
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDroplet-based microfluidic synthesis of nanogels for controlled drug delivery: tailoring nanomaterial properties via pneumatically actuated flow-focusing junction†
Sara Maria
Giannitelli‡
 a,
Emanuele
Limiti‡
a,
Pamela
Mozetic
bc,
Filippo
Pinelli
d,
Xiaoyu
Han
e,
Franca
Abbruzzese
a,
Francesco
Basoli
a,
Danila
Del Rio
f,
Stefano
Scialla
ag,
Filippo
Rossi
d,
Marcella
Trombetta
a,
Laura
Rosanò
f,
Giuseppe
Gigli
ch,
Zhenyu Jason
Zhang
e,
Emanuele
Mauri
*a and
Alberto
Rainer
*ac
a,
Emanuele
Limiti‡
a,
Pamela
Mozetic
bc,
Filippo
Pinelli
d,
Xiaoyu
Han
e,
Franca
Abbruzzese
a,
Francesco
Basoli
a,
Danila
Del Rio
f,
Stefano
Scialla
ag,
Filippo
Rossi
d,
Marcella
Trombetta
a,
Laura
Rosanò
f,
Giuseppe
Gigli
ch,
Zhenyu Jason
Zhang
e,
Emanuele
Mauri
*a and
Alberto
Rainer
*ac
aDepartment of Engineering, Università Campus Bio-Medico di Roma, via Álvaro del Portillo 21, 00128 Rome, Italy. E-mail: e.mauri@unicampus.it; a.rainer@unicampus.it; Fax: +39 06225419419; Tel: +39 06225419627 Tel: +39 06225419214
bDivision of Neuroscience, Institute of Experimental Neurology, San Raffaele Scientific Institute, Via Olgettina, 60, 20132, Milan, Italy
cInstitute of Nanotechnology (NANOTEC), National Research Council, via Monteroni, 73100, Lecce, Italy
dDepartment of Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico di Milano, via L. Mancinelli 7, 20131 Milan, Italy
eSchool of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
fInstitute of Molecular Biology and Pathology, National Research Council (CNR), via Degli Apuli 4, 00185 Rome, Italy
gNational Institute of Chemical Physics and Biophysics, Akadeemia tee 23, 12618 Tallinn, Estonia
hDepartment of Mathematics and Physics “Ennio De Giorgi”, Università del Salento, via per Arnesano, 73100 Lecce, Italy
First published on 29th July 2022
Abstract
Conventional batch syntheses of polymer-based nanoparticles show considerable shortcomings in terms of scarce control over nanomaterials morphology and limited lot-to-lot reproducibility. Droplet-based microfluidics represents a valuable strategy to overcome these constraints, exploiting the formation of nanoparticles within discrete microdroplets. In this work, we synthesized nanogels (NGs) composed of hyaluronic acid and polyethyleneimine using a microfluidic flow-focusing device endowed with a pressure-driven micro-actuator. The actuator achieves real-time modulation of the junction orifice width, thereby regulating the microdroplet diameter and, as a result, the NG size. Acting on process parameters, NG hydrodynamic diameter could be tuned in the range 92–190 nm while preserving an extremely low polydispersity (0.015); those values are hardly achievable in batch syntheses and underline the strength of our toolbox for the continuous in-flow synthesis of nanocarriers. Furthermore, NGs were validated in vitro as a drug delivery system in a representative case study still lacking an effective therapeutic treatment: ovarian cancer. Using doxorubicin as a chemotherapeutic agent, we show that NG-mediated release of the drug results in an enhanced antiblastic effect vs. the non-encapsulated administration route even at sublethal dosages, highlighting the wide applicability of our microfluidics-enabled nanomaterials in healthcare scenarios.
1. Introduction
In recent years, nanomedicine has developed innovative therapeutic strategies based on polymeric nanoparticles (NPs).1,2 The rational combination of different polymers through physical or chemical crosslinking has led to the formulation of nano-sized materials eligible as platforms for drug delivery, gene therapy or nanotheranostics.3–5 In particular, the orthogonal functionalization of polymeric chains with specific biomolecules (such as small peptides and ligands) or chemical groups has enabled the synthesis of nanocarriers for targeted therapies in a variety of clinical scenarios.6–10 However, the clinical application of polymeric NPs still remains challenging due to the constraints occurring in conventional batch-synthesis processes, which are characterized by limited scalability, scarce control over reaction parameters, extensive NP polydispersity, unsatisfactory batch-to-batch reproducibility, and use of large volumes of chemicals and therapeutics.11,12 Furthermore, these factors profoundly affect the encapsulation efficiency of the payload and its release profile, preventing the treatment from reaching optimal performance. To overcome these limitations, microfluidics has been recently demonstrated as an enabling technology to achieve highly controllable and scalable production yields, opening new scenarios for advanced NP design.13–15Microfluidics enables the manipulation of small volumes of fluids with precise control of molecule concentrations in space and time, thus achieving more controlled synthesis conditions and higher throughput.16 Microfluidic reactors offer a continuous in-flow production of NPs that can lead to narrow size distribution, increased encapsulation efficiency and sustained drug release profiles.17,18 Among the available microfluidic strategies, nanoprecipitation and self-assembly methods are commonly used to produce polymeric NPs. In the former, the mixing time between solvent and non-solvent phases represents the key parameter to control NP nucleation and growth,19,20 whereas in the latter, the pH- or temperature-responsive behavior of amphiphilic derivatives causes the generation of self-assembled structures, due to the diffusion-mediated pH or temperature gradient into the focused polymer stream.21–23 However, both approaches are subject to channel clogging due to undesired interactions between reagents and solvents.24,25 This does not concern droplet-based microfluidics: in this case, the microfluidic platform uses immiscible phases to create droplets as discrete volumes where the NP formation occurs. Each droplet works as a micro-reactor, ensuring rapid heat and mass transfer within the confined microvolume and enabling faster reaction kinetics. Moreover, this technique gives precise control over process parameters such as size and composition of the droplets, that are produced in series as multiple identical micro-reactor units.26
To date, droplet-based approaches have been largely documented for the synthesis of inorganic NPs based on reduction and oxidation reactions,27,28 and hybrid organic–inorganic nanocarriers via a combination of photo-crosslinking with hydrolysis/condensation reactions.29 Conversely, the use of droplet-based microfluidics for polymeric particles has been largely tied to microscale systems. As an example, Rezvantalab and Moraveji30 analyzed the literature for PLGA-based particles, pointing out that segmented microfluidics, such as the droplet-based strategy, could be applied to the synthesis of microparticles in the 10–1000 μm range, while nanosized particles required the implementation of continuous phase flow microfluidics approaches. Similar findings are reported in other works, for a wide range of polymeric materials.12,32,33 Only recently, Visaveliya and Köhler have reported a droplet-based microfluidic process for the synthesis of poly(methyl methacrylate) NPs via emulsion polymerization.31 However, the polymerization reaction occurs at 97 °C and requires a crosslinking agent and a thermal initiator, thus narrowing the range of eligible polymer systems, due to the increased complexity and the potential degradation of the polymeric chains under those reaction conditions. Hence, the challenge remains to bring droplet-based microfluidics into organic chemistry reaction protocols with the aim to advance the formulation of NPs showing improved properties for nanomedicine.
Here, we propose the synthesis of polymeric nanocarriers via droplet-based microfluidics using a chip endowed with a pneumatic micro-actuator, enabling the active tuning of the hydrodynamic flow-focusing (HFF) geometry, thereby modulating the diameter of the microdroplets.34,35 The rationale underlying this chip configuration is two-fold: first, to achieve the covalent crosslinking of polymer chains in each droplet to produce highly monodispersed NPs; and second, to actively control NP dimensions by acting on the tunable HFF junction. In detail, we have adapted the microfluidic process to the synthesis of a specific class of polymeric nanomaterials, i.e., nanogels (NGs). NGs have received increasing attention thanks to their unique features which address many of the shortcomings of other classes of nanocarriers. Indeed, NGs appear as very promising candidates for nanomedicine, especially in terms of enhanced drug loading (mainly mediated by NG swelling behavior36), efficient encapsulation of both hydrophilic and lipophilic payloads, and internalization by the target cells, lowering the therapeutic dosage and minimizing harmful side effects.37–39 Specifically, hyaluronic acid (HA) and linear polyethyleneimine (LPEI) have been chosen as starting polymers to synthesize NGs potentially aimed to cancer therapy: the combination of the well-known features of HA in cancer therapy scenarios40–43 and the unique proton-sponge effect of PEI can act synergistically improving the drug delivery performance of the nanocarriers.44,45 The proposed pressure-actuated microfluidic system produces tunable water-in-oil (W/O) microemulsions, where the polymers are dissolved in the aqueous dispersed phase to form NGs. The validation of the synthetized nanoscaffolds was performed in vitro demonstrating the benefits of the NG-mediated drug delivery in a representative cancer scenario still demanding innovative therapeutic strategies: ovarian cancer.
2. Experimental
2.1 Materials
Hyaluronic acid sodium salt (HA, ultra-low molecular weight, Mw = 8–15 kDa, Biosynth Carbosynth, Compton, UK) and linear polyethyleneimine (LPEI, Mw = 2.5 kDa, Polysciences, Warrington, PA) were used as reagents for nanogel formulation. SU-8 photoresist (MicroChem, Newton, MA) and polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, Midland, MI) were used for chip fabrication. Doxorubicin (DOX) was purchased from Teva Italia S.r.l. (Milan, Italy). All other chemicals were purchased from Merck KGaA (Darmstadt, Germany) and used as received, without any further purification. Solvents were of analytical-grade purity. The Cy5-conjugated products were stored at −20 °C.2.2 Chip microfabrication
The microfluidic device, featuring a HFF junction flanked by dead-end pressurizable channels to act as a pneumatic micro-actuator modulating the junction width, was designed using a CAD suite (Layout Editor, juspertor GmbH, Unterhaching, Germany) and fabricated by soft-lithography starting from a photolithographically obtained silicon master. Master mold was fabricated in SU-8 2050 negative photoresist on a 3′′ silicon wafer following a standard photolithographic process.46 The master was silanized overnight in a chamber saturated with trimethylchlorosilane (TMCS) vapors to facilitate demolding.Replicas were obtained by casting PDMS (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w prepolymer to curing agent ratio) onto the silicon master, followed by thermal curing (2 h at 70 °C). Once cured, replicas were carefully peeled off and fluidic ports were created using a biopsy puncher prior to oxygen plasma bonding to 1′′ × 3′′ microscope glass slides (Femto Plasma Etcher, Diener electronic, Ebhausen, Germany).
