
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manipulation of the crystalline phase diagram of hydrogen through nanoscale confinement effects in porous carbons†
Lui R.
Terry
 *a,
Stephane
Rols
b,
Mi
Tian
c,
Ivan
da Silva
d,
Simon J.
Bending
e and
Valeska P.
Ting
*a
*a,
Stephane
Rols
b,
Mi
Tian
c,
Ivan
da Silva
d,
Simon J.
Bending
e and
Valeska P.
Ting
*a
aDepartment of Mechanical Engineering, University of Bristol, BS8 1TR UK. E-mail: v.ting@bristol.ac.uk; lt7006@bristol.ac.uk
bInstitut Laue-Langevin (ILL), Grenoble, France
cCollege of Engineering, Mathematics and Physical Sciences, University of Exeter, EX4 4QF, UK
dISIS Neutron Source, Rutherford Appleton Laboratory, Harwell Oxford, Didcot OX11 0QX, UK
eDepartment of Physics, University of Bath, Bath BA2 7AY, UK
First published on 6th May 2022
Abstract
Condensed phases of molecular hydrogen (H2) are highly desired for clean energy applications ranging from hydrogen storage to nuclear fusion and superconductive energy storage. However, in bulk hydrogen, such dense phases typically only form at exceedingly low temperatures or extremely high (typically hundreds of GPa) pressures. Here, confinement of H2 within nanoporous materials is shown to significantly manipulate the hydrogen phase diagram leading to preferential stabilization of unusual crystalline H2 phases. Using pressure and temperature-dependent neutron scattering at pressures between 200–2000 bar (0.02–0.2 GPa) and temperatures between 10–77 K to map out the phase diagram of H2 when confined inside both meso- and microporous carbons, we conclusively demonstrate the preferential stabilisation of face-centred cubic (FCC) solid ortho-H2 in microporous carbons, at temperatures five times higher than would be possible in bulk H2. Through examination of nanoconfined H2 rotational dynamics, preferential adsorption and spin trapping of ortho-H2, as well as the loss of rotational energy and severe restriction of rotational degrees of freedom caused by the unique micropore environments, are shown to result in changes to phase behaviour. This work provides a general strategy for further manipulation of the H2 phase diagram via nanoconfinement effects, and for tuning of anisotropic potential through control of confining material composition and pore size. This approach could potentially provide lower energy routes to the formation and study of more exotic non-equilibrium condensed phases of hydrogen that could be useful for a wide range of energy applications.
1. Introduction
The urgent global need for alternative and clean energy sources has driven demand for compact or condensed forms of H2, as a sustainable, zero-carbon fuel.1 Furthermore, the promise of room-temperature superconductivity in metallic hydrogen2–4 has also driven interest in the creation of high-density hydrogen solids and other condensed non-equilibrium hydrogen phases. The phase diagram of H2 is indeed rich, with eight solid phases of pure hydrogen being thought to exist,4–11 ranging from phase I hexagonally close-packed (HCP) H2,5,12 metastable FCC ortho-H2,11,13 the broken symmetry phase (II)10 all the way up to Cmca![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 2 metallic hydrogen.4 However, one of the most challenging aspects of the practical use of H2 stems from its existence as a very low-density gas across most accessible temperatures and pressures, with the formation of solid phases requiring temperatures below 13.8 K (at 1 bar)14 or pressures up to millions of times atmospheric pressure to form at ambient temperature.12 The practical limitations of working at such high pressures or low temperatures make the application and study of condensed H2 experimentally demanding, necessitating the exploration of alternative routes toward the formation of compact and condensed hydrogen phases at more accessible temperatures or pressures.
2 metallic hydrogen.4 However, one of the most challenging aspects of the practical use of H2 stems from its existence as a very low-density gas across most accessible temperatures and pressures, with the formation of solid phases requiring temperatures below 13.8 K (at 1 bar)14 or pressures up to millions of times atmospheric pressure to form at ambient temperature.12 The practical limitations of working at such high pressures or low temperatures make the application and study of condensed H2 experimentally demanding, necessitating the exploration of alternative routes toward the formation of compact and condensed hydrogen phases at more accessible temperatures or pressures.
The adsorption of gases into nanoporous materials has been studied as a method of densifying gaseous hydrogen to higher gravimetric and volumetric densities.15–17 Recently, experimental studies of H2 adsorbed in highly microporous carbons with pore diameters <2 nm have demonstrated the formation of H2 with solid-like characteristics at atmospheric pressure and temperatures above hydrogen's critical temperature – conditions where, classically, bulk H2 should be gaseous. Results have shown severely restricted mobility of molecules,18,19 increased densities,15,16,20,21 signature quantum rotational transitions of solid-like H2 at temperatures up to 100 K (ref. 16 and 17) and have allowed observation of possible phase transitions22 when pore diameters are smaller than 1 nm. If appreciable manipulation of the H2 phase diagram can be achieved through nanoconfinement in porous materials such as these, lower energy routes to more exotic non-equilibrium condensed phases of H2 could be identified and potentially exploited for applications such as H2 fuel storage, superconductivity, or fusion pellets. Furthermore, such porous materials may be readily scalable, which is key for any envisaged future large-scale technological applications of solid hydrogen. Thus, a better understanding of what happens to nanoconfined H2 is necessary to help us design and model better H2 confinement materials for future applications.
