
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-surface synthesis of Mn-phthalocyanines with optically active ligands†
Amelia
Domínguez-Celorrio
 ab,
Carlos
Garcia-Fernandez
cd,
Sabela
Quiroga
e,
Peter
Koval
f,
Veronique
Langlais
b,
Diego
Peña
e,
Daniel
Sánchez-Portal
cd,
David
Serrate
*agh and
Jorge
Lobo-Checa
*ag
ab,
Carlos
Garcia-Fernandez
cd,
Sabela
Quiroga
e,
Peter
Koval
f,
Veronique
Langlais
b,
Diego
Peña
e,
Daniel
Sánchez-Portal
cd,
David
Serrate
*agh and
Jorge
Lobo-Checa
*ag
aInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza 50009, Spain. E-mail: serrate@unizar.es; jorge.lobo@csic.es
bCentre d'Elaboration de Materiaux et d'Etudes Structurales – Centre National de la Recherche Scientifique, Toulouse, France
cCentro de Física de Materiales CSIC/UPV-EHU-Materials Physics Center, Manuel Lardizabal 5, E-20018 San Sebastián, Spain
dDonostia International Physics Center, Paseo Manuel de Lardizabal 4, E-20018 San Sebastian, Spain
eCentro de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, Santiago de Compostela 15782, Spain
fSimune Atomistics S.L., Avenida Tolosa 76, 20018, San Sebastian, Spain
gDepartamento de Física de la Materia Condensada, Universidad de Zaragoza, E-50009 Zaragoza, Spain
hLaboratorio de Microscopías Avanzadas, Universidad de Zaragoza, E-50018, Zaragoza, Spain
First published on 27th April 2022
Abstract
The synthesis of novel organic prototypes combining different functionalities is key to achieve operational elements for applications in organic electronics. Here we set the stage towards individually addressable magneto-optical transducers by the on-surface synthesis of optically active manganese-phthalocyanine derivatives (MnPc) obtained directly on a metallic substrate. We created these 2D nanostructures under ultra-high vacuum conditions with atomic precision starting from a simple phthalonitrile precursor with reversible photo-induced reactivity in solution. These precursors maintain their integrity after powder sublimation and coordinate with the Mn ions into tetrameric complexes and then transform into MnPcs on Ag(111) after a cyclotetramerization reaction. Using scanning tunnelling microscopy and spectroscopy together with DFT calculations, we identify the isomeric configuration of two bi-stable structures and show that it is possible to switch them reversibly by mechanical manipulation. Moreover, the robust magnetic moment brought by the central Mn ion provides a feasible pathway towards magneto-optical transducer fabrication. This work should trigger further research confirming such magneto-optical effects in MnPcs both on surfaces and in liquid environments.
I. Introduction
Organic electronic devices are nowadays an industrial reality due to their high efficiency, low-cost and synthetic design tunability.1–3 Among them, phthalocyanines (Pcs) and porphyrins stand out as prototypical examples of organic semiconductor building units due to their electric, magnetic and optical adjustability by metallation of their central cores.4 These metal–organic complexes are used as pigment catalysts and photo-sensitive semiconductors besides being promising candidates for molecular spintronic applications.5,6When organic complexes are adsorbed on surfaces, important interfacial interactions emerge that influence their electronic and magnetic properties.5–7 In the case of metallated Pcs and porphyrins with first-row transition-metal (TM) ions, significant conformational and spin state modifications have been reported.7–10 These are strongly dependent on the local adsorption sites of metal centers and the arrangement of their external ligands, which alter the relative position of the metal center with respect to the π-conjugated macrocyclic plane. Similarly, selective adsorption of external and axial ligands on the metal centers modify these metal–organic complex properties.11–14 Such local variability limits the scalability of single-molecule device production since building blocks require dynamical control and operational reversibility while maintaining their overall chemical and physical environments.
A way to overcome these local conformation limitations could be inserting active molecular switches into molecular complexes so that morphological control can be exerted using external stimuli, in the form of photons,15,16 focused electric fields17,18 or electric currents.19,20 Light illumination is particularly simple to technically control, but demands that the spin centers of molecular complexes are strongly coupled to switchable ligands whose conformations can be changed by photon adsorption. The synthesis and deposition on the surfaces of such magnetic and optically active molecules by direct thermal evaporation is still pending, as it is compromised by the structural fragility of these metal–organic complexes. Such molecular integrity limitation has prevented its fabrication and implementation into magneto-optical transducers.
