
Temperature-dependent CO2 sorption and thermal-reduction without reactant gases on BaTiO3 nanocatalysts at low temperatures in the range of 300–1000 K†
 a
and
Tomonori
Ohba
*a
a
and
Tomonori
Ohba
*a
Abstract
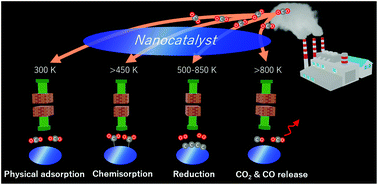
Carbon utilization techniques to mitigate the impact on global warming are an important field in environmental science. CO2 reduction is a significant step for carbon utilization. However, CO2 reduction with less energy consumption has major challenges. In this study, CO2 thermal reduction was demonstrated using nanocatalysts at temperatures lower than 1000 K, and the CO2 sorption and reduction mechanisms within the temperature range of 300–1000 K were evaluated. The physical adsorption on nanocatalysts with a crystal size of 7.4 ± 0.4 nm (10 nm-nanocatalysts) majorly occurred at 300 K and was considerably decreased beyond that temperature. CO2 chemisorption occurred above 450 K and subsequent CO2 reduction occurred above 500 K, which was expected based on the temperature-programmed reaction. CO2 reduction decreased above 900 K by the deactivation of the 10-nm nanocatalyst as a result of its crystal growth. The transmission electron microscopy images also indicated the complete reduction of CO2 into carbon products at 600 and 800 K. Therefore, an optimal condition of CO2 reduction in the temperature range of 500–800 K. The highly active thermocatalyst achieved CO2 reduction into CO and carbon products without any reducing agents even at an extremely low temperature (500 K). In summary, temperature-dependent CO2 sorption and reduction were observed on the 10-nm nanocatalyst; CO2 physical adsorption at 300–500 K, CO2 chemisorption above 450 K, CO2 reduction at 500–850 K, and CO2 and CO releases above 800 K.
- This article is part of the themed collection: CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