:1 w/w prepolymer to curing agent ratio) onto the silicon master, followed by thermal curing (2 h at 70 °C). Once cured, replicas were carefully peeled off and fluidic ports were created using a biopsy puncher prior to oxygen plasma bonding to 1′′ × 3′′ microscope glass slides (Femto Plasma Etcher, Diener electronic, Ebhausen, Germany).
Finally, the microfluidic channels were silanized by flushing with a stream of nitrogen saturated with TMCS for 15 min followed by thermal treatment at 140 °C for 10 min.
2.3 Polymer functionalization
HA carboxyl groups were activated through carbodiimide/hydroxysuccinimide coupling, and LPEI was functionalized with the far-red dye Cy5 (Fig. 1). In detail, HA (6.8 mg, 0.016 mmol of monomer) was dissolved in deionized water (DIW, 1.5 mL) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, 15.3 mg, 0.08 mmol) and N-hydroxysuccinimide (NHS, 3.76 mg, 0.032 mmol) were added to the polymer solution, resulting in a 1:5:2 molar ratio for HA carboxyl groups, EDC and NHS, respectively. The reaction system was stirred for 3 h at room temperature (RT), and directly used in the microfluidic NG synthesis. On the other side, the Cy5 conjugation of LPEI was performed by slightly modifying the protocol described in our previous work:47 briefly, LPEI was first functionalized with alkyne moieties through nucleophilic substitution with propargyl bromide (LPEI:C3H3Br molar ratio = 1:10) in methanol, and then used in a copper-catalyzed alkyne–azide cycloaddition (CuAAC) with a 2 mg mL−1 solution of Cy5-N3 in DMSO (LPEI:Cy5-N3 molar ratio = 1:0.01) with CuSO4 as the catalyst and sodium ascorbate as the reducing agent. The click reaction was performed in water, at 50 °C for 36 h, in the dark and under stirring. The final product was collected as a freeze-dried polymer after dialysis (membrane MWCO = 100–500 Da) against DIW.
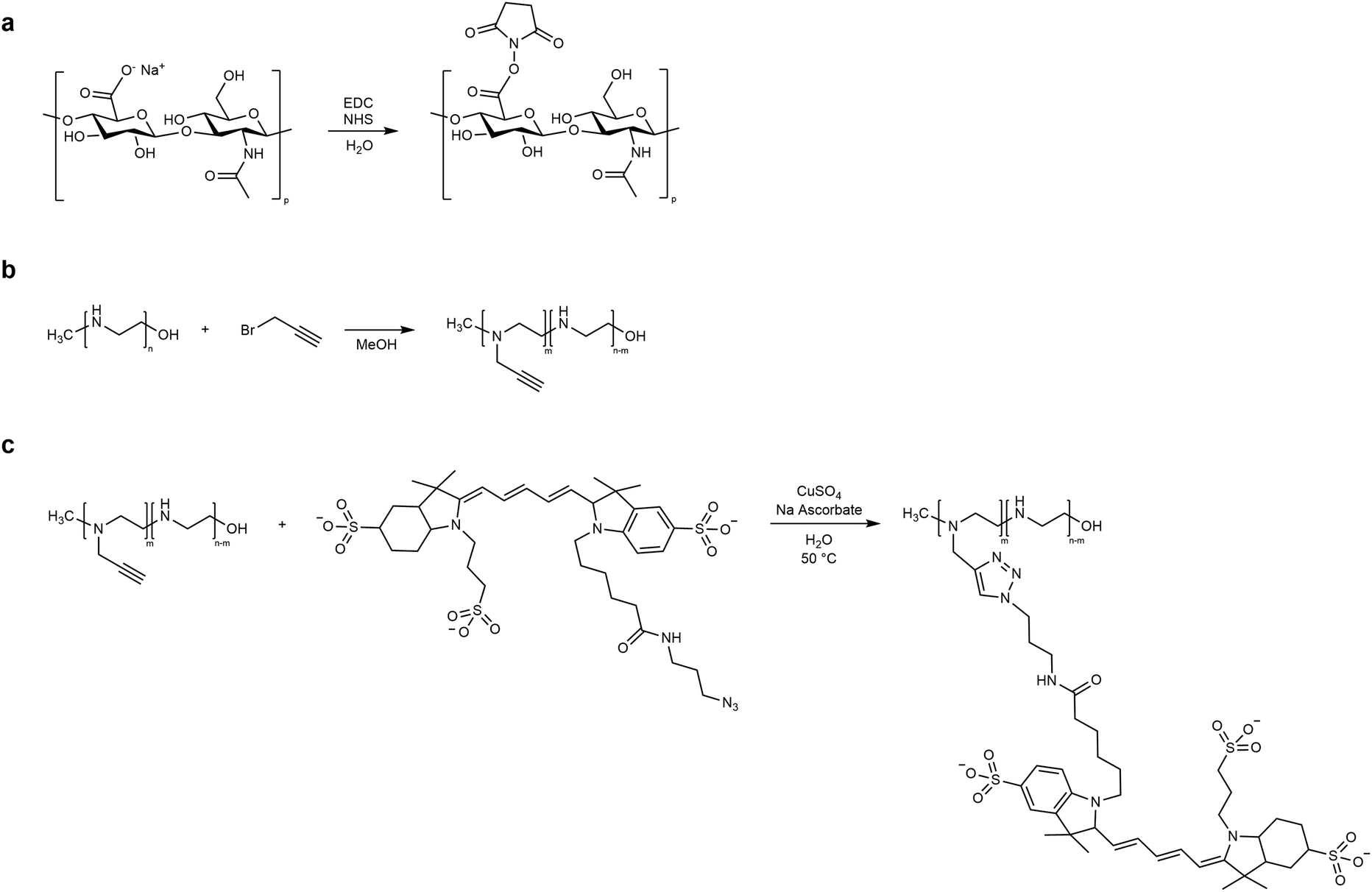 | ||
| Fig. 1 Reaction scheme of HA and LPEI functionalization: (a) HA activation; (b) LPEI modification with alkyne groups; (c) CuAAC reaction between LPEI and Cy5-azide. | ||
2.4 Nanogel synthesis through droplet-based microfluidics
The nanogel synthesis required the preparation of an aqueous and an organic solution as the dispersed and continuous phase, respectively. The aqueous phase was obtained as follows: LPEI–Cy5 (6.8 mg, 2.7 μmol) was dissolved in DIW (1.5 mL), then the same amount of pristine LPEI was added, and the pH of the solution was adjusted to 4.5 with 1 M HCl, to enhance the solubility of the pristine polymer in the aqueous medium. The resulting mixture was added dropwise to the solution of activated HA and vortexed for 1 min; finally, the obtained blend was loaded into a 2.5 mL gas-tight glass syringe (Hamilton Company, Reno, NV) connected to the chip inlet for the dispersed phase. The organic solution consisted of 3% w/v Span 80 in mineral oil (5 mL) and was loaded into a 5 mL gas-tight glass syringe (Hamilton) connected to the chip inlet for the continuous phase. The two liquid phases were injected into the chip channels through a programmable syringe pump (Nemesys, CETONI GmbH, Korbussen, Germany): the flow rates were set to 2 μL min−1 for the polymer solution and to 10 μL min−1 for the organic phase. To avoid the precipitation of pristine LPEI during the experiments, the whole set-up was kept at 37 °C. The on-chip micro-actuator was pneumatically driven using a programmable pressure controller (OB-1 MK3, Elveflow, Paris, France) fed by compressed air. Pressurization caused the deflection of the thin PDMS walls separating the pressurized channels from the flow-focusing junction. Droplet generation occurred at the junction, forming a W/O microemulsion which was collected at the end of the output channel. A preliminary evaluation of the droplet size as a function of orifice deformation was conducted in the pressure range compatible with chip operation, and the two process conditions stably generating the largest (i.e., 0 bar) and smallest (i.e., 2 bar) droplets, without transient jetting phenomena, were chosen for further investigation. Consistently, the resulting NGs were labeled NG_0 and NG_2, respectively. Microdroplet formation was monitored under a motorized inverted optical microscope (Eclipse Ti-E, Nikon Instruments, Tokyo, Japan) equipped with a high-sensitivity camera (Neo 5.5, Andor Technology, Belfast, UK) and a dedicated control software (NIS Elements AR, Nikon). Full frame micrographs were acquired at 10 μs exposure time, while videos of 128 × 128 pixel ROIs were grabbed at 1600 fps.The collected microemulsions were broken by adding diethyl ether (10 mL), and the separated aqueous phase containing the NGs was further extracted with diethyl ether (2 × 5 mL) and purified via dialysis (MWCO = 6–8 kDa) against DIW, with daily changes for 2 days. The NG_0 and NG_2 specimens were finally obtained as solid powders after freeze-drying.
