0/6690–1 | 0.9 | |||
n-BG and p-R are dissolved at high temperature (T > 80 °C) in n-butanol, DMSO or a mixture thereof (both 2.0 mM in 1 mL) and cooled down to room temperature (22–23 °C) to induce self-assembly. Microscopy (HS-CFM) provides the fluorescence spectrum (spectral resolution Δλ < 3 nm) for each pixel and was therefore used to characterize the orthogonality of the self-assembly with optical diffraction limited spatial resolution. The typically 40 × 40 μm images allow to easily visualize the distribution of the molecules since for each pixel the spectra are converted into RGB colour code. Fig. 1b–e clearly show that two sets of nanoribbons form in n-butanol, emitting with a narrow distribution of distinguishable colours (see CIE graph in Fig. S4†). The green- and red-emitting ribbons are attributed to n-BG and p-R, respectively. A spectral filtering highlights each set of objects (Fig. 1c and d), showing that they are constituted of continuous nanoribbons of similar size and aspect ratio, 200–1500 nm wide (bundles can form). The spectral shifts, as compared to solution, are typically observed for self-assembly of n-acenes and are discussed below. In addition, the two ribbon-sets emit with very different fluorescence decay times (distributions at 23.6 ± 6.2 ns and 0.9 ± 0.7 ns for n-BG and p-R, respectively) as shown by fluorescence lifetime imaging microscopy (FLIM, Fig. 1e and S5†), a technique also previously used to differentiate orthogonal self-assembly.5 HS-CFM further reveals that the emission of the n-BG ribbons does not contain measurable contribution of structured red emission (Fig. 1a). This demonstrates that the n-BG ribbons are not contaminated by p-R. Indeed, previous studies on similar nano-ribbons or -fibres have shown that even small amounts of dopants (∼0.5%mol equivalents), in this case p-R, would act as efficient acceptors in an energy transfer process (Fig. S4b†) and emit significantly.36,40 Approximatively similar amounts of green and red ribbons are typically observed, the red ribbons being attributed to p-R. These do not present any measurable n-BG emission under UV excitation, and comparison with previous studies also suggests that if present in large amounts, residual emission of unquenched n-BG should be observed. Whereas the presence of small amounts of n-BG, quenched by energy transfer, in p-R ribbons cannot be completely excluded, it crucially shows no impact on fluorescence properties. This constitutes thereby a significant case of quantitative narcissistic self-sorting of n-BG and p-R, a case without H-bonds, only sporadically described in the literature.
To obtain further insight on the dynamics of the orthogonal self-assembly, each type of nanoribbon's growth was discriminated in two spectral ranges and followed by simultaneous imaging using fluorescence video-microscopy. The final stage is shown in Fig. 2b. In pure n-butanol, the ribbons grow and ripen within minutes, the red emissive p-R ribbons (λdetection > 720 nm) being ripe slightly before the n-BG ribbons (λdetection: 510–550 nm). Changing to dimethylsulfoxide (DMSO), a non-protic solvent with similar polarity but larger permanent dipole (ET(BM)DMSO = 50.3 vs. ET(BM)BuOH = 53.6, μDMSO = 3.96 vs. μBuOH = 1.66),41,42 the time necessary to obtain ripe red ribbons increases to several tens of minutes and the ripening order is inverted. Mixing the two solvents induces intermediate growth speeds, and thereby ripe n-BG and p-R ribbons are obtained after a similar time in a 3:1 n-butanol:DMSO blend (the onset of nucleation could not be observed due to a lack of contrast of brightness). As seen in Fig. 2b, in all cases, both sets of ribbons grow orthogonally, suggesting that the self-sorting mechanism does not rely on differences of growth kinetics.