In this work, we achieved an adequate strategy to create metal–organic complexes by on-surface synthesis (OSS) featuring optically active moieties. In particular, we generated active Pcs with a central magnetic atom by means of cyclotetramerization of a functionalized phthalonitrile precursor on the Ag(111) surface. We chose Mn atoms as metallic centers based on their expected magnetic response that is evidenced in their reported Kondo signals on Ag substrates.21,22 We find that the produced manganese phthalocyanines (MnPcs) have the central magnetic ions coupled to four diarylethylene (DAE) moieties. These MnPcs on Ag(111) show at each branch two possible conformations that we image and study electronically by scanning tunneling microscopy (STM) and spectroscopy (STS) techniques. Density functional theory (DFT) calculations allow us to identify the relevant configurations of the open and closed DAE isomers on the Ag(111) surfaces, and confirm the robust magnetic moment localized at the Mn ion. Notably, we induce reversible changes between the open and closed conformations by lateral tip-molecule manipulation. Although magneto-optical experiments were not attempted here, this OSS process ensures that each MnPc we formed contains four switchable branches.
II. Results and discussion
Optical activity of precursor 1
As a molecular precursor we designed and synthesized 4,5-bis(benzo[b]thiophen-3-yl)phthalonitrile that we name hereafter compound 1a. This compound was obtained in two steps by solution chemistry from commercially available compounds (see the ESI† for details). Compound 1a includes two vicinal nitrile (CN) groups to induce the on-surface cyclotetramerization reaction leading to phthalocyanine. It presents two vicinal benzothiophene groups forming a diarylethene moiety that should be involved in a reversible electrocyclization reaction leading to compound 1b (closed isomer), i.e. these moieties are introduced as an active switchable structure.23When a UV light of 365 nm wavelength irradiates a solution of compound 1a in CH2Cl2, the optical activity is photochromatically evident: the initial colorless solution corresponding to the open isomer 1a changes into blue as these molecules transform into the closed isomer 1b (Fig. 1a). The photochemically induced electrocyclic reaction involves six π-electrons, and therefore, according to the Woodward–Hoffmann rules the cyclization should be conrotatory, giving product trans-1b. The isomerization is reversible since the solution changes back to colorless once the UV irradiation is stopped (see ESI Fig. S1†).
 | ||
| Fig. 1 Molecular precursors and MnPc complexes of this work. (a) Scheme of the reversible reaction between open 1a and closed 1b isomers. (b) Molecular structure of the fully conjugated and non-switchable precursor dicyanonaphthalene 2 used for verification. (c) Molecular structure of the MnPcs synthesized by cyclotetramerization reactions. Complex 3 is generated from 1 and presents four switchable DAE moieties, whereas 4 is a rigid MnPc produced from 2. Green shading marks the identical phthalocyanine rings containing the central Mn, while blue denotes the bisbenzothiophene moieties. | ||
Dehydrogenation of the closed isomer 1b leads to the formation of conjugated dicyanonaphthalene 2 (Fig. 1b), which does not vary its color in solution when irradiated with the same UV light. Its unique isomeric configuration and planar geometry will be used as a control to ensure the presence of the optically active ligands of 1.
Integrity and isomeric configurations of 1 and 2 upon Mn co-deposition on Ag(111)
Isomeric molecules exhibiting active reversible switching have already been reported on metal surfaces responding to different stimuli, e.g. photons,15,16 electrons19,20 or electric fields.17,18 These delicate ligand groups can withstand UHV evaporation whenever the molecules are small, since this permits sublimation at low enough temperatures that preserve their integrity. By contrast, larger molecules containing heavier metal centers tend to break apart at the active (and weakest) end groups.The pathway that could lead to the generation of magneto-optical transducers requires to fulfill two conditions: first, deposition of optically active precursors that preserve their moiety integrity and, second, their combination with magnetic atoms. To achieve this, we evaporated small, optically active precursor molecules under UHV conditions so they self-assemble with Mn atoms on Ag(111). This surface is then used to catalyze a more complex structure by OSS. In order to prevent the Mn from clustering, we simultaneously deposited Mn atoms and precursor 1a on a Ag(111) surface kept at 50 °C (see the Methods section in the ESI†). As shown in Fig. 2a, squared-lattice islands are formed by the self-assembly of tetramer units that consist of Mn atoms (imaged as dark depressions) coordinated to four molecules by their cyano groups.24,25 These metal–organic tetramers are quite stable since they conserve their integrity after lateral atomic manipulation with an STM tip (see the inset of Fig. 2a). The islands of Mn + four 1a ligands are stabilized by the formation of hydrogen bonds with adjacent molecules (see Fig. S2†). In the coordinated islands, STM topography reveals two types of asymmetric ligands with different external lobes, also found for other diaryl derivatives on Ag(111).26 For convenience, we will refer to the brighter lobes as A-configuration and the darker lobes as B-configuration (cf. red and green arrows in the inset of Fig. 2a). These lobes are randomly distributed and for an island with 452 molecules of 1 we find an A/B ratio of 0.72, as if the interactions between neighboring tetramer units were independent of their external conformations. This randomness suggests that both configurations are structurally very similar, as expected from the two isomers 1a and 1b sketched in Fig. 1a.