2.5 Nanogel characterization techniques
2.6 Drug loading
Encapsulation efficiency (EE%) was assessed using doxorubicin (DOX) as a model drug. 30 μL of a 100 μM DOX solution were added to the lyophilized specimens (3 mg), followed by centrifugation at 6000 rpm for 15 s to maximize the contact between the NGs and the drug. The system was then left at RT for 15 min to complete the uptake of DOX. The resulting loaded nanocarriers were dialyzed (MWCO = 3.5 kDa) against PBS for 30 min to remove the unabsorbed drug. EE% was estimated as (eqn (1)):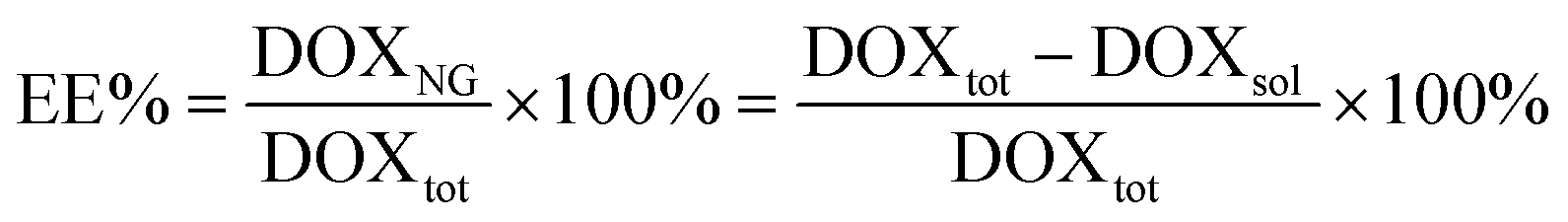 | (1) |
2.7 Drug release
DOX-loaded NGs were diluted to a final concentration of 10 mg mL−1 and 100 μL aliquots of the resulting suspension were allowed to exchange against PBS (0.6 mL) at 37 °C. At defined time points, aliquots (3 × 100 μL) were withdrawn and DOX concentration was determined (λex = 488 nm, λem = 590 nm; TECAN Infinite M200 Pro) against a DOX calibration curve. At each withdrawal, elution buffer was replenished with fresh PBS to avoid mass transfer equilibrium between the nanosystem and the surrounding solution. The experimental data were collected from three independent replicates and reported as mean ± standard deviation (SD).2.8 Cell culture
The human epithelial ovarian cancer cell line OVCA433 was obtained from the American Type Culture Collection (ATCC, Manassas, VA). OVCA433 cells were cultured in Dulbecco's modified Eagle's medium (DMEM low glucose, GIBCO, Thermo Fisher Scientific), supplemented with 10% fetal calf serum, 100 units per mL penicillin and 100 μg mL−1 streptomycin. Cells were incubated at 37 °C in a humidified atmosphere with 5% CO2.2.9 Nanogel cytocompatibility: G6PD assay
Cytotoxicity associated to the administration of NGs to OVCA433 was assessed using Vybrant Cytotoxicity Assay (Thermo Fisher Scientific) as reported in a previous work.48 Cells were seeded at the density of 1.5 × 104 cells per cm2 into 96-well plate for 24 h and then supplemented with NGs at a concentration of 20 μg mL−1. The release of the cytosolic enzyme glucose 6-phosphate dehydrogenase (G6PD) from damaged cells into the surrounding medium was quantified after 24 h of incubation with NGs. 50 μL of supernatant were transferred into a 96-well plate and, after 10 min of incubation with 50 μL of resazurin/reaction mixture at 37 °C in 5% CO2, the fluorescent metabolite of resazurin (resorufin) was detected (λex = 530 nm, λem = 590 nm; TECAN Infinite M200-Pro). Cytotoxicity was calculated as the fluorescence intensity ratio between NG-treated cells and fully-lysed control cells (positive control), previously background-corrected by subtracting from each reading the value of healthy untreated cells (negative control). Experiments were performed in triplicate.2.10 Flow cytometric analysis for nanogel internalization
OVCA433 cells were seeded at a density of 1.5 × 104 cells per cm2 into 12-well plates for 24 h. Then, a NG suspension at a concentration of 20 μg mL−1 was added. At selected timepoints (0.5, 1 and 2 h), cells were analyzed by flow cytometry (CytoFLEX flow cytometer, Beckman Coulter, Brea, CA) with CytExpert software (Beckman Coulter). Live cells were gated based on their forward and side scatter parameters; NG signal was recorded in the allophycocyanin (APC-A700) channel and quantified as the median intensity value at each time point.2.11 Confocal microscopy
The cellular internalization of NG_0 and NG_2 specimens was also visualized through fluorescence microscopy. Cells (1.5 × 104 cells per cm2) were seeded into 8-well glass bottom μ-Slide chambers (Ibidi GmbH, Gräfelfing, Germany). After 24 h, cells were incubated with NGs at a concentration of 20 μg mL−1 for 30 min. Cells were then fixed with 4% buffered paraformaldehyde (PFA) for 15 min at RT and incubated for 5 min in Triton X-100 (0.1% in PBS) to permeabilize cell membranes. After washing in PBS (3 times), cells were incubated with 1% bovine serum albumin in PBS (blocking buffer) for 30 min, stained with FITC-labeled phalloidin (Acti-stain 488, Cytoskeleton Inc., Denver, CO; 1:200 dilution in blocking buffer for 40 min) and counterstained with DAPI (Thermo Fisher Scientific; 1:1000 dilution in PBS for 10 min). Representative micrographs were collected using a Nikon A1R + laser scanning confocal microscope (Nikon Instruments, Tokyo, Japan) with a 20× NA1.0 air objective.
2.12 Biological activity of NG-mediated DOX delivery
NGs were validated with a cell-based assay, measuring cell metabolic activity following DOX administration. OVCA433 were seeded at a density of 1.5 × 104 cells per cm2 in growth medium and incubated for 24 h. Then, DOX was encapsulated in NG_0 and NG_2 specimens (23 ng DOX per mg NG) and the resulting systems (labeled as DOX/NG_0 and DOX/NG_2, respectively) were separately added to cells at a concentration of 20 μg mL−1 and incubated for 30 min, followed by medium exchange. A reference group was represented by OVCA433 cells supplemented with pristine (i.e., non-encapsulated) DOX at the same final concentration of 0.8 μM in culture medium. MTT assay was used according to the manufacturer's instructions. Briefly, activity of mitochondrial dehydrogenases in living cells was measured in terms of absorbance at 570 nm (TECAN Infinite M200-Pro) after 3 h exposure to a 0.5 mg mL−1 MTT solution in PBS, at 37 °C and 5% CO2. Absorbance values obtained in the absence of cells were used for background subtraction. Untreated cells were used as a control for normalization. Experiments were performed in triplicate at different time points (24 h, 48 h, 72 h and 1 week).2.13 Statistical analysis
Data were analyzed using Prism ver. 9.2.0 (GraphPad Software, San Diego, CA) and reported as mean ± SD unless otherwise specified. One-way analysis of variance (ANOVA) followed by Tukey's multiple comparisons test was used to assess statistical significance, which was set at the 0.05 level.3. Results and discussion
3.1 Nanogel formation and physico-chemical characterization
NG synthesis was carried out through a droplet-based microfluidic approach, using a chip endowed with micro-actuation features (Fig. 2a). Two immiscible fluids were supplied to the device to generate a W/O microemulsion: the polymer solution constituted the aqueous dispersed phase, and mineral oil with a nonionic surfactant represented the organic continuous phase (Fig. 2b). The formation of the microemulsion was obtained via hydrodynamic flow focusing, exploiting the flow rates, the shear force and the interfacial tension between the two phases, and the geometry of the microfluidic channels.32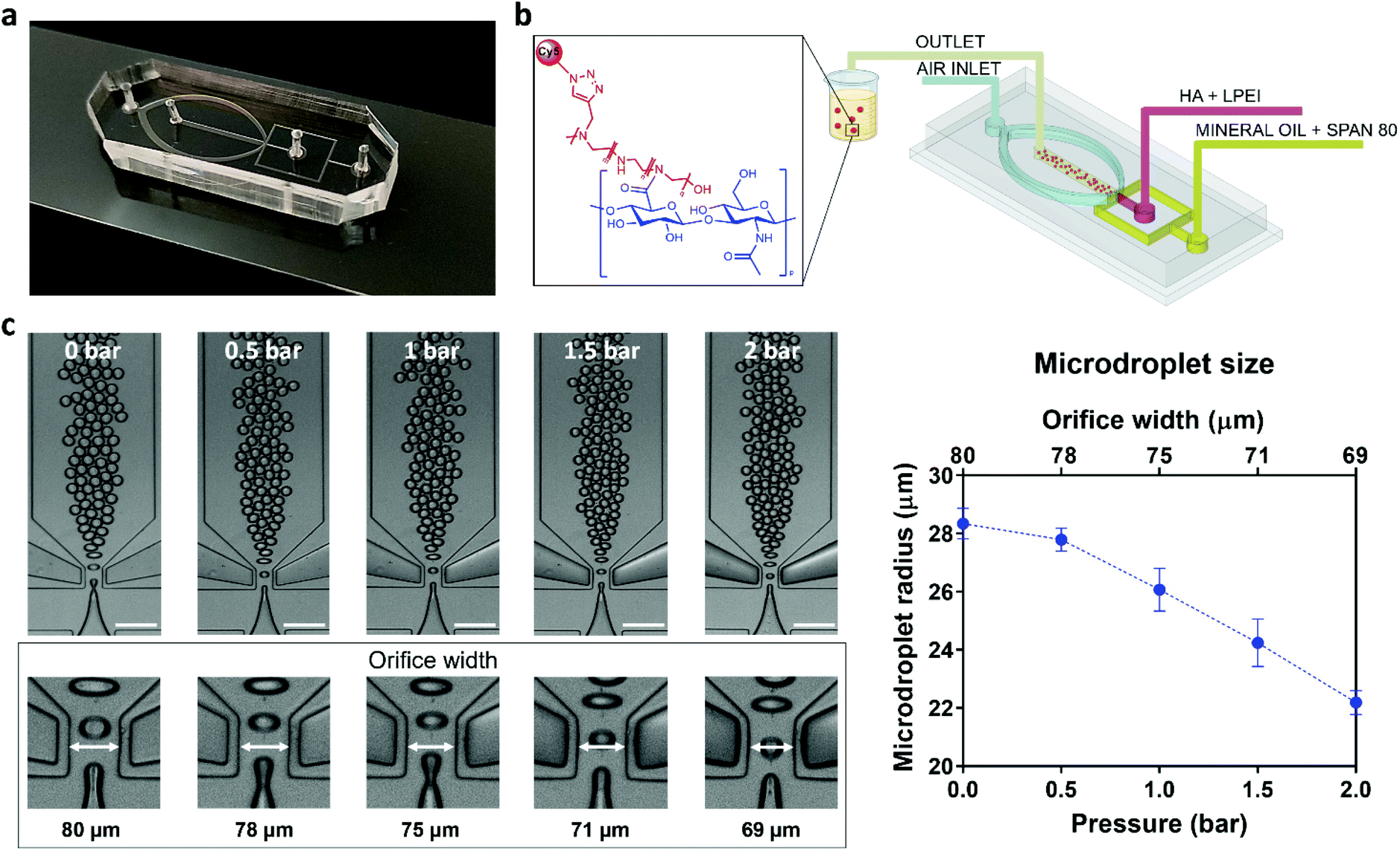 | ||
| Fig. 2 PDMS microfluidic device (a) and schematic representation of the droplet-based microfluidic approach for NGs production (b). Representative micrographs demonstrating the active control over microdroplet size distribution were collected under a brightfield optical microscope and the values of microdroplet radii vs. actuation pressure and orifice width are plotted in (c). Measurements of the orifice width were performed in triplicate on distinct microfluidic devices. Scale bar: 200 μm. | ||
In particular, a flow ratio of 0.2 was set between the flow rates of the aqueous and the organic phases, in order to avoid jetting mode during the droplet generation. The resulting microdroplets were monodisperse and stable in the continuous phase, without coalescence phenomena. The design of a pneumatic actuator to stretch the flow-focusing junction enabled the active tuning of the flow-focusing geometry (representative 3D volume reconstructions in Fig. S2, ESI†), thereby modulating the diameter of the formed microdroplets (Fig. 2c). We report microdroplets ranging from 56.7 ± 0.4 μm at 0 bar to 44.2 ± 0.3 μm at 2 bar, indicating a volume reduction of ca. 53% between the two conditions. The pivotal role of the actuator lays in the dynamic control of the orifice geometry, resulting in on-demand variation of the droplet size without changing the input flow rates of the two immiscible phases. This also results in improved flexibility, with a single chip design allowing end-users to actively tune the microdevice geometrical parameters, adjusting them to the application requirements. High-frame-rate videos of the droplet generation process were recorded to determine the stability, the accessible range of droplet diameters and the process reproducibility. Each microdroplet was conceived as a self-contained reaction chamber, where the crosslinking reaction between the HA-NHS ester derivatives and the LPEI amine groups occurred, forming the 3D nanoscaffold (see also Fig. S3, ESI†). The covalent bonding between the polymers is proportional to the volume concentration of the polymeric chains: considering a homogeneous distribution of the reagents in the disperse phase, a lower concentration of reactive polymers, which should be ascribed to the smaller microdroplets, will lead to the formation of smaller NGs. This can be linked to the limited reactive sites available in the droplet volume that will generate smaller nanoarchitectures, as discussed below.