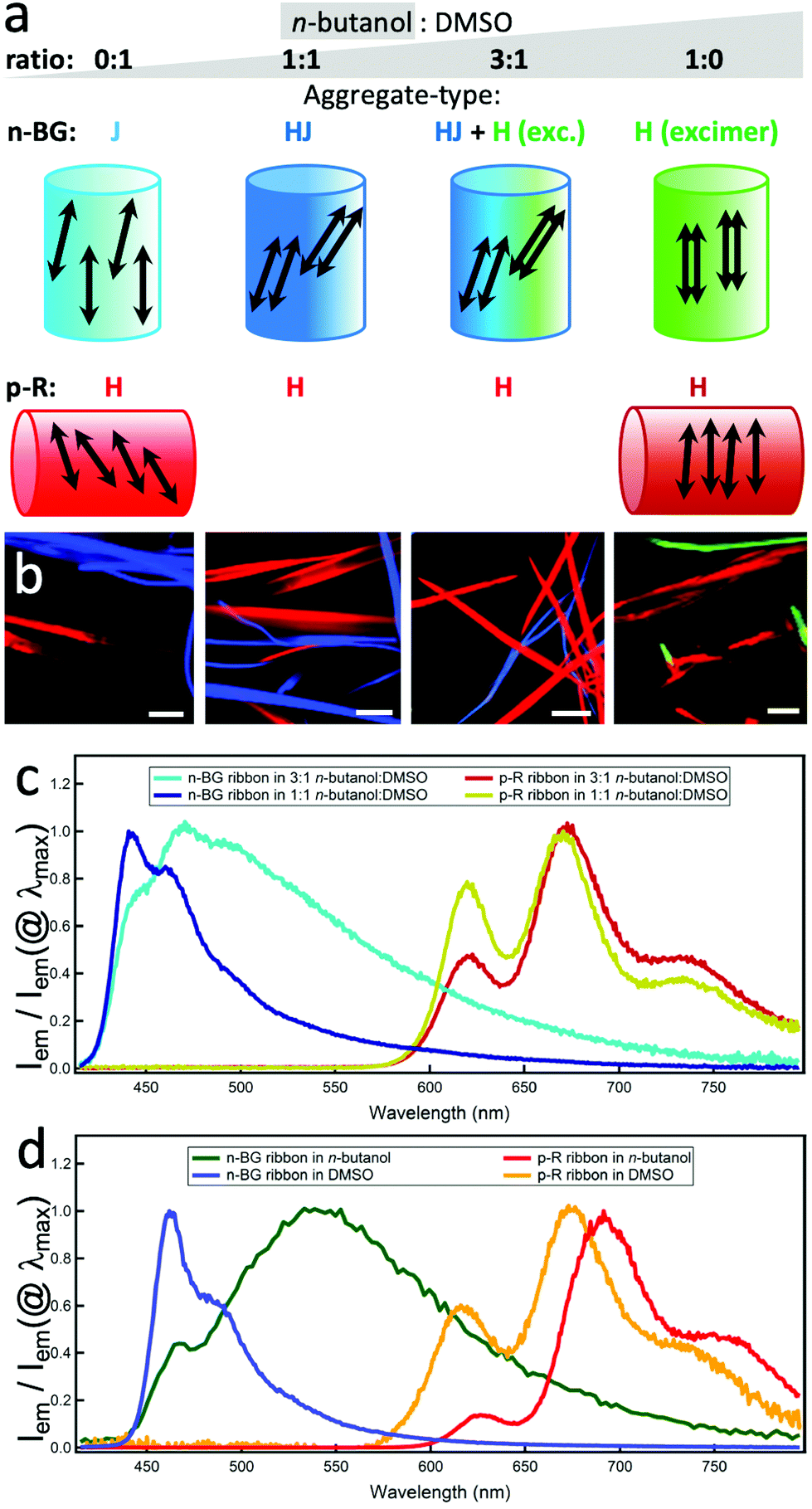 | ||
| Fig. 2 (a) Solvent mixtures for (a) and (b). Attributions of the main emission features to the aggregate types and simplified representation of possible packing within a ribbon of four molecules (each represented by the emission dipole oriented along the short axis of the n-acene, additional axial symmetry is probable). (b) Hyperspectral images of self-sorted ribbons in the four solvent conditions. Red-emissive ribbons are attributed to p-R, the other to n-BG (scale bars: 5 μm). (c) and (d) Relative emission intensity spectra of each ribbon type in the four solvents conditions; spectral data taken from the ribbons of the hyperspectral images (b). λex = 375 nm. | ||
The molecular packing determines the emission colours and affects the charge mobility in the solid state, and can be deduced from the spectral properties.43 The emission can originate from different states, and the lower energy state can act as an energy trap populated by Förster type resonance energy transfer (FRET).34 In n-BG ribbons, significant solvent-dependent spectral changes (Fig. 2c and d) are attributed to variations in packing geometries.37 In pure n-butanol, the emission is mostly constituted of a long-lived broad excimer-like emission at 545 nm (Fig. 2d), attributed to co-planar stacked n-BG molecules (same geometry as a “H-type” aggregate, Fig. 2a).38,44 This excimer emits with a typical reduced radiative constant45 and lifetimes much longer than those observed for the ICT state emission in solution (Tables 1 and S2†), although possibly displaying a CT-character.46 The non-radiative processes are reduced due to restricted conformational relaxation. With progressive increase of DMSO proportion, the excimer band decreases (3:1 mix) and disappears (1:1 mix) in favour of a structured emission at 442 nm, weakly shifted compared to that observed in an apolar solvent. This contrasts with the more pronounced red-shifted structured emission in pure DMSO (λ0–0max = 462 nm), typically attributed to J-type dipolar coupling of slip-stacked n-BG molecules.47 Hence, the 442 nm emission could be attributed to HJ-aggregates (or “null” aggregates),48 in which the J-type coulombic long distance coupling is compensated by CT-mediated short-distance H-type coupling.48 In summary, the molecular packing evolves from “J” to “HJ”, “H” + “HJ”, and “H”, changing the solvent mixture from DMSO to n-butanol (Fig. 2a). The excimer emission polarization is remarkably high along the ribbons’ long axis (Pmax > 0.7, Fig. S5b and f†), and the same orientation is deduced for the short axis of the anthracenes in n-butanol ribbons (Fig. 2a). Indeed, considering the large Pmax value and rare previous studies,49–52 it can be proposed that the main polarization component is oriented parallel to the excimer pair's short axis, with minor components along the two other orthogonal axes. The J-coupled molecules in DMSO also emit with a polarization oriented along the short axis. However, the lower Pmax (0.15 < Pmax < 0.5, Fig. S5c and f†) suggests the presence of a P component along the long axis of the ribbon, resulting from the predominant J-coupling of dipoles that are non-parallel and arranged along the long axis.43
The spectral changes observed for p-R emission are also indicative of coulombic dipolar coupling, favoured by a larger oscillator strength over CT-mediated coupling. A blue-shift and a lower energy tail appear in the absorption (Fig. S2a,† the latter should not lead to strong emission-reabsorption). The red-shift of the emission in the ribbons in DMSO, even more pronounced in n-butanol, can be due to stabilization of the excited state in a polar environment created by the large dipoles of distorted conformers (see below and previous studies34). Although a red-shift is often attributed to J-aggregates, which usually present a dominant 0–0 emission band, the absorption features and the significantly lower intensity of the 0–0 band emission (600–650 nm) relative to the 0–1 band (at 650–700 nm) suggest however H-type aggregates which emit due to local disorder-induced symmetry-breaking.53 In n-butanol, the weakness of the 0–0 band is also in agreement with such H-aggregates with a larger coupling correlation length.54 The main polarization component of the 0–1 band emission is essentially oriented perpendicular to the long axis of the ribbons (Fig. 2a, S5 and S6†), indicating the same orientation for the tetracene short axis. The 0–0 band, although only weak in n-butanol, shows a different orientation, in agreement with the expectation to be perpendicular to the 0–1 case and to reflect the direction of the chain of coupling dipoles (out-of-plane contributions could be at the origin of attenuated polarization values). This confirms a H-type molecular packing, which is likely to promote the charge mobility necessary for electro-luminescence (see below).