 | ||
| Fig. 2 Representative STM images of the metal–organic structures. (a) Survey and closeups of precursors 1a (a) and 2 (b) co-evaporated with Mn atoms on Ag(111). Each Mn atom is surrounded by four ligands forming stable metal–organic coordination units. In contrast to (b), the Mn + 1 units cluster into islands (see the molecular model in Fig. S2†) where each complex with four ligands can be manipulated and extracted without damage (cf. inset). The DAE moieties present two types of lobes (named A and B) which are marked by red and green arrows in the inset. Post-annealing the above structures to ∼300 °C and ∼350 °C induces cyclotetramerization of the ligands forming the MnPcs 3 (c) and 4 (d), respectively. As evidenced in the insets, the Pc core in 3 displays branches with two distinct appearances, whereas 4 is fully conjugated and structurally flat in comparison (see bond-resolved STM images in Fig. S3†). STM images: (a) 40 × 40 nm2, 0.6 V, 60 pA, inset (a) 3.5 × 3.5 nm2, 0.6 V, 60 pA; (b) 40 × 40 nm2, −1 V, 100 pA, inset (b) 3.5 × 3.5 nm2, −1 V, 100 pA; (c) 50 × 50 nm2, 10 mV, 50 pA, inset (c) 2.5 × 2.5 nm2, 0.4 V, 100 pA; and (d) 50 × 50 nm2, 10 mV, 50 pA, inset (d) 2.5 × 2.5 nm2, −0.4 V, 20 pA. | ||
An identical process of co-depositing Mn + compound 2 on Ag(111) leads to very different structures. The similarity with the previous metal–organic complex is restricted to the formation of coordinated units of Mn atoms and four molecules by their cyano groups24,25 (cf.Fig. 2b). However, these complexes are monodispersed and do not self-assemble into islands. Moreover, the ligands do not present visible differences in the external groups. We assign the absence of island aggregation to the lack of the two additional hydrogen atoms at their external DAE moieties compared to 1a and to the enhanced structural conjugation of the 2 precursors. All these findings confirm the molecular integrity of both 1 and 2 on the Ag(111) surface and the coordination with Mn into tetramer units that sets the stage for the MnPc formation by OSS.
MnPc formation by cyclotetramerization of metal–organic complexes
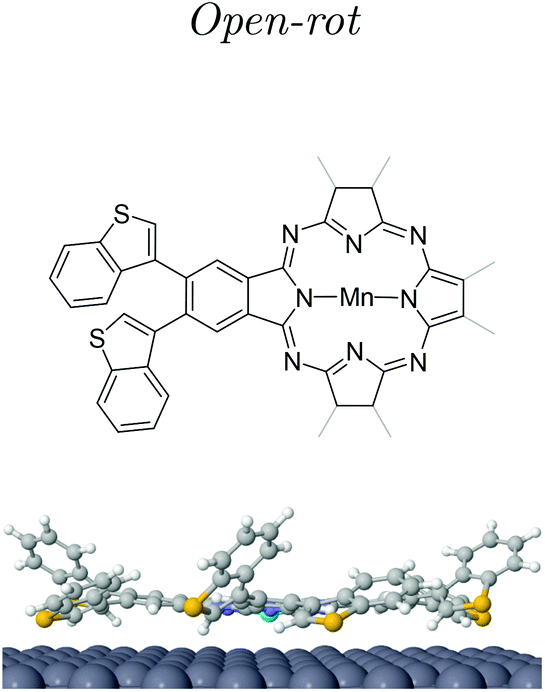
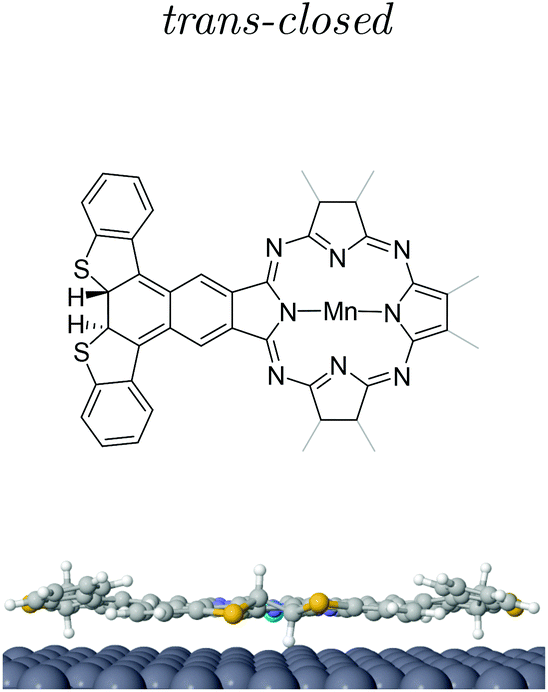
Post-annealing of these Mn complexes leads to the cyclotetramerization process of the ligands around the Mn atom24,25,27 (see the Methods section in the ESI†). The cross shape observed in STM (cf. inset Fig. 2c) follows the MnPc structure sketched in Fig. 1c with four branches associated with the precursor molecules. Interestingly, the central Mn is now imaged as a bright protrusion, implying a significant electronic reconfiguration after the annealing process. The synthesized switchable MnPcs (3) still arrange into squared self-assembled islands (Fig. 2c), whereas the conjugated MnPcs (4) continue to be spread out on the surface (Fig. 2d). This difference should relate once again to the presence or absence of hydrogen atoms at the bisbenzothiophene