Active droplet generation in microfluidics represents a milestone for different approaches reported in the literature:34,49 for example, a valve-based flow-focusing junction was used to generate foams as templates for graded porous structures and 3D-printed constructs as bone models,46 and cross-junctions and T-junctions were designed to generate droplets containing the precursors species by shear force or electro-pneumatic regulators to synthesize inorganic nanoparticles.28,50,51 However, the use of pressure-actuated microfluidic platforms to produce polymer-based nanoparticles via microdroplet generation has not been comprehensively investigated yet;52,53 indeed, these nanomaterials are usually fabricated by microfluidic nanoprecipitation and the procedure does not involve a chemical crosslinking reaction between different polymers in each individual droplet.25,54–57 Furthermore, the application of the methodology in the design of NGs is still unexplored. These considerations highlight the relevance of the present work, which may represent a significant breakthrough.
The formation of NGs was confirmed by NMR and FT-IR analyses. 1H-NMR spectra of both NG_0 and NG_2 showed the chemical shifts of polymer moieties related to the amide bonding between HA and LPEI. In detail, the signal ascribable to the methylene protons (N–CH2–CH2, d and f in Fig. 3) of LPEI monomer shifted to 3.39 ppm (at 2.93 ppm in the spectrum of neat LPEI), whereas the methine group (CH–COOH, e and g in Fig. 3) of the HA ring moiety involved in the reaction was recognizable at 3.62 ppm (shifting from 3.72 ppm in the spectrum of pristine HA). Moreover, in both specimens, the characteristic peak of HA backbone and methyl groups (respectively, range 4.60÷3.25 ppm and 2.00 ppm) and of LPEI backbone (range 3.06÷2.70 ppm) were clearly detectable.58,59 An estimation of NG composition, in terms of molar ratio between the two polymers, could be carried out considering the integration of the peaks attributable to each polymer: as a result, NG_0 specimen was approximately characterized by a LPEI:HA 2.3:1 molar ratio, whereas NG_2 presented a LPEI:HA ratio of 1.8:1.
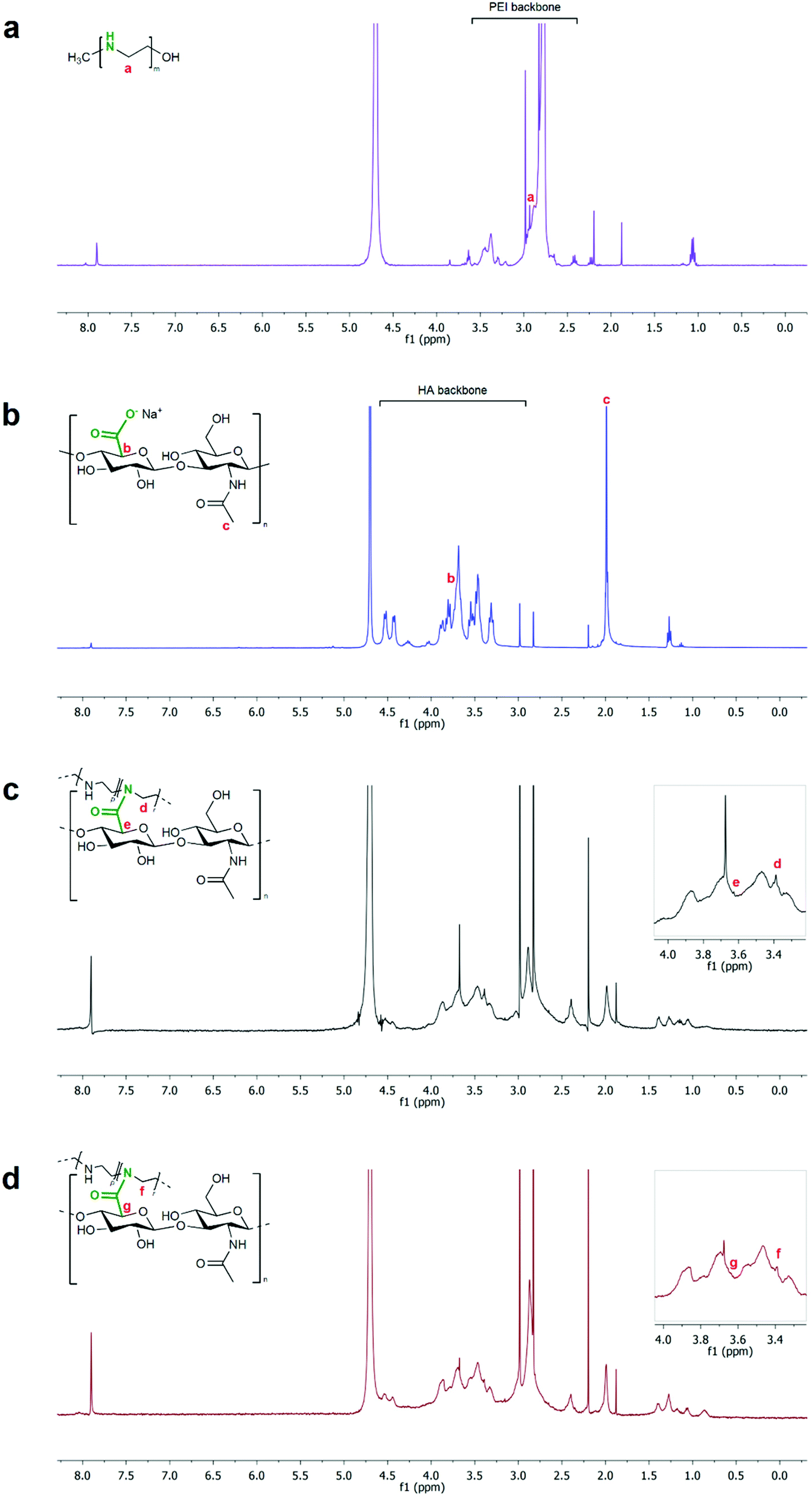 | ||
| Fig. 3 1H-NMR spectra of starting polymers and NG specimens: LPEI (a, violet; due to the different protonation degrees of amino groups in the polymer backbone, signal a is indicative of the methylene protons subsequently involved in the crosslinking reaction); HA (b, blue); NG_0 (c, black) and NG_2 (d, red). The characteristic signals of LPEI and HA and the relative shifts (e and d in NG_0; g and f in NG_2) after crosslinking reaction are highlighted. | ||
FT-IR spectra of both NG specimens (Fig. 4) showed the characteristic signal of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1724 cm−1, indicative of the crosslinking between HA and LPEI via amide bond. Furthermore, both spectra presented the peculiar peaks ascribable to the starting polymers. In detail, HA signals were recognizable at 3288 cm−1 (–OH stretching), 1718 cm−1 (carbonyl stretching of the carboxylic group), 1600 cm−1 (amide II, CO stretching), 1400 cm−1 and 1367 cm−1 (C–H bending), 1295 cm−1 (stretching of the C–CH3 amide), and 1040 cm−1 (skeletal vibration of the C–O stretching).60,61 LPEI showed its signals at 3376 cm−1 (–OH stretching), 2879 cm−1 (stretching vibration of the aliphatic C–H), 1610 cm−1 (N–H bending) and in the wavenumbers range 1450÷900 cm−1 (C–H bending and C–N stretching vibrations).