In addition to aromatic core interactions and phase separation of aromatics and alkyl chains at the molecular scale,34 directional self-assembly in ribbons in the absence of H-bonds can be dominated by dipole–dipole interactions.55 Recent work on 9,10-disubstituted anthracene nanoribbons unravelled the contribution in the anisotropic growth mechanism of dipole–dipole interactions originating from thermally-activated asymmetric conformers.34 Therefore, by analogy, the same type of conformers of p-R and n-BG were studied by theoretical calculations using DFT (density functional theory) (Fig. 3 and S8†). Although the most stable conformer is symmetric with a moderate dipole (1.22 D and 1.93 D for p-R and n-BG, respectively, Table S3†), in a higher energy meta-stable conformer with an asymmetric conformation of the alkyl-chains, a larger dipole moment can be formed mostly located on the aryl(–O–CH2–)2 area of the molecule (Fig. 3). For p-R, the dipole μp-R can reach 3.46 Debye. n-BG displays however a more complex charge distribution. Indeed, not only can the asymmetry-induced dipole moment reach μn-BG = 4.11 Debye, but additionally, the electron-withdrawing cyano groups contribute with strong dipoles (μCN ∼ 3.5 Debye)56 and lead to an approximatively quadrupolar character of the molecule. The observed polymorphism of the ribbons could be related to the solvent-dependent stabilization of the asymmetric conformers, in solution (∼1.5–2.5% of population, Table S3†), but also in the nuclei and in the final ribbons (population up to ∼100%).34 This could influence self-assembly kinetics, the final packing geometry, but also the increased polarity of the micro-environment in the ribbons34 and related stabilization of CT-character and red-shift of emission. A finer understanding of this interplay would certainly prove of interest in the field of organic opto-electronics with an ever-growing interest in excited states displaying a CT-character.
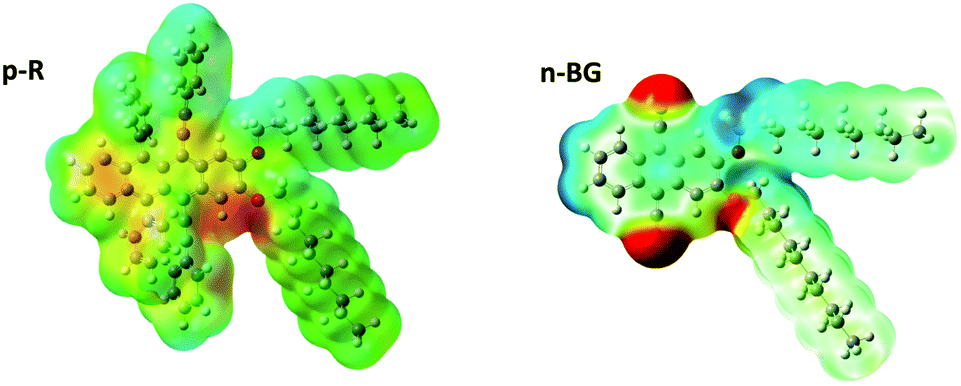 | ||
| Fig. 3 Calculated charge density of p-R and n-BG in n-butanol solution of meta-stable asymmetric conformers, obtained by energy minimization upon rotation of the C(ar)–O–C–C(alk) torsional angle (9 steps of 20°). Both molecules display a large dipolar moment of 3.46 and 4.11 Debye, respectively. | ||
Concerning the origin of the self-sorting, one of the most remarkable differences between n-BG and p-R resides thus in the fact that n-BG has a strong quadrupolar character. Indeed, thermodynamic parameters determining nucleation and growth kinetics have been levelled out in the solvent mixture 3:1. In addition, no specific binding functions are present, such as H-bond donor or acceptor, or apolar/polar side-chains.57 Another explanation could be related to the size of the molecules or of crystallographic unit cells. As previously shown, a blending of two components can be favoured by the match of their respective crystallographic unit cells.58 A mismatch could inhibit the blending of nanoaggregates of n-BG and p-R during growth, possibly involving the oriented-attachment mechanism proposed recently for similar nanoribbons.34 At the molecular level, steric hindrance could also lead to self-sorting, but it was shown that DDOA, a molecule with a size comparable to n-BG, and the bulkier p-R blend into mixed nanoobjects (Fig. S7†). Similarly, p-R does not self-sort with a 9,10-diphenylethnyl-anthracene37 analogue (Fig. S7†), and forms mixed nanoribbons. Therefore, this work suggests that the mismatch of dipolar and quadrupolar character of n-BG and p-R, respectively, plays an essential role in the exclusive narcissistic self-sorting.