moieties.Focusing on 3, from the high resolution images of individual molecules extracted from the island by atomic manipulation (Fig. 3a–d), we still identify two apparent configurations of the peripheral DAE groups. We keep naming A for the brighter lobes (e.g. top branch in Fig. 2c inset) and B for the darker ones. These lobes must correspond to different benzothiophene arrangements since these two configurations are randomly present in the MnPc branches. These MnPcs (3) are far from being planar, with A lobes being further corrugated than B lobes. Indeed, recording constant-height and bond-resolved images with a functionalised CO-tip (see Fig. S3†) is only feasible by vertically adjusting the tip height for each kind of lobe. Interestingly, the external branches in 3 do not show mirror symmetry with respect to their adjacent pyrrole ring (see Fig. 3). The superimposed chemical structure on a switchable MnPc with two A and two B branch configurations duplicated in Fig. 3c and d includes the branch configurations with the lowest energies (see DFT calculations in Table 1). The open-rot configuration fails when fitting the underlying topographic STM footprints both for A and B lobes at the red arrows (cf. Fig. S4†). In contrast, the molecular models for open and closed configurations in Fig. 3d satisfactorily reproduce the observed topography.
 | ||
| Fig. 3 High resolution imaging and DFT structural models of 3. (a)–(c) STM images with a functionalized tip (2.5 × 2.5 nm2, −50 mV, 20 pA) of three different switchable MnPcs (3) isolated by lateral atomic manipulation (manipulation set point 5 mV and 15–40 nA). (d) is duplicated from (c) to compare two different superimposed molecular models from the three DFT branch configurations (see Table 1 and Fig. S4†). The positions where the model does not fit their underlying topographic STM footprints are indicated by red arrows in (c). This corresponds to the open-rot, which does neither fit A nor B lobes (cf. Fig. S4†). The correct molecular structure is overlaid in (d). The perpendicular dotted lines in (a) and (b) indicate the symmetrical axis expected for square-planar molecules. | ||
| Isomeric MnPc | Total energy (eV) | Total magnetic moment (μB) | Δhmax (Å) | ΔzMn (Å) |
|---|---|---|---|---|
| Open | 0 | 1.91 | 0.00 | 0.00 |
| Open-rot | 0.02 | 1.95 | 0.90 | 0.01 |
| trans-Closed | 0.67 | 1.89 | −1.50 | −0.04 |

|
|
|
||
By contrast, the cyclotetramerized molecules 4 in Fig. 2d† present four similar and symmetric branches with a rather homogenous intensity distribution. The formation of a cyclodehydrogenated MnPc with its expected planar structure is confirmed by bond resolved STM images (see Fig. S3†), corroborating the cyclotetramerization reaction process of these MnPcs on the surface. Moreover, the differences in the STM images of 3 confirms the integrity of DAE moieties in the latter, based on the A/B branch lobe detection that maintains a bimodal distribution.