O stretching at 1724 cm−1, indicative of the crosslinking between HA and LPEI via amide bond. Furthermore, both spectra presented the peculiar peaks ascribable to the starting polymers. In detail, HA signals were recognizable at 3288 cm−1 (–OH stretching), 1718 cm−1 (carbonyl stretching of the carboxylic group), 1600 cm−1 (amide II, CO stretching), 1400 cm−1 and 1367 cm−1 (C–H bending), 1295 cm−1 (stretching of the C–CH3 amide), and 1040 cm−1 (skeletal vibration of the C–O stretching).60,61 LPEI showed its signals at 3376 cm−1 (–OH stretching), 2879 cm−1 (stretching vibration of the aliphatic C–H), 1610 cm−1 (N–H bending) and in the wavenumbers range 1450÷900 cm−1 (C–H bending and C–N stretching vibrations).
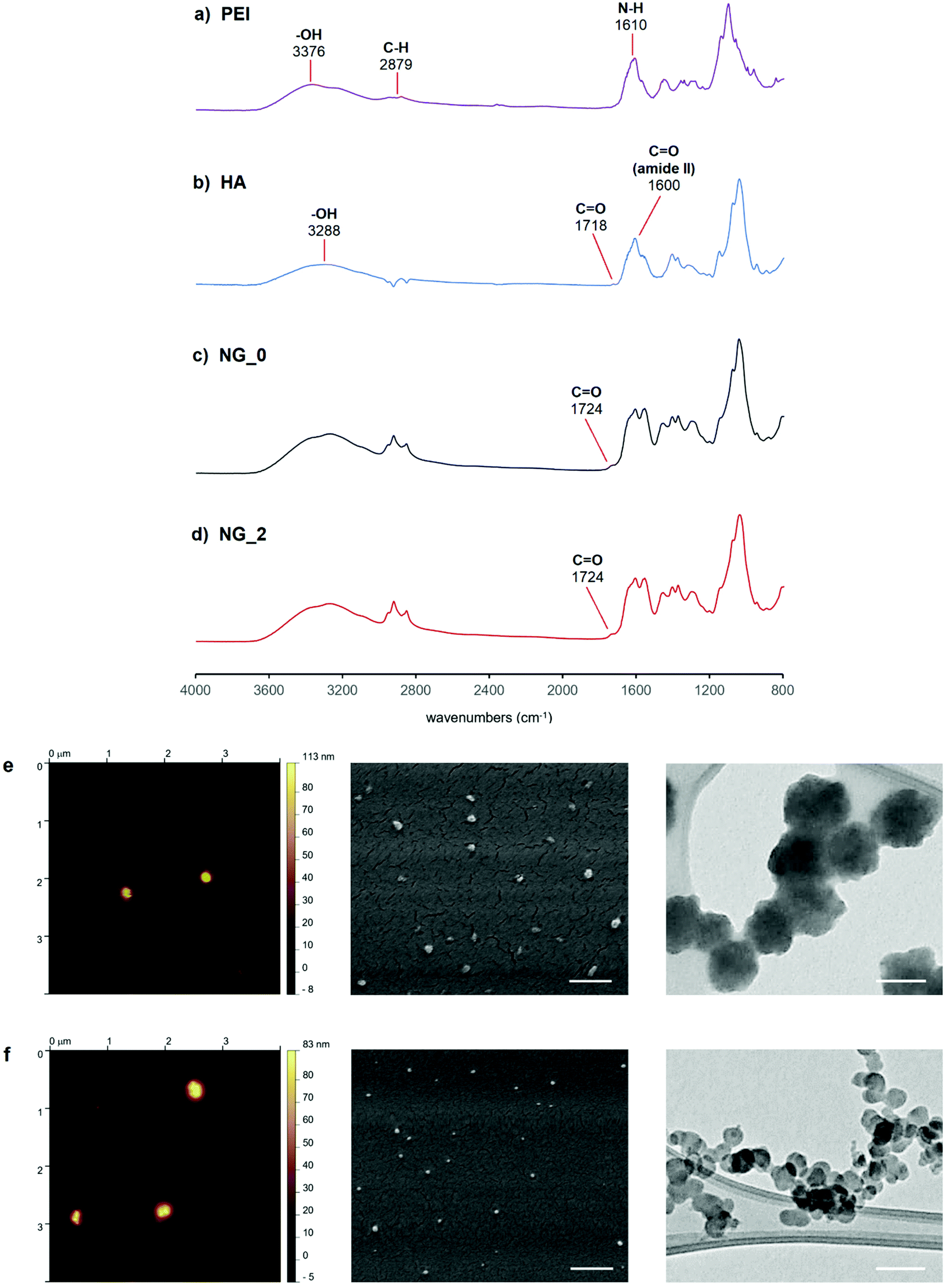 | ||
| Fig. 4 (a–d) FT-IR spectra of LPEI (a, violet), HA (b, blue), NG_0 (c, black) and NG_2 (d, red). (e and f) AFM, SEM (scale bar = 500 nm) and TEM (scale bar = 100 nm) micrographs of NG_0 (e) and NG_2 (f). | ||
NG specimens were also evaluated in terms of size and morphology through DLS, AFM, SEM and TEM analyses, in order to define a correlation between the active tuning of the flow focusing geometry and the physical features of the resulting nanoscaffolds. DLS measurements gave a mean hydrodynamic diameter of 188.3 nm for NG_0 and 92.4 nm for NG_2, indicating a volume reduction of ca. 88% between the two specimens, and a monodispersity level (PDING_0 = 0.023; PDING_2 = 0.015) far higher than that of HA-LPEI NGs produced via conventional W/O emulsion-based batch synthesis (Fig. S4 and Table S1, ESI†). Additionally, both specimens were characterized by a slightly negative ζ-potential value (NG_0: −2.2 mV; NG_2: −2.13 mV) and showed good dimensional stability in aqueous environment (Fig. S5, ESI†).
AFM analysis, conducted in air on hydrated NGs, gave NG diameter values of ca. 130 nm and 73 nm for NG_0 and NG_2, respectively. The discrepancy between DLS and AFM measurements was attributable to the different sample processing: DLS analysis was performed on fully solvated NGs and the recorded dimensions corresponded to the NG hydrodynamic diameters, whereas AFM samples did not reflect the thickness of the adsorbed water layer.62 Moreover, during sample preparation for AFM, the formation of colloidal nano-agglomerates can occur, due to the variation of the surface tension gradient during evaporation of the laid droplet (Marangoni effect).63–65 In particular, the presence of colloidal clusters was more noticeable in the AFM image of NG_2.
SEM analysis, performed on dehydrated specimens, gave diameters of ca. 92 nm for NG_0 and 57 nm for NG_2, indicating a dynamic behavior of the nanoscaffold during the forced transition from hydrated to dry state, under SEM vacuum. This confirmed the capability of the synthesized nanosystems to absorb and retain water, when dissolved in aqueous medium, which is typical of hydrogel matrices.66,67 TEM analysis showed that NGs were rough surfaced with a quasi-spherical morphology, with nanoparticle size values comparable to those observed by SEM.
Collectively, results from the physico-chemical characterization emphasize that the proposed microfluidic chip design allowed us to tune the morphological features of the resulting NGs via active modulation of the FFJ during microdroplet generation.
3.2 Drug release
DOX was chosen as a candidate drug to validate NGs as a controlled drug delivery system. DOX is an anthracycline and an effective anticancer drug, widely used in the chemotherapeutic treatment of various cancers, including lung,68 breast,69 skin (melanoma)70 and ovarian cancer.71As measured by fluorescence spectroscopy, the two NG classes showed similar drug encapsulation efficiency levels (77.7% for NG_0 and 77.5% for NG_2). Conversely, they were characterized by different drug release profiles. As reported in Fig. 5, DOX release from NG_0 specimens was almost completed after 2 days (ca. 98% of released drug), whereas NG_2 presented a more sustained release, up to 10 days. Specifically, NG_2 exhibited a slightly higher initial DOX release than NG_0, whereas after 8 h this trend was reversed. This implies that NG size and composition could affect the drug distribution between the inner core and the surface of the polymeric nano-network and, consequently, the drug diffusion regime, mainly driven by DOX–polymer interactions.