p–n junctions
The interfacial interactions between self-sorted ribbons were emphasized by two experiments: inter-ribbon energy transfer and p–n junction electroluminescence. In the first case, the p–n junctions were highlighted by HS-CFM (Fig. 4a) and analysed by FLIM (Fig. 4b and c). The spectral overlap of n-BG emission and p-R absorption (Fig. S4†) is ideal for excitation energy transfer (ET) by either resonant (Förster, FRET) or electron exchange (Dexter) mechanism, or trivial emission-reabsorption. Emission intensity studies do not clearly reveal the ET, mostly because the emission of the acceptor p-R resulting from direct excitation cannot be avoided and overwhelms the variations in intensities. However, FLIM performed in the green spectral region (470–550 nm) reveals that the average decay-time of n-BG emission drops at a junction from 5.24 ns to 3.38 ns, corresponding to 35% of quenching (Fig. 4b and c). Decay times are intensity-independent, and change only if ET occurs (not for emission-reabsorption). This significant quenching of n-BG can be attributed to ET, most probably by FRET mechanism which can occur between singlet states at contact and at nanometric distances. Since this ribbon-to-ribbon ET occurs efficiently even though the ribbons are at least tens of nm thick, it can be proposed that an efficient exciton hopping, i.e. homo-ET, occurs within the ribbons. To the best of our knowledge this constitutes a rare case of efficient optical communication at junctions of nanofibers or ribbons of this size through ET mechanisms, and could be interesting for the development of photonic elements.59–61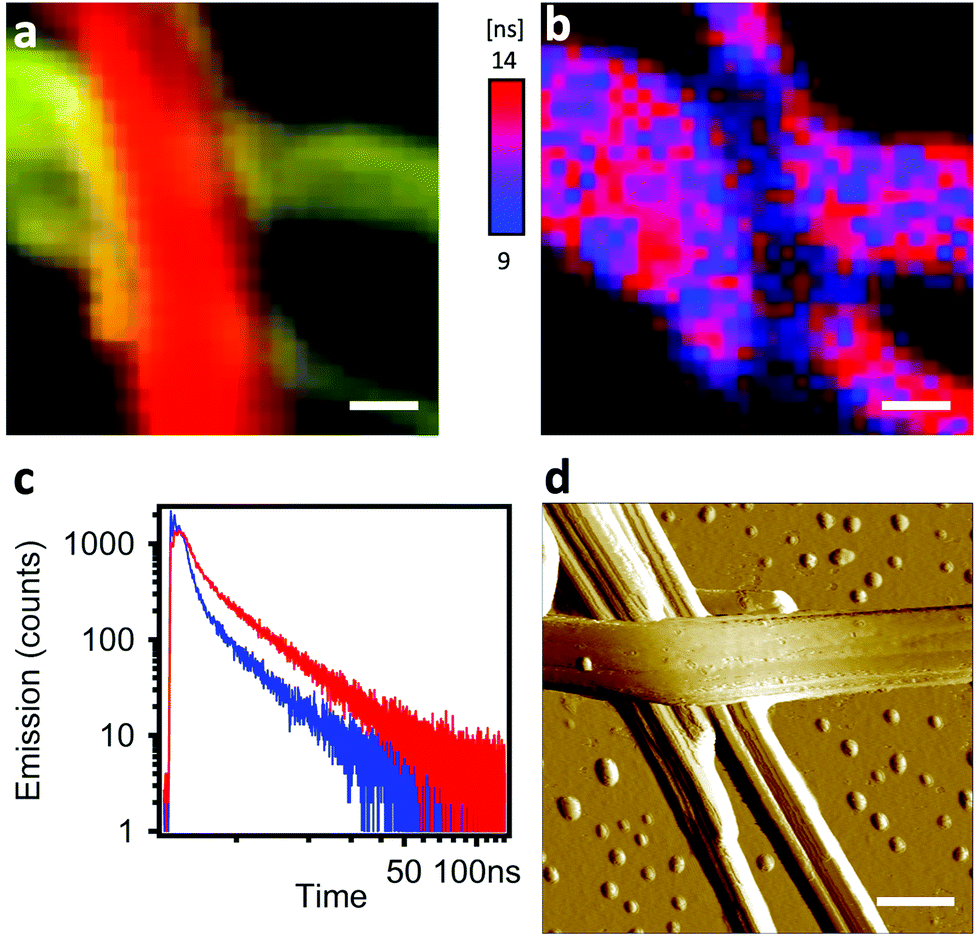 | ||
| Fig. 4 (a) Hyperspectral image of a n-BG/p-R ribbon p–n-junction grown in n-butanol (scale bar: 500 nm). (b) FLIM (average fluorescence lifetime) image using a 510/80 bandpass filter to retain n-BG emission only (scale bar: 500 nm). (c) Decays of n-BG emission of an isolated ribbon (red curve) and of the junction in (b) (blue curve). Red decay tri-exponential fit according to ΣAiexp(−t/τi): τ1 = 0.6 ns (A1 = 21%), τ2 = 3.4 ns (A2 = 60%), τ3 = 15.9 ns (A3 = 19%); blue decay: τ1 = 0.3 ns (A1 = 16%), τ2 = 1.9 ns (A2 = 63%), τ3 = 10.2 ns (A3 = 21%). λex = 375 nm. (d) AFM amplitude image of a p–n junction grown in n-butanol, deposited and dried on a glass cover slip. The “horizontal” ribbon is n-BG, and the “vertical” ribbon is p-R (scale bar: 500 nm). | ||
A remarkable ‘light ON’-switching occurs at the p–n junctions in an electroluminescent device studied by microscopy (Fig. 5a and b). Self-sorted n-BG and p-R ribbons grown in n-butanol are spin-coated on an ethylene glycol treated PEDOT:PSS thin-film, acting as hole-transport and hole-injection layer, and dried. The ITO-free, non-optimized electroluminescent device was completed with the electron-injection and electron-transport layer TBPi (2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole)), and an Li/Al cathode, both evaporated on top of the ribbons according to published procedures.62 Fluorescence microscopy correlated with HS-CFM demonstrated that electro-luminescence (EL) was generated at some p–n junctions (Fig. 5a and S10†), whereas isolated ribbons do not emit. EL demonstrates charge transport in both ribbons, n-BG acting as electron transporting n-type and p-R as hole transporting p-type semiconductors, respectively.
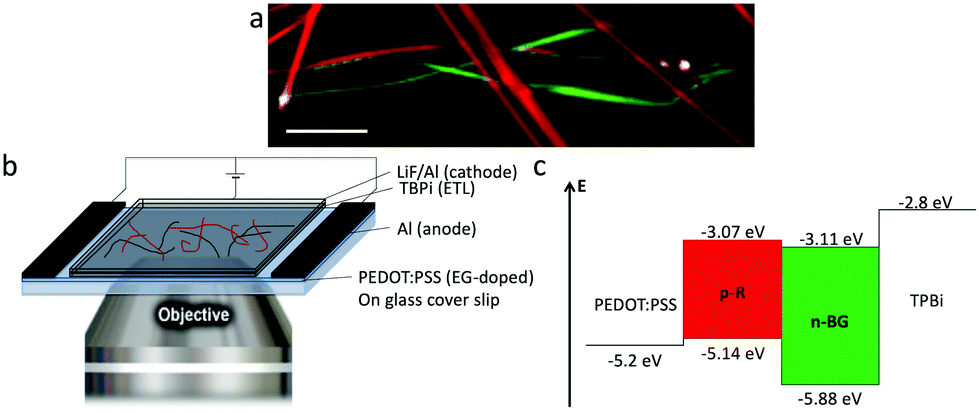 | ||
| Fig. 5 (a) Overlaid images of emission of self-sorted ribbons in a device, (red and green) fluorescence hyperspectral and (white) electroluminescence intensity at certain p–n junctions (λem > 405 nm, see also ESI,† scale bar: 10 μm). (b) Scheme of the device, placed on a microscope objective. (c) Energy level diagram of the EL-device. | ||
The absence of EL from certain junctions could be due to an insufficient charge transport resulting from an inappropriate order of the successive layers (inversion of n-BG and p-R relative to those observed by AFM in Fig. 4d, and the energy levels depicted in Fig. 5c), or an excessive thickness of certain ribbons. The anisotropic shape of the emissive spots and their orientation (Fig. S10†) could be related to some exciton hopping or waveguiding along the p-R ribbons, attributing thus the EL to p-R, although insufficient photons are emitted per spot to obtain a spectrum. Cyclic voltammetry of n-BG and p-R in solution show that hole injection from PEDOT:PSS could be possible into p-R, but not n-BG (Fig. 5c and S9†). In contrast, electron injection from TBPi could occur into both n-BG and p-R. The rarely observed and very weak EL from some isolated p-R ribbons (Fig. S11†) suggests that electron injection is inefficient. Therefore, the combination of n-BG and p-R in a p–n junction is required herein to transport both holes and electrons, with a recombination that can only occur in p-R due to an energy gap between the HOMO levels of p-R and n-BG that cannot be overcome. EL also demonstrates that self-sorting allows interfacial interactions that are sufficiently intimate for electron injection to occur from n-BG ribbons into p-R ribbons and charge recombination therein. p–n junctions are not a requisite for EL of organic nanoobjects,63 but this allows to have an EL switch-on at specific sites in an interpenetrated network. In perspective, p–n junctions in hierarchically organized and patterned nanoribbon electroluminescent devices could be of interest. The methodology developed for the photo-induced hierarchical self-assembly and patterning recently demonstrated for anthracene nanofibers33 could be useful. In addition, EL of p-R could be expected to be significantly linearly polarized (photo-luminescence P value can reach ∼0.5), an important feature for EL-devices such as OLEDs.