Electronic structure by STS of the switchable MnPc
The identified A/B conformers in the four branches of 3 must have associated with different electronic states, even if they are isomers. To confirm this, we acquired constant height differential tunneling conductance (dI/dV) spectra at selected positions on several individual molecules extracted from a compact island (see Fig. 4a). For the B lobe (darker branch), we observed two occupied states at −1.3 V and −0.3 V, an increase of the conductance around 0.41 V and two clearly resolved peaks at 1.6 V and 2.3 V. This is in contrast to the rather featureless A branch spectrum in this energy range, which presents its dominant peak at 3.1 V (see ESI Fig. S5†). Such electronic differences between branches are evident in the topographic images and the dI/dV map at −1.3 V (Fig. 4b and e). The HOMO–LUMO gap is significantly smaller for the B branch (0.7 eV) than for the A branch, as can be expected for the closed isomer. Interestingly, the Mn spectrum is independent of the configuration of the branches and presents a sharp peak right below the Fermi level with a ∼50 mV linewidth and a broad resonance at 2.3 V. | ||
| Fig. 4 Experimental and calculated electronic structures of 3. (a) Position dependent dI/dV spectra (Vmod = 10 mV) recorded at representative points on several isomeric MnPcs: Mn is shown in violet, the A lobe in red, the B branch in green and the substrate reference in gray. (b) Constant current STM image (V = −50 mV, I = 20 pA, 3.5 × 3.5 nm2) with color dots indicating the reference positions of the STS in (a). (c) and (d) LDOS calculated isosurfaces of 3 for the highest fully occupied molecular orbital in the energy window (−1, −0.5) eV with the four branches in open (c) and trans-closed (d) atomic arrangements. The electronic density becomes more delocalized over the external branches on the trans-closed, indicative of conjugation at the external branches in comparison to the open configuration. (e) dI/dV map at −1.3 V (Vmod = 10 mV) corresponding to (b), showing the spatial distribution of the occupied states. | ||
DFT calculations of isomeric MnPc (3) adsorbed on Ag(111)
At this point we must correlate the A and B electronic structures of the DAE moieties of 3 with their atomic arrangement. To do so, we performed DFT calculations of fully relaxed isomeric MnPc on a 9 × 10 × 3 Ag(111) supercell setting an equal atomic arrangement to the four branches (see the Methods section in the ESI†). Table 1 shows the relative energies of the three most stable configurations, where the minimum corresponds to the open arrangement (one sulfur atom pointing towards the surface and the other away from it). The second most favorable configuration is the open-rot that features a benzothiophene group rotated around its σ-bond by about 195° so that both sulphur atoms point to the surface. The third most stable configuration is the trans-closed, where the external hydrogen atoms are vertically opposed with respect to the MnPc central plane (one pointing towards the surface and the second away). Another identified possible configuration is the cis-closed isomer (see Fig. S8†), where both hydrogen atoms point away from the surface. Note that this structure confers a distinct mirror symmetry to the branch, which was experimentally absent.These DFT atomic arrangements must agree with the STM/STS datasets. We can start by discarding the symmetric cis-closed arrangement shown in Fig. S8† since none of the branches of 3 exhibits mirror symmetry with respect to the pyrrole ring (cf.Fig. 3a–d). In a similar way, the more compressed structural model of the open-rot isomer (superimposed to the MnPc in Fig. 3c) does not match the topographic STM footprint neither for A nor B lobes. Not only is energetically less stable than the open configuration, but it also shows almost identical LDOS for the molecular orbitals near the Fermi level (see ESI Fig. S9†). Thus, it is justified to rule out the presence of the open-rot isomer and restrict our external atomic arrangements to the trans-closed and open configurations. Their sideview comparison in Table 1 shows a larger apparent height and a more pronounced asymmetry for the latter, which agrees with the corrugation found in the constant-current images in Fig. 3. Consequently, we associate the open arrangement with the A configuration and the trans-closed with the B lobes.
To further increase the confidence in the assignment of the atomic arrangements of A/B lobes we calculated the local density of states (LDOS) in the energy window (−1, −0.5) eV corresponding to the highest fully occupied molecular orbital of 3 with four identical branches. The resulting LDOS for the open and trans-closed MnPcs is shown in Fig. 4c and d. The open branched MnPc exhibits a higher electronic density mainly localized at the pyrrole rings, whereas the trans-closed shows more weight towards the external branches. Moreover, our calculated PDOS (see ESI Fig. S9†) support a shift to lower energies in the HOMO position for the trans-closed structure due to a higher conjugation of its branches with the pyrrole ring center. Then, based on this feature of the HOMO, we correlated the calculated molecular orbital of Fig. 4d for the trans-closed geometry with the experimental dI/dV signal observed at −1.3 V of Fig. 4e. In contrast to constant current images, the darker B branches (number 4 in Fig. 4b) display a dominant character in the charge density distribution in the dI/dV maps of occupied states. Indeed, the trans-closed intensity maximizes closer to the sulfur atoms for the HOMO (cf.Fig. 4e). In contrast, the A branches (numbers 1, 2 and 3 in Fig. 4b) featuring brighter signals in the constant current, become practically irrelevant and only reflect the higher corrugation inherent to the open configuration in the dI/dV map. The LDOS of this occupied molecular orbital qualitatively agree with the features described for the calculated HOMO shown in Fig. 4c.