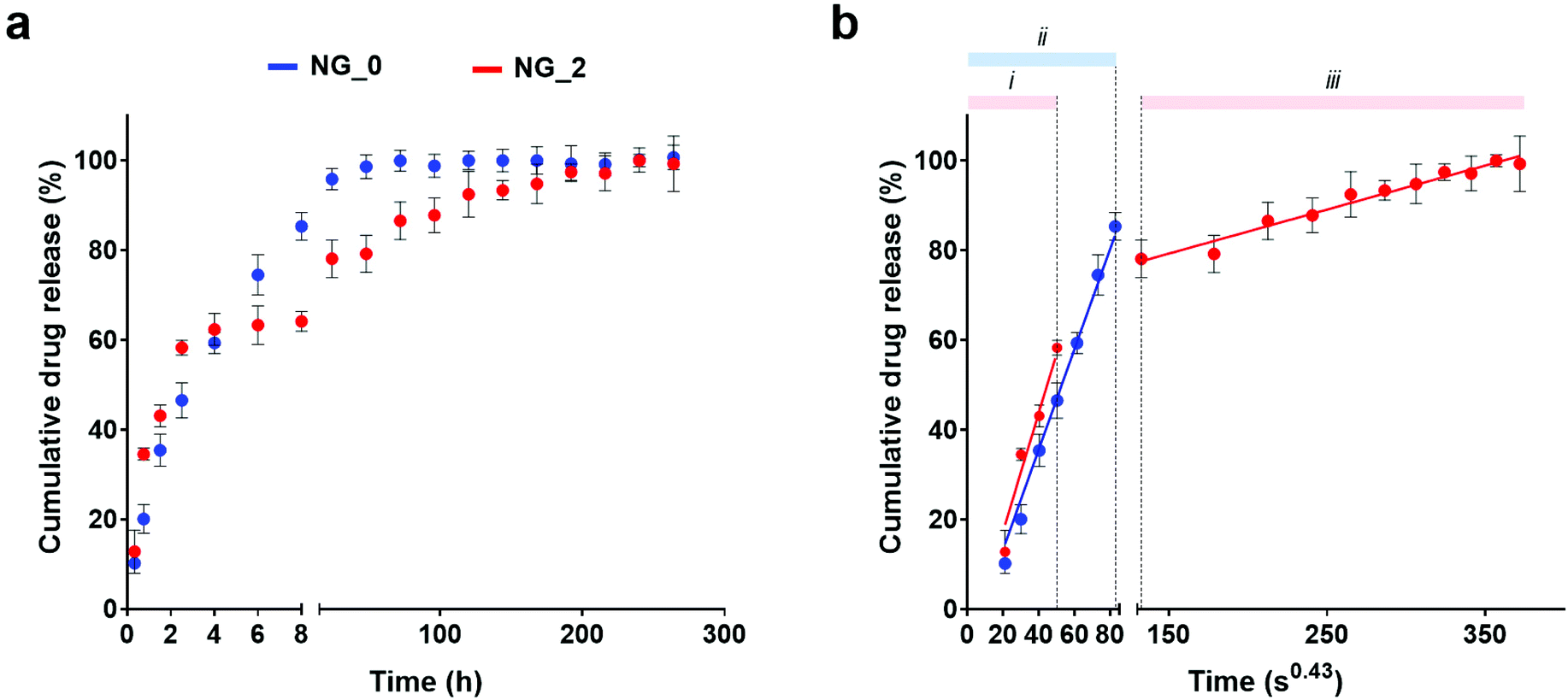 | ||
| Fig. 5 (a) Release profile of DOX from NG_0 (blue) and NG_2 (red). (b) Drug release against time expressed as t1/2.3. Good linearity of data is indicative of a Fickian diffusion process, with the slope representing the empirical kinetic constant for the DOX/NG system. Cumulative drug release is represented as a percentage of the total drug payload (mean value ± SD is plotted). | ||
An estimation of the drug diffusion regime could be performed by plotting the cumulative drug release against time to the power of 0.43 (i.e., t1/2.3, exponent typically used to approximate the spherical geometry of the nanocarriers), where a linear relationship is indicative of a Fickian diffusion. Drug release by both NG_0 and NG_2 could be described in terms of a Fickian regime, with no initial burst release in either sample. Moreover, in NG_2, data defined a double diffusion regime with different slopes, whereas in NG_0 a single linear trend was detected. The main interactions tuning the drug release are reliably attributable to the drug adsorption at the NG surface and the aliphatic–aromatic stacking occurring between DOX and HA.72–74 The smaller dimensions of NG_2 reliably led to a higher amount of DOX adsorbed at the NG outer layer, which was released initially (steeper slope of regime (i), corresponding to DOX adsorption at the NG/water interface), whereas the residual amount, entrapped in the nanomeshes, diffused more slowly (regime (iii)). This could be ascribed to the aliphatic–aromatic stacking which provided a stacked arrangement of the DOX molecules within the NG architecture, characterized by a higher amount of HA compared to NG_0, and delayed the drug release.75 On the other hand, NG_0 showed a single diffusion regime (ii) due to the larger size, which did not give a net transition between the two diffusion mechanisms: in this case, a higher DOX fraction was reliably encapsulated within NG meshes, and a lower amount of drug adsorbed at the NG surface was subjected to a quick diffusion process.
3.3 Cell viability and NG internalization
The cytocompatibility analysis and the cellular uptake of NG_0 and NG_2 were characterized using OVCA433, chosen as a representative cancer cell line. Accordingly, we tested our HA-LPEI NGs as candidate nanomaterials for the definition of a therapeutic approach for ovarian cancer, with the aim to demonstrate an improved effect compared to conventional anticancer therapies.76 A suspension of pristine NGs (without drug payload) was directly added to the cell medium and the cell viability, measured by G6PD assay on culture supernatants, was close to 97.5% after 24 h for both NGs, indicating good biocompatibility. In detail, Fig. 6a reports the high normalized viability levels of NG-treated cells showing no significant differences between the two groups. Once proven that NGs were permissive for cell survival, the internalization process of NG_0 and NG_2 was assessed by flow cytometric analysis of OVCA433 at different time points (30 min, 1 h, 2 h), after the administration of the nanomaterial at the concentration of 20 μg mL−1. We report an increase in median fluorescence intensity (MFI) levels over time, confirming the progressive NG uptake by cells. In particular, as reported in Fig. 6b, after just 30 min exposure to NGs, the distribution of cell fluorescence shifted to higher values (3.7-fold change in MFI for NG_0 and 3.5-fold change for NG_2) as a consequence of nanocarrier uptake.77 At 1 h and 2 h, further increases in MFI measurements were observed, up to ca. 13- and 17-fold change for NG_0 and NG_2 specimens, respectively.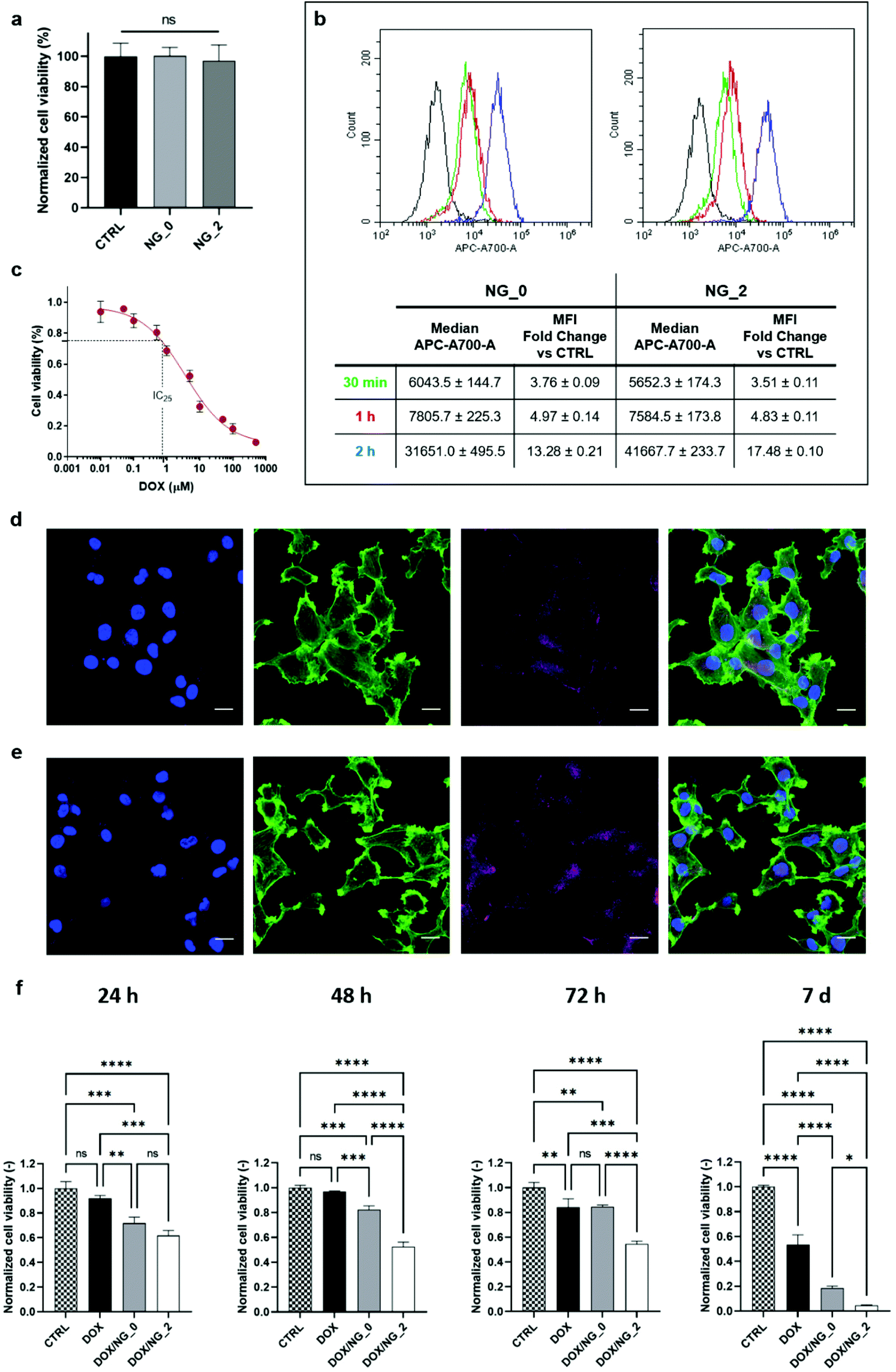 | ||
| Fig. 6 (a) Cytocompatibility of pristine NG_0 and NG_2 by G6PD assay. Viability levels have been normalized to CTRL group and expressed as mean ± SD. (b) Flow cytometric analysis of NG internalization by OVCA433 cells after 30 min (green), 1 h (red) and 2 h (blue); plot reports the shift of the allophycocyanin (APC-A700) signal for each time point in the target gate (median fluorescence intensity values are specified in the table, and fold change is calculated vs. control cells). (c) Dose–response curve of DOX using the MTT assay. The solid line represents the fitted curve according to four-parameter logistic curve, defining an IC25 of 0.8 μM. (d and e) Representative confocal micrographs of NG_0 (d, in purple, false color) and NG_2 (e, in purple, false color) internalization in OVCA433 after 30 min. Actin cytoskeleton was stained with FITC-labeled phalloidin (in green) and nuclei were counterstained with DAPI (in blue). Scale bar = 20 μm. (f) DOX-induced cytotoxicity in OVCA433 at 24 h, 48 h, 72 h and 7 days, after the administration of free-drug (DOX) and drug-loaded NGs (DOX/NG_0 and DOX/NG_2). All groups refer to a 30 min incubation with the indicated treatment. The therapeutic effect is expressed in terms of cell viability levels normalized against their internal controls, measured through the MTT assay. Results are the mean ± SD of three independent experiments. Statistical analysis was performed using one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant. | ||
Further validation of the internalization process was provided by fluorescence microscopy (Fig. 6d and e), which showed the NG distribution in the cytosol, with a progressive perinuclear localization. Overall, these results demonstrated the efficient NG uptake and indicated the potential of these nanocarriers as controlled drug delivery systems in the treatment of ovarian carcinoma. For these reasons, we evaluated the in vitro therapeutic effects of DOX delivered by both formulated NGs: in order to select an ideal sublethal drug concentration, we first estimated a preliminary dose–response curve at 24 h on OVCA433 cultured in adhesion using the MTT assay (Fig. 6c) and the value of 0.8 μM was identified as the IC25 level (i.e., the concentration at which the drug produces a 25% viability reduction). This level was chosen to set the drug payload for DOX/NG_0 and DOX/NG_2, and the NG-associated treatments were compared to the administration of the same DOX concentration in a non-encapsulated form. Fig. 6f shows the evaluation of the OVCA433 metabolic activity through MTT assay, up to 7 days: NG-mediated delivery of DOX resulted in enhanced therapeutic effect of the drug. In detail, after 24 h, both DOX/NG_0 and DOX/NG_2 effectively reduced cell viability (p < 0.001 and p < 0.0001 vs. CTRL for DOX/NG_0 and DOX/NG_2, respectively), outperforming the non-encapsulated drug (p < 0.01 and p < 0.001 vs. DOX). Moreover, the DOX release mediated by NG_2 ensured a sustained and prolonged effect on cell metabolism over time, with 5% residual viability at 1 week (p < 0.0001 vs. CTRL). A promising trend could also be observed in the case of DOX/NG_0, where cell viability decreased to 18% after 1 week (p < 0.0001 vs. CTRL). Both nanoformulations