Conclusions
In conclusion, narcissistic self-sorting can be obtained for two polyaromatic molecules di-substituted identically in 2,3-positions with decyloxy side-chains. Herein, self-sorting does not rely on the commonly accepted strategies such as H-bonds, differentiated side-chains, self-assembly kinetics, or steric hindrance. In particular, the discrepancy of quadrupolar and dipolar characters of n-BG and p-R, respectively, is proposed to contribute significantly to the self-sorting by inhibiting blending at the molecular level. In addition, the molecular packing affects strongly the emissive properties, within a wider colour range in n-BG ribbons, and favours charge transport, thus EL. Despite efficient self-sorting, the interfaces at p–n junctions are sufficiently intimate to lead to inter-ribbon FRET and electroluminescence switch-on. In perspective, this type of molecular nanoribbons show an interesting combination of properties: narcissistic self-sorting, polarized emission, optical communication and electroluminescence.Experimental
All methods are described in more details in the ESI.†The mixed nanoribbons were prepared by adding 2 mM of both n-BG and p-R in 1 ml of n-butanol or DMSO, or mixture thereof, at r.t., then heating at least at 80 °C in a water bath in the dark with continuous N2 purging, and finally leaving at 22 °C in a temperature-controlled room to allow cooling down. The procedures were reproduced systematically to obtain similar cooling rates. Pristine nanoribbon dispersions were used for bulk spectroscopic measurements. However, the optical density of the p-R nanoribbon dispersion was not appropriate for an accurate Φem measurement, and addition of solvent, to reduce the density, lead to partial dissolution of the ribbons. Pristine n-BG nanoribbons for Φem measurement were obtained by the injection method, and the final solvent composition included n-butanol and dichloromethane. In order to perform microscopy studies, the nanoribbon dispersions were drop-casted or spin-coated on standard microscopy glass coverslips, dried by quick spin-coating in 1–3 seconds, and placed under gentle N2 flow. No impact of solvent drying on nanoribbon morphology was observed during sample preparation. For electroluminescence experiments, the nanoribbons were spin-coated onto the solvent post-treated PEDOT:PSS layer, and dried. The electron transport layer TBPi is thermally evaporated under vacuum, and bilayer cathodes of lithium fluoride and Al are then evaporated through a shadow mask. The voltage sweeps are driven manually (15 V max) until luminescence is observed by the CCD-camera under microscopy.
Confocal fluorescence microscopy was realized on a Picoquant Microtime 200 (polarization, FLIM and HS-CFM). Due to wavelength dependence of the two SPAD detectors used for polarization microscopy, the G-factor was measured for several narrow spectral regions applying bandpass filters (455–480 nm, 510–550 nm, 608–650 nm) and a 720 long-pass filter on a broadband emissive dilute dyes solution. I∥ and I⊥ are the intensities of the components of the fluorescence emission parallel and perpendicular, respectively, to the polarization of the excitation beam. The polarization P of each pixel is given by P = (I∥ − G × I⊥)/(I∥ + G × I⊥). P varies with the orientation of the long axis of the ribbon, between −1 and +1, and therefore Pmax corresponds to the highest value of the absolute value |P| measured for a straight section of a ribbon. For HS-CFM hyperspectral imaging the emission light was detected by an Andor SR300i spectrometer equipped with a Newton EMCCD. A high resolution spectrum was taken, with a 6 ms per pixel scan, for each pixel and then converted into CIE coordinates and RGB-colours. The emission spectra were intensity corrected by using an ARGOLIGHT calibration slide. The same microscope was used to measure electroluminescence through a Hamamatsu video CCD camera, and therefore superposition with HS-CFM was possible on the same sample area.
Author contributions
P. S.: conceptualization, spectroscopy, microscopy, data analysis, writing. C. d. V.: synthesis. L. G.-R. spectroscopy, theoretical studies. G. R. microscopy, software. M.-T. K. spectroscopy. C. S. synthesis. L. R. and B. P. anodes for EL. Y.-T. T., L. H. and D. B. cathodes, expertise and funding for EL. A. D. G. conceptualization, supervision, data analysis, writing, funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the European Research Council Marie Curie Actions (FP7-PEOPLE-2012-ITN SMARTNET Grant agreement Nr 316656); the CNRS; the French Ministry of Education and Research; the Région Aquitaine; the ANR-06-JCJC-0030; the Department of Education, Science and Universities of the Basque Country Government (postdoctoral grant and project IT1639-22); the “Arina” informatic cluster of UPV/EHU; the facilities ELORGA of UB; and the ANR-17-CE24-0033-01 RESOLVE. The authors thank Dr A. Mendez-Ardoy for the CV measurements and CESAMO for structural analyses (ISM, Univ. Bordeaux).Notes and references
- X. Y. Hu, T. Xiao, C. Lin, F. Huang and L. Wang, Acc. Chem. Res., 2014, 47, 2041–2051 CrossRef CAS PubMed.