Isomer branch switching by mechanical manipulation
After identifying these two A/B configurations, we must demonstrate the feasibility of switching between the open and trans-closed configurations without destroying the molecule. We can do so by lateral atomic manipulation over the Ag surface. We observed frequent switching of the branches during manipulation events triggered by the tip-molecule-substrate forces, as shown in Fig. 5a and b. A double switch from open to trans-closed and the back-switch of trans-closed to open is shown in Fig. 5c. This reversible switching confirms the full active functionality of 3 on the Ag(111) surface. Importantly, the dI/dV spectra of the switched forms have the same spectroscopic features as those shown in Fig. 4a, confirming the MnPc capability to undergo open/close isomerization while keeping the ligands structurally intact. | ||
| Fig. 5 Mechanical switching of the DAE moieties by tip manipulation. STM images recorded before and after the controlled lateral drag of 3 over the Ag surface (−100 mV, 50 pA, 8.75 × 8.75 nm2). The arrows point to the switched branches. In the case of (a) and (b) one DAE branch changes from the open to trans-closed configuration. Alternatively, the sequence in (c) shows a reversible switch. | ||
We also tested the use of bias pulses and slow ramps in the current or electric field to drive the molecular switches.17,18,28,29 However, we discarded these methods since no changes were observed below a 3V threshold, whereas biasing the molecules above this value systematically led to non-reversible structural changes of the branches, in the form of dehydrogenation with the appearance of 4-type branches (see ESI Fig. S6†).
Magnetic fingerprint of isomeric MnPcs
The second condition that we previously mentioned for the pathway to generate magneto-optical transducers is that our isomeric MnPc carries a well-defined magnetic moment. Based on previous work,21,22 such properties can be validated through the detection of a Kondo effect on the Mn central atom. In MnPcs, the fingerprint of the Kondo effect has been identified by other groups as a resonance at the Fermi level with a linewidth of 13 meV convoluted with the Mn dxz/yz orbitals (characteristic of the D4h squared planar symmetry group) appearing at −50 mV.9,21,22 Therefore, using high-resolution dI/dV spectra in proximity of the Fermi energy we should verify the existence of the Kondo feature. The experimental data are shown in the inset of Fig. 4a. Indeed, our Mn STS reproduces the expected deconvolution analysis (see ESI Fig. S7d†), which can also be considered a further evidence of the correct MnPc formation by OSS.DFT theory sheds light on the origin of this Kondo resonance that we locate at the pyrrole ring center, in agreement with previous work.21,22 The calculated density of states projected onto the Mn d orbitals indicate that the dx2−y2 orbital is emptied, whereas the other four d orbitals are partially filled (see ESI Fig. S10†). This electronic arrangement results in a total magnetic moment slightly less than 2μB where the main contribution is associated with the Mn center but with an opposite magnetic contribution from the molecular framework. Previously, the Kondo resonance was associated with the many-body screening of the dz2 orbital by the conduction electrons of the substrate,21 which in our case is substantiated by the nearly half-filling observed for this orbital. Also, the degenerated dxz/yz orbitals exhibit a strong contribution just below the Fermi level, accounting for the tail to the left of the Kondo resonance.
A critical parameter for the Kondo state is the screening strength, which affects the Kondo temperature and resonance line shape when, for instance, altering the distance of the central Mn to the metallic substrate. Despite the possibility of switching the branch configurations between open and trans-closed, we experimentally find for our switchable MnPcs that the low energy Mn spectra do not change (cf. ESI Fig. S7†). This can be understood if the Mn vertical distance turns out to be insensitive to the branch configuration, as deduced by theory (cf. ΔzMn in Table 1). Furthermore, the identical planar environment of the Mn atoms in both configurations leads to nearly the same d orbital decomposition (ESI Fig. S10†). The use of bulkier groups at the hydrogen positions that are still optically active could lead to significant Mn height changes when switching between different open/closed isomers, thereby bearing the prospect of fulfilling the magnetic transducer functionality.