confirmed a significant cytotoxic effect vs. the free drug administration at the same time point (p < 0.0001 vs. DOX for both DOX/NG_0 and DOX/NG_2). The different results between the two NGs could be ascribed to the diverse drug release profiles in NG_0 and NG_2: indeed, considering the similar MFI fold change for both specimens, NG_2 were characterized by a prolonged DOX release mediated by the aliphatic–aromatic stacking, which led to a more sustained intracellular drug delivery over time, improving the DOX cytotoxic effects. Overall, our results confirmed the efficacy of NG-encapsulated DOX, demonstrating a clear advantage of NGs as carriers of the chemotherapeutic agent. As a further validation activity, we also proved that our microfluidics-enabled NGs outperformed control HA-LPEI NGs produced via conventional W/O batch emulsion process (Fig. S6 and S7, ESI†) both in terms of sustained drug release and therapeutic efficacy. This strengthens the concept that the tunable microfluidic process disclosed in the present work augments the control over NG formulations beyond the bare physical and morphological features, also dictating their drug release and therapeutic performances.
4. Conclusion
Several bulk methods have been described in the literature to synthesize polymeric nanocarriers for controlled drug delivery purposes. However, these procedures are generally characterized by significant constraints, such as limited control over the reaction parameters, difficult optimization of the colloidal properties of the nanosystems, and scarce lot-to-lot reproducibility, which have hampered their potential eligibility as therapies in nanomedicine. In this scenario, microfluidic platforms represent a valid alternative to modulate and control the dimensions and the physico-chemical properties of the nanocarriers toward a more precise and versatile nanomaterial design. In this work, we propose the synthesis of nanogels in a microfluidic chip endowed with a pneumatic micro-actuator, exploiting the active formation of a W/O microemulsion: each generated microdroplet acted as a reaction micro-chamber, where the polymer crosslinking occurred, giving rise to the NG architecture. Thanks to the pneumatic actuator integrated in the chip design, we ensured an active tuning of the flow-focusing geometry, resulting in an on-demand modulation of the microdroplet diameter, which in turn resulted in augmented control over the NG size and monodispersity. The effects of our droplet-based microfluidic process positively impacted the NG drug delivery performances, since modulation of NG size affected the resulting drug release profile, with smaller NGs showing a more sustained DOX release.Microfluidics-enabled NGs resulted in a promising nanocarrier for cancer therapy: as a preliminary validation, we evaluated the in vitro administration of DOX-loaded NGs to ovarian cancer cells, and we demonstrated a high cell internalization of the nanomaterials, which boosted the therapeutic effects of the drug compared to the administration of non-encapsulated DOX. Overall, our results testify an advancement in NG design, and represent a preliminary basis for the definition of a continuous in-flow production of nanogel carriers, which could support an effective clinical translation and meet the needs for ameliorated drug release performances in healthcare scenarios still lacking an efficient treatment.
Author contributions
S. M. G. and E. L.: investigation, formal analysis, visualization, writing – original draft; P. M., F. P. and X. H.: investigation; formal analysis; F. A., F. B. and D. D. R.: investigation; S. S.: supervision, writing – review & editing; L. R.: supervision, funding acquisition; F. R., M. T., G. G. and Z. J. Z.: supervision, resources; E. M.: conceptualization, methodology, validation, formal analysis, visualization, writing – original draft, writing – review & editing; A. R.: supervision, methodology, validation, writing – original draft, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been partially supported by Regione Lazio – POR FESR Lazio 2014–2020 “Gruppi di Ricerca 2020”, proposal ID A0375-2020-36596 (ORGANOVA), CUP: B85F21001500002. P. M., G. G. and A. R. are thankful to “Tecnopolo per la medicina di precisione” (TecnoMed Puglia) – Regione Puglia: DGR no. 2117 dated 21/11/2018, CUP: B84I18000540002 and “Tecnopolo di Nanotecnologia e Fotonica per la Medicina di Precisione” (TECNOMED) – FISR/MIUR – CNR: delibera CIPE no. 3449 dated 07/08/2017, CUP: B83B17000010001.References
- B. L. Banik, P. Fattahi and J. L. Brown, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 271–299 Search PubMed.
- S. Sur, A. Rathore, V. Dave, K. R. Reddy, R. S. Chouhan and V. Sadhu, Nano-Struct. Nano-Objects, 2019, 20, 100397 CrossRef CAS.
- S. Indoria, V. Singh and M.-F. Hsieh, Int. J. Pharm., 2020, 582, 119314 CrossRef CAS PubMed.
- Y. Li, T. Thambi and D. S. Lee, Adv. Healthcare Mater., 2018, 7, 1700886 CrossRef PubMed.
- M.-Q. Wang, H. Zou, W.-B. Liu, N. Liu and Z.-Q. Wu, ACS Macro Lett., 2022, 11, 179–185 CrossRef CAS PubMed.
- R. Sun, Q. Luo, X. Li, X. Huang, L. Teng, Z. Shen and W. Zhu, Mater. Today Nano, 2021, 14, 100115 CrossRef CAS.
- E. Mauri, S. Papa, M. Masi, P. Veglianese and F. Rossi, Expert Opin. Drug Delivery, 2017, 14, 1305–1313 CrossRef CAS PubMed.
- W. Song, M. Das, Y. Xu, X. Si, Y. Zhang, Z. Tang and X. Chen, Mater. Today Nano, 2019, 5, 100029 CrossRef.
- W.-B. Liu, S.-M. Kang, X.-H. Xu, L. Zhou, N. Liu and Z.-Q. Wu, ACS Appl. Bio Mater., 2020, 3, 5620–5626 CrossRef CAS PubMed.
- S.-Q. Zhao, G. Hu, X.-H. Xu, S.-M. Kang, N. Liu and Z.-Q. Wu, ACS Macro Lett., 2018, 7, 1073–1079 CrossRef CAS PubMed.
- Y. Liu and X. Jiang, Lab Chip, 2017, 17, 3960–3978 RSC.
- C. Wischke, Int. J. Pharm., 2020, 584, 119401 CrossRef CAS PubMed.
- J. Ahn, J. Ko, S. Lee, J. Yu, Y. Kim and N. L. Jeon, Adv. Drug Delivery Rev., 2018, 128, 29–53 CrossRef CAS PubMed.
- Q. Feng, J. Sun and X. Jiang, Nanoscale, 2016, 8, 12430–12443 RSC.
- Q. Ma, J. Cao, Y. Gao, S. Han, Y. Liang, T. Zhang, X. Wang and Y. Sun, Nanoscale, 2020, 12, 15512–15527 RSC.
- E. Mauri, S. M. Giannitelli, M. Trombetta and A. Rainer, Gels, 2021, 7, 36 CrossRef CAS PubMed.
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Nano Lett., 2008, 8, 2906–2912 CrossRef CAS PubMed.
- S. Garg, G. Heuck, S. Ip and E. Ramsay, J. Drug Targeting, 2016, 24, 821–835 CrossRef CAS PubMed.
- W. Li, Q. Chen, T. Baby, S. Jin, Y. Liu, G. Yang and C.-X. Zhao, Chem. Eng. Sci., 2021, 235, 116468 CrossRef CAS.
- S. I. Hamdallah, R. Zoqlam, P. Erfle, M. Blyth, A. M. Alkilany, A. Dietzel and S. Qi, Int. J. Pharm., 2020, 584, 119408 CrossRef CAS PubMed.
- E. Dashtimoghadam, H. Mirzadeh, F. A. Taromi and B. Nyström, Polymer, 2013, 54, 4972–4979 CrossRef CAS.
- F. Peng, L. K. Månsson, S. H. Holm, S. Ghosh, G. Carlström, J. J. Crassous, P. Schurtenberger and J. O. Tegenfeldt, J. Phys. Chem. B, 2019, 123, 9260–9271 CrossRef CAS PubMed.