- A. Pal, S. Karthikeyan and R. P. Sijbesma, J. Am. Chem. Soc., 2010, 132, 7842–7843 CrossRef CAS PubMed.
- N. Hosono, M. A. J. Gillissen, Y. Li, S. S. Sheiko, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501–510 CrossRef CAS PubMed.
- Y.-C. Yeh, R. Tang, R. Mout, Y. Jeong and V. M. Rotello, Angew. Chem., Int. Ed., 2014, 53, 5137–5141 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, M. C. A. Stuart, L. Florusse, G. Raffy, A. Del Guerzo and J. H. van Esch, Chem. Sci., 2016, 7, 6021–6031 RSC.
- H. Sato, Y. Miura, N. Saito, K. Kobayashi and O. Takai, Biomacromolecules, 2007, 8, 753–756 CrossRef CAS PubMed.
- N. C. Wickremasinghe, V. A. Kumar and J. D. Hartgerink, Biomacromolecules, 2014, 15, 3587–3595 CrossRef CAS PubMed.
- K. Jakab, C. Norotte, F. Marga, K. Murphy, G. Vunjak-Novakovic and G. Forgacs, Biofabrication, 2010, 2, 1–14 CrossRef PubMed.
- T. J. Gardner, C. D. Frisbie and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 6927–6933 CrossRef CAS.
- B. R. Walker, R. A. Wassel, D. M. Ştefanescu and C. B. Gorman, J. Am. Chem. Soc., 2004, 126, 16330–16331 CrossRef CAS PubMed.
- H. Xu, R. Hong, T. Lu, O. Uzun and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 3162–3163 CrossRef CAS PubMed.
- J. van Herrikhuyzen, A. Syamakumari, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2004, 126, 10021–10027 CrossRef CAS PubMed.
- Y. Zhang, H. Dong, Q. Tang, S. Ferdous, F. Liu, S. C. B. Mannsfeld, W. Hu and A. L. Briseno, J. Am. Chem. Soc., 2010, 132, 11580–11584 CrossRef CAS PubMed.
- K. Sugiyasu, S. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS.
- A. Heeres, C. van der Pol, M. Stuart, A. Friggeri, B. L. Feringa and J. van Esch, J. Am. Chem. Soc., 2003, 125, 14252–14253 CrossRef CAS PubMed.
- P. E. Laibnis, J. J. Hickman, M. S. Wrighton and G. M. Whitesides, Science, 1989, 245, 845–847 CrossRef PubMed.
- A. M. Johnson, C. A. Wiley, M. C. Young, X. Zhang, Y. Lyon, R. R. Julian and R. J. Hooley, Angew. Chem., Int. Ed., 2015, 54, 5641–5645 CrossRef CAS PubMed.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- M. R. Molla, A. Das and S. Ghosh, Chem. – Eur. J., 2010, 16, 10084–10093 CrossRef CAS PubMed.
- Y. T. Tsai, G. Raffy, H. F. Liu, B. J. Peng, K. P. Tseng, L. Hirsch, A. Del Guerzo, D. M. Bassani and K. T. Wong, Mater. Chem. Front., 2020, 4, 845–850 RSC.
- R. Kubota, S. Liu, H. Shigemitsu, K. Nakamura, W. Tanaka, M. Ikeda and I. Hamachi, Bioconjugate Chem., 2018, 29, 2058–2067 CrossRef CAS PubMed.
- S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS PubMed.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 316–318 RSC.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- F. Würthner, C. Bauer, V. Stepanenko and S. Yagai, Adv. Mater., 2008, 20, 1695–1698 CrossRef.
- S. Prasanthkumar, S. Ghosh, V. C. Nair, A. Saeki, S. Seki and A. Ajayaghosh, Angew. Chem., Int. Ed., 2015, 54, 946–950 CrossRef CAS PubMed.
- N. Fujita, S. Shinkai and R. V. January, Chem. Mater., 2008, 20, 3871–3873 CrossRef.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- E. R. Cross, S. Sproules, R. Schweins, E. R. Draper and D. J. Adams, J. Am. Chem. Soc., 2018, 140, 8667–8670 CrossRef CAS PubMed.
- D. Genovese, A. Aliprandi, E. A. Prasetyanto, M. Mauro, M. Hirtz, H. Fuchs, Y. Fujita, H. Uji-I, S. Lebedkin, M. Kappes and L. De Cola, Adv. Funct. Mater., 2016, 26, 5271–5278 CrossRef CAS.
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- C. de Vet, L. Gartzia-Rivero, P. Schäfer, G. Raffy and A. Del Guerzo, Small, 2020, 16, 1906723 CrossRef CAS PubMed.