III. Conclusions
We have demonstrated that the branches of individual MnPc synthesized on Ag(111) can be mechanically switched between open and trans-closed isomeric forms. Preparation of the molecular complex has been possible by the OSS of a smaller precursor (1), which exhibits optical activity in solution, but most importantly maintains its integrity upon UHV sublimation. Indeed, the assignment of the two distinct configurations (open and closed atomic arrangements) was possible by combining STM images, STS spectroscopy and DFT calculations. Notably, we demonstrate the prospect of active functionality of these DAE branches by the configuration switching using tip manipulation processes. We also find that the Mn ions of 3 exhibits a significant magnetic fingerprint, so an entirely new playground to fabricate individually addressable magneto-optical transducers at the atomic scale is envisioned and requires further experimental validation.The flexibility of the OSS process to produce complex structures, where the optical activity is brought by diarylethylene (DAE) moieties and the magnetism by metal centers exchangeable by metallation, opens vast opportunities. For instance, we foresee the implementation of different readout signals changing the magnetic state of the ion, the magneto crystalline anisotropy,30,31 the energy resolved spin polarization of the local DOS,32,33 or the Kondo screening.21,22 Also, we envision the use of bulkier ligands to alter the adsorption geometry and height of the pyrrole ring around the metal center, thereby correlating the Kondo resonance to the different combinations of isomer ligands. All these possibilities expand the well-stablished spin-crossover photo-magnetism approach at the individual molecular level.
Author contributions
A. D.-C. and D. S. conducted the experiment and its data analysis; C. G.-F., P. K. and D. S.-P. performed the DFT calculations; S. Q. and D. P. designed and synthesized and purified the precursor molecules; and A. D.-C., D. S. and J. L.-C. wrote the manuscript. All authors contributed to the revision and final discussion of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We sincerely thank Marten Piantek and Carlos Casas for their contribution to this work. Dr Piantek is also acknowledged for providing ideas for this work and for in-depth discussions. We gratefully acknowledge financial support from the Spanish Ministries of Economy, Industry and Competitiveness (MINECO, grants MAT2016-78293-C6-4-R and MAT2016-78293-C6-6) and of Science and Innovation (MICINN, grant no. PID2019-107338RB-C62, PID2019-107338RB-C64, PID2019-107338RB-C66/AEI/10.13039/501100011033 and RED2018-102833-T), the regional Governments of Aragon (E13-20R and E12-20R), the Xunta de Galicia (Centro de Investigación de Galicia acreditation 2019–2022, ED431G 2019/03), the Dept. of Education of the Basque Government and UPV/EHU (grant no. IT1246-19), the Spanish Research Agency (AEI), the European Regional Development Fund (ERDF) under the program Interreg V-A España-Francia-Andorra (contract no. EFA194/16 TNSI) and the European Union through Horizon 2020 (FET-Open project SPRING grant. no. 863098). Finally, we acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- V. A. Dediu, L. E. Hueso, I. Bergenti and C. Taliani, Nat. Mater., 2009, 8, 707–716 CrossRef CAS PubMed.
- W. J. M. Naber, S. Faez and W. G. van der Wiel, J. Phys. D: Appl. Phys., 2007, 40, R205–R228 CrossRef CAS.
- H. Lu and N. Kobayashi, Chem. Rev., 2016, 116, 6184–6261 CrossRef CAS PubMed.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- N. Tsukahara, K.-i. Noto, M. Ohara, S. Shiraki, N. Takagi, Y. Takata, J. Miyawaki, M. Taguchi, A. Chainani, S. Shin and M. Kawai, Phys. Rev. Lett., 2009, 102, 167203 CrossRef PubMed.
- L. Gao, W. Ji, Y. B. Hu, Z. H. Cheng, Z. T. Deng, Q. Liu, N. Jiang, X. Lin, W. Guo, S. X. Du, W. A. Hofer, X. C. Xie and H.-J. Gao, Phys. Rev. Lett., 2007, 99, 106402 CrossRef CAS PubMed.
- K. J. Franke, G. Schulze and J. I. Pascual, Science, 2011, 332, 940–944 CrossRef CAS PubMed.
- J. Kügel, M. Karolak, A. Krönlein, D. Serrate, M. Bode and G. Sangiovanni, npj Quantum Mater., 2018, 3, 53 CrossRef.
- C. Wäckerlin, D. Chylarecka, A. Kleibert, K. Müller, C. Iacovita, F. Nolting, T. A. Jung and N. Ballav, Nat. Commun., 2010, 1, 61 CrossRef PubMed.