- N. E. Clay, J. J. Whittenberg, J. Leong, V. Kumar, J. Chen, I. Choi, E. Liamas, J. M. Schieferstein, J. H. Jeong, D. H. Kim, Z. J. Zhang, P. J. A. Kenis, I. W. Kim and H. Kong, Nanoscale, 2017, 9, 5194–5204 RSC.
- J. Ma, S. M.-Y. Lee, C. Yi and C.-W. Li, Lab Chip, 2017, 17, 209–226 RSC.
- S. J. Shepherd, D. Issadore and M. J. Mitchell, Biomaterials, 2021, 274, 120826 CrossRef CAS PubMed.
- S. Sohrabi, N. Kassir and M. Keshavarz Moraveji, RSC Adv., 2020, 10, 27560–27574 RSC.
- M. V. Bandulasena, G. T. Vladisavljević and B. Benyahia, Chem. Eng. Sci., 2019, 195, 657–664 CrossRef CAS.
- J. W. Choi, Y. J. Kim, J. M. Lee, J.-H. Choi, J.-W. Choi and B. G. Chung, BioChip J., 2020, 14, 298–307 CrossRef CAS.
- D.-Y. Kim, S. H. Jin, S.-G. Jeong, B. Lee, K.-K. Kang and C.-S. Lee, Sci. Rep., 2018, 8, 8525 CrossRef PubMed.
- S. Rezvantalab and M. K. Moraveji, RSC Adv., 2019, 9, 2055–2072 RSC.
- N. Visaveliya and J. M. Köhler, ACS Appl. Mater. Interfaces, 2014, 6, 11254–11264 CrossRef CAS PubMed.
- P. Shrimal, G. Jadeja and S. Patel, Chem. Eng. Res. Des., 2020, 153, 728–756 CrossRef CAS.
- E. Chiesa, R. Dorati, S. Pisani, B. Conti, G. Bergamini, T. Modena and I. Genta, Pharmaceutics, 2018, 10, 267 CrossRef CAS PubMed.
- Z. Z. Chong, S. H. Tan, A. M. Gañán-Calvo, S. B. Tor, N. H. Loh and N.-T. Nguyen, Lab Chip, 2016, 16, 35–58 RSC.
- C.-Y. Lee, Y.-H. Lin and G.-B. Lee, Microfluid. Nanofluid., 2009, 6, 599–610 CrossRef CAS.
- G. Soni and K. S. Yadav, Saudi Pharm. J., 2016, 24, 133–139 CrossRef PubMed.
- M. A. Qureshi and F. Khatoon, J. Sci.: Adv. Mater. Devices, 2019, 4, 201–212 Search PubMed.
- E. Mauri, G. Perale and F. Rossi, ACS Appl. Nano Mater., 2018, 1, 6525–6541 CrossRef CAS.
- Y. Wang, S.-L. Qiao and H. Wang, ACS Appl. Nano Mater., 2018, 1, 785–792 CrossRef CAS.
- J. H. Kim, M. J. Moon, D. Y. Kim, S. H. Heo and Y. Y. Jeong, Polymers, 2018, 10, 1133 CrossRef PubMed.
- J. M. Rios de la Rosa, P. Pingrajai, M. Pelliccia, A. Spadea, E. Lallana, A. Gennari, I. J. Stratford, W. Rocchia, A. Tirella and N. Tirelli, Adv. Healthcare Mater., 2019, 8, 1901182 CrossRef CAS PubMed.
- A. Tirella, K. Kloc-Muniak, L. Good, J. Ridden, M. Ashford, S. Puri and N. Tirelli, Int. J. Pharm., 2019, 561, 114–123 CrossRef CAS PubMed.
- Z. Luo, Y. Dai and H. Gao, Acta Pharm. Sin. B, 2019, 9, 1099–1112 CrossRef PubMed.
- Y. Zou, D. Li, M. Shen and X. Shi, Macromol. Biosci., 2019, 19, 1900272 CrossRef CAS PubMed.
- T. Bus, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2018, 6, 6904–6918 RSC.
- M. Costantini, J. Jaroszewicz, Ł. Kozoń, K. Szlązak, W. Święszkowski, P. Garstecki, C. Stubenrauch, A. Barbetta and J. Guzowski, Angew. Chem., Int. Ed., 2019, 58, 7620–7625 CrossRef CAS PubMed.
- E. Mauri, P. Veglianese, S. Papa, A. Mariani, M. De Paola, R. Rigamonti, G. M. F. Chincarini, I. Vismara, S. Rimondo, A. Sacchetti and F. Rossi, RSC Adv., 2017, 7, 30345–30356 RSC.
- M. Gori, S. M. Giannitelli, M. Torre, P. Mozetic, F. Abbruzzese, M. Trombetta, E. Traversa, L. Moroni and A. Rainer, Adv. Healthcare Mater., 2020, 9, 2001163 CrossRef CAS PubMed.
- A. R. Abate, M. B. Romanowsky, J. J. Agresti and D. A. Weitz, Appl. Phys. Lett., 2009, 94, 023503 CrossRef.
- P. Stolzenburg, T. Lorenz, A. Dietzel and G. Garnweitner, Chem. Eng. Sci., 2018, 191, 500–510 CrossRef CAS.
- L. Xu, J. Peng, M. Yan, D. Zhang and A. Q. Shen, Chem. Eng. Process., 2016, 102, 186–193 CrossRef CAS.
- L. Zhang, Q. Chen, Y. Ma and J. Sun, ACS Appl. Bio Mater., 2020, 3, 107–120 CrossRef CAS PubMed.
- L. Capretto, D. Carugo, S. Mazzitelli, C. Nastruzzi and X. Zhang, Adv. Drug Delivery Rev., 2013, 65, 1496–1532 CrossRef CAS PubMed.
- L. Wang and J. Wang, Nanoscale, 2019, 11, 16708–16722 RSC.
- L.-H. Hung, S.-Y. Teh, J. Jester and A. P. Lee, Lab Chip, 2010, 10, 1820–1825 RSC.
- Z. Mahdavi, H. Rezvani and M. Keshavarz Moraveji, RSC Adv., 2020, 10, 18280–18295 RSC.
- C. B. Roces, D. Christensen and Y. Perrie, Drug Delivery Transl. Res., 2020, 10, 582–593 CrossRef CAS PubMed.
- G. M. Son, H. Y. Kim, J. H. Ryu, C. W. Chu, D. H. Kang, S. B. Park and Y.-I. Jeong, Int. J. Mol. Sci., 2014, 15, 16057–16068 CrossRef PubMed.
- B. Zhou, L. Zheng, C. Peng, D. Li, J. Li, S. Wen, M. Shen, G. Zhang and X. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 17190–17199 CrossRef CAS PubMed.
- J. P. Quiñones, O. Brüggemann, C. P. Covas and D. A. Ossipov, Carbohydr. Polym., 2017, 173, 157–169 CrossRef PubMed.
- C. Muzzarelli, V. Stanic, L. Gobbi, G. Tosi and R. A. A. Muzzarelli, Carbohydr. Polym., 2004, 57, 73–82 CrossRef CAS.
- C. M. Maguire, M. Rösslein, P. Wick and A. Prina-Mello, Sci. Technol. Adv. Mater., 2018, 19, 732–745 CrossRef CAS PubMed.
- J. Ren, A. Crivoi and F. Duan, Langmuir, 2020, 36, 15064–15074 CrossRef CAS PubMed.
- R. Su, S. H. Park, Z. Li and M. C. McAlpine, in Robotic Systems and Autonomous Platforms, ed. S. M. Walsh and M. S. Strano, Woodhead Publishing, 2019, pp. 309–334 Search PubMed.
- C. Zhang and P. Akcora, RSC Adv., 2017, 7, 18321–18326 RSC.
- K. Horigome, T. Ueki and D. Suzuki, Polym. J., 2016, 48, 273–279 CrossRef CAS.
- M. J. Garcia-Salinas and A. M. Donald, J. Colloid Interface Sci., 2010, 342, 629–635 CrossRef CAS PubMed.
- Y. Hong, S. Che, B. Hui, Y. Yang, X. Wang, X. Zhang, Y. Qiang and H. Ma, Biomed. Pharmacother., 2019, 112, 108614 CrossRef CAS PubMed.
- G. Zheng, M. Zheng, B. Yang, H. Fu and Y. Li, Biomed. Pharmacother., 2019, 116, 109006 CrossRef CAS PubMed.
- N. S. V. Capanema, I. C. Carvalho, A. A. P. Mansur, S. M. Carvalho, A. P. Lage and H. S. Mansur, ACS Appl. Nano Mater., 2019, 2, 7393–7408 CrossRef CAS.
- S. Eetezadi, J. C. Evans, Y.-T. Shen, R. De Souza, M. Piquette-Miller and C. Allen, Mol. Pharm., 2018, 15, 472–485 CrossRef CAS PubMed.
- D. B. Ninković, D. Z. Vojislavljević-Vasilev, V. B. Medaković, M. B. Hall, E. N. Brothers and S. D. Zarić, Phys. Chem. Chem. Phys., 2016, 18, 25791–25795 RSC.
- P. K. C. Paul, Cryst. Eng., 2002, 5, 3–8 CrossRef CAS.
- E. M. Cabaleiro-Lago and J. Rodríguez-Otero, ACS Omega, 2018, 3, 9348–9359 CrossRef CAS PubMed.
- E. Limiti, P. Mozetic, S. M. Giannitelli, F. Pinelli, X. Han, D. Del Rio, F. Abbruzzese, F. Basoli, L. Rosanò, S. Scialla, M. Trombetta, G. Gigli, Z. J. Zhang, E. Mauri and A. Rainer, ACS Appl. Nano Mater., 2022, 5, 5544–5557 CrossRef CAS.
- A. Spadea, J. M. Rios de la Rosa, A. Tirella, M. B. Ashford, K. J. Williams, I. J. Stratford, N. Tirelli and M. Mehibel, Mol. Pharm., 2019, 16, 2481–2493 CrossRef CAS PubMed.
- G. Kannan, S. P. Kambhampati and S. R. Kudchadkar, J. Controlled Release, 2017, 263, 192–199 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr00827k |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2022 |