- P. Schäfer, L. Gartzia-Rivero, M. T. Kao, C. Schäfer, S. Massip, C. De Vet, G. Raffy and A. Del Guerzo, J. Mater. Chem. C, 2021, 9, 136–147 RSC.
- T. Brotin, R. Utermohlen, F. Fages, H. Bouas-Laurent and J.-P. Desvergne, J. Chem. Soc., Chem. Commun., 1991, 416–418 RSC.
- C. Giansante, G. Raffy, C. Schäfer, H. Rahma, M. T. Kao, A. G. L. Olive and A. Del Guerzo, J. Am. Chem. Soc., 2011, 133, 316–325 CrossRef CAS PubMed.
- M. T. Kao, C. Schäfer, G. Raffy and A. Del Guerzo, Photochem. Photobiol. Sci., 2012, 11, 1730–1736 CrossRef CAS PubMed.
- A. F. Olea, D. R. Worrall, F. Wilkinson, S. L. Williams and A. A. Abdel-Shafi, Phys. Chem. Chem. Phys., 2002, 4, 161–167 RSC.
- S. J. Strickler and R. A. Berg, J. Chem. Phys., 1962, 37, 814–822 CrossRef CAS.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- M. A. do R. Silva, D. C. da Silva, V. G. Machado, E. Longhinotti and V. L. A. Frescura, J. Phys. Chem. A, 2002, 106, 8820–8826 CrossRef.
- D. C. da Silva, I. Ricken, M. A. do R. Silva and V. G. Machado, J. Phys. Org. Chem., 2002, 15, 420–427 CrossRef CAS.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- S. Samanta, S. K. Ray, S. Deolka, S. Saha, K. R. Pradeep, R. Bhowal, N. Ghosh and D. Chaudhuri, Chem. Sci., 2020, 11, 5710–5715 RSC.
- H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC.
- M. S. Myong, J. Zhou, R. M. Young and M. R. Wasielewski, J. Phys. Chem. C, 2020, 124, 4369–4377 CrossRef CAS.
- J. Xiao, Z. Yin, B. Yang, Y. Liu, L. Ji, J. Guo, L. Huang, X. Liu, Q. Yan, H. Zhang and Q. Zhang, Nanoscale, 2011, 3, 4720–4723 RSC.
- N. J. Hestand and F. C. Spano, J. Chem. Phys., 2015, 143, 244707 CrossRef PubMed.
- R. L. Barnes and J. B. Birks, Proc. R. Soc. London, Ser. A, 1966, 291, 570–582 CAS.
- J. Tanaka and M. Shibata, Bull. Chem. Soc. Jpn., 1968, 41, 34–41 CrossRef CAS.
- R. M. Hochstrasser and A. Malliaris, J. Chem. Phys., 1965, 42, 2243–2244 CrossRef CAS.
- B. Lu, X. Fang and D. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 31940–31951 CrossRef CAS PubMed.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- F. C. Spano, J. Chem. Phys., 2005, 122, 234701 CrossRef PubMed.
- X. X. Zhang, X. X. Zhang, K. Zou, C.-S. S. Lee and S.-T. T. Lee, J. Am. Chem. Soc., 2007, 129, 3527–3532 CrossRef CAS PubMed.
- H. S. Nalwa and S. Miyata, Nonlinear optics of organic molecules and polymers, CRC Press, 1997 Search PubMed.
- M. J. Mayoral, C. Rest, J. Schellheimer, V. Stepanenko and G. Fernández, Chem. – Eur. J., 2012, 18, 15607–15611 CrossRef CAS PubMed.
- S. Banerjee, R. K. Das, P. Terech, A. De Geyer, C. Aymonier, A. Loppinet-Serani, G. Raffy, U. Maitra, A. Del Guerzo and J. P. Desvergne, J. Mater. Chem. C, 2013, 1, 3305–3316 RSC.
- C. Zhang, Y. Yan, Y. S. Zhao and J. Yao, Acc. Chem. Res., 2014, 47, 3448–3458 CrossRef CAS PubMed.
- Y. Yan, C. Zhang, J. Yao and Y. S. Zhao, Adv. Mater., 2013, 25, 3627–3638 CrossRef CAS PubMed.
- A. Camposeo, D. B. Granger, S. R. Parkin, D. Altamura, C. Giannini, J. E. Anthony and D. Pisignano, Chem. Mater., 2019, 31, 1775–1783 CrossRef CAS PubMed.
- K. P. Tseng, F. C. Fang, J. J. Shyue, K. T. Wong, G. Raffy, A. Del Guerzo and D. M. Bassani, Angew. Chem., Int. Ed., 2011, 50, 7032–7036 CrossRef CAS PubMed.
- Y.-T. Tsai, K.-P. Tseng, Y.-F. Chen, C.-C. Wu, G.-L. Fan, K.-T. Wong, G. Wantz, L. Hirsch, G. Raffy, A. Del Guerzo and D. M. Bassani, ACS Nano, 2016, 10, 998–1006 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, spectroscopy, microscopy, calculations, methods. See DOI: https://doi.org/10.1039/d2nr01017h |
| This journal is © The Royal Society of Chemistry 2022 |