- K. Seufert, W. Auwärter and J. V. Barth, J. Am. Chem. Soc., 2010, 132, 18141–18146 CrossRef CAS PubMed.
- C. F. Hermanns, M. Bernien, A. Krüger, W. Walter, Y.-M. Chang, E. Weschke and W. Kuch, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 104420 CrossRef.
- A. Köbke, F. Gutzeit, F. Röhricht, A. Schlimm, J. Grunwald, F. Tuczek, M. Studniarek, D. Longo, F. Choueikani, E. Otero, P. Ohresser, S. Rohlf, S. Johannsen, F. Diekmann, K. Rossnagel, A. Weismann, T. Jasper-Toennies, C. Näther, R. Herges, R. Berndt and M. Gruber, Nat. Nanotechnol., 2020, 15, 18–21 CrossRef PubMed.
- N. Katsonis, T. Kudernac, M. Walko, S. J. van der Molen, B. J. van Wees and B. L. Feringa, Adv. Mater., 2006, 18, 1397–1400 CrossRef CAS.
- S. V. Snegir, P. Yu, F. Maurel, O. L. Kapitanchuk, A. A. Marchenko and E. Lacaze, Langmuir, 2014, 30, 13556–13563 CrossRef CAS PubMed.
- M. Alemani, M. V. Peters, S. Hecht, K.-H. Rieder, F. Moresco and L. Grill, J. Am. Chem. Soc., 2006, 128, 14446–14447 CrossRef CAS PubMed.
- G. Reecht, C. Lotze, D. Sysoiev, T. Huhn and K. J. Franke, ACS Nano, 2016, 10, 10555–10562 CrossRef CAS PubMed.
- P. Liljeroth, J. Repp and G. Meyer, Science, 2007, 317, 1203–1206 CrossRef CAS PubMed.
- B.-Y. Choi, S.-J. Kahng, S. Kim, H. Kim, H. W. Kim, Y. J. Song, J. Ihm and Y. Kuk, Phys. Rev. Lett., 2006, 96, 156106 CrossRef PubMed.
- J. Kügel, M. Karolak, J. Senkpiel, P.-J. Hsu, G. Sangiovanni and M. Bode, Nano Lett., 2014, 14, 3895–3902 CrossRef PubMed.
- R. Tuerhong, F. Ngassam, S. Watanabe, J. Onoe, M. Alouani and J.-P. Bucher, J. Phys. Chem. C, 2018, 122, 20046–20054 CrossRef CAS.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- M. Piantek, D. Serrate, M. Moro-Lagares, P. Algarabel, J. I. Pascual and M. R. Ibarra, J. Phys. Chem. C, 2014, 118, 17895–17899 CrossRef CAS.
- M. Koudia and M. Abel, Chem. Commun., 2014, 50, 8565–8567 RSC.
- J. Wirth, N. Hatter, R. Drost, T. R. Umbach, S. Barja, M. Zastrow, K. Rück-Braun, J. I. Pascual, P. Saalfrank and K. J. Franke, J. Phys. Chem. C, 2015, 119, 4874–4883 CrossRef CAS.
- S. Kezilebieke, A. Amokrane, M. Abel and J.-P. Bucher, J. Phys. Chem. Lett., 2014, 5, 3175–3182 CrossRef CAS PubMed.
- L. Grill, K.-H. Rieder, F. Moresco, S. Stojkovic, A. Gourdon and C. Joachim, Nano Lett., 2006, 6, 2685–2689 CrossRef CAS PubMed.
- X. H. Qiu, G. V. Nazin and W. Ho, Phys. Rev. Lett., 2004, 93, 196806 CrossRef CAS PubMed.
- A. F. Otte, M. Ternes, K. von Bergmann, S. Loth, H. Brune, C. P. Lutz, C. F. Hirjibehedin and A. J. Heinrich, Nat. Phys., 2008, 4, 847–850 Search PubMed.
- C. F. Hirjibehedin, C.-Y. Lin, A. F. Otte, M. Ternes, C. P. Lutz, B. A. Jones and A. J. Heinrich, Science, 2007, 317, 1199–1203 CrossRef CAS PubMed.
- F. Meier, L. Zhou, J. Wiebe and R. Wiesendanger, Science, 2008, 320, 82–86 CrossRef CAS PubMed.
- D. Serrate, P. Ferriani, Y. Yoshida, S.-W. Hla, M. Menzel, K. von Bergmann, S. Heinze, A. Kubetzka and R. Wiesendanger, Nat. Nanotechnol., 2010, 5, 350–353 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr00721e |
| This journal is © The Royal Society of Chemistry 2022 |