To understand how confinement could be extended to produce and study condensed phases of hydrogen, a detailed investigation of which solid phases of H2 form under confinement and why was conducted, using confinement inside both mesoporous (pore diameters >2 nm) and microporous (pores <2 nm in diameter) carbons. Here we used a powerful combination of variable-temperature and variable-pressure neutron diffraction and inelastic neutron scattering (INS) studies. The large neutron scattering cross-section of the hydrogen nucleus makes neutrons highly sensitive to hydrogen atoms, meaning that the structural arrangement and dynamic properties of the multiple phases of H2 confined within a carbon nanomaterial can be easily distinguished.23 Our results uncover considerable manipulation of the H2 phase diagram by nanoconfinement. For the first time, the crystal structures of H2 confined in microporous activated carbons have been revealed and the driving forces behind preferential adsorption and spin trapping of solid ortho-H2, when confined in highly microporous carbon, have been identified, furthering our understanding of H2 phase manipulation and stabilisation by nanoconfinement effects.
2. Experimental
2.1 Materials and preparation
To investigate the effects of nanoscale confinement on the crystal structures and phase diagram of H2, two porous carbons (a microporous “TE7” carbon and a mesoporous onion like carbon, “OLC”) were chosen due to their amorphous graphitic carbon frameworks of slit-shaped pores and their differing pore size distributions. TE7 is a highly microporous disordered activated carbon derived from phenolic resin, sourced from MAST Carbon International Ltd (Basingstoke, UK). OLC is a highly mesoporous activated carbon synthesised by vacuum annealing of detonation nanodiamond powder24 and was supplied by Prof. Volker Presser (INM, Saarland University, Saarbrücken, Germany). All other chemicals used were sourced from Sigma Aldrich and used as received unless otherwise stated.All experiments used room temperature “normal H2” gas, comprised of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 equilibrium mixture of the two nuclear spin isomers of hydrogen, para-H2 and ortho-H2, respectively. para-H2 is the ground state of H2, whereby nuclear spins on each hydrogen atom are antisymmetric. It is characterized by an even rotational quantum number (J = 0) where its rotation and charge distribution are spherical/isotropic (see HCP in Fig. 1). ortho-H2 is the first excited state and is therefore metastable, having symmetric nuclear spins characterized by an odd rotational quantum number (J = 1) where its rotations and charge distribution are anisotropic (see FCC in Fig. 1). A minimum of 99.999% purity “normal H2” was used for all experiments.
:3 equilibrium mixture of the two nuclear spin isomers of hydrogen, para-H2 and ortho-H2, respectively. para-H2 is the ground state of H2, whereby nuclear spins on each hydrogen atom are antisymmetric. It is characterized by an even rotational quantum number (J = 0) where its rotation and charge distribution are spherical/isotropic (see HCP in Fig. 1). ortho-H2 is the first excited state and is therefore metastable, having symmetric nuclear spins characterized by an odd rotational quantum number (J = 1) where its rotations and charge distribution are anisotropic (see FCC in Fig. 1). A minimum of 99.999% purity “normal H2” was used for all experiments.
 | ||
| Fig. 1 Neutron powder diffraction of confined crystalline H2 at low temperature. Diffraction data from (a) detector Bank 5 and (b) Bank 3 of the GEM diffractometer for microporous TE7 and mesoporous OLC carbons under vacuum (0 bar H2) at 77 K and dosed with 200 or 2 kbar H2 at 10 K. The Bragg peaks reveal the presence of two distinct crystalline phases of H2 (HCP ★ and FCC ■) in the porous carbons at 10 K. Reflections from the aluminium sample holder are shaded in grey and patterns are offset in the y-direction, for clarity. (c) H2 crystal structures for the orientationally disordered HCP H2 lattice and the orientationally ordered FCC H2 lattice.12 | ||
Paramagnetic metal impurities are known to catalyse the conversion of ortho-H2 to para-H2. In preparation for physical characterization, all carbon materials were powdered using an agate mortar and pestle, then washed in HCl to remove any possible metal contaminants. Samples were suspended in aqueous 1 mol dm−3 HCl (ultra-trace analysis) at 100 mg mL−1, then sonicated for 15 minutes before filtering and a second rinse with 1 M HCl. This process was then repeated with an excess of acetone (ultra-trace) followed by methanol (ultra-trace) and finally ultra-pure deionised water. Samples were completely dried at 623 K under dynamic vacuum (P < 0.001 bar) for a minimum of 8 hours before any physical characterisation.
2.2 Gas sorption analysis
The specific surface area and internal pore structure of both samples were obtained from N2 adsorption at 77 K and CO2 adsorption at 298 K on a Micromeritics 3Flex volumetric gas sorption analyser. Samples (∼120 mg) were degassed under vacuum prior to the experiment (623 K, ∼8 h, 1 × 10−6 mbar). Helium was used for free-space determination following isotherm collection. N2 and helium were supplied by Air Liquide and of a purity of 99.999%. Pore volume distribution as a function of pore width was calculated from combined CO2 and N2 adsorption data using the advanced combination non-local density functional theory (NL-DFT) method and an asymmetrical slit-shaped pore model. The BET surface area was calculated according to the procedure from ISO 9277 (see ESI – Table S1 and Fig. S1†).2.3 Powder X-ray diffraction
Information on the structure of the carbons was obtained by powder X-ray diffraction (PXRD) using a Bruker D8 diffractometer (Cu Kα source, λ = 1.54 Å), with a step size of 0.05 2θ and an integration time interval of 3 seconds. Diffraction data of the silicon sample holder was background subtracted for clarity, due to the low diffraction intensities of the carbon samples (see ESI – Fig. S2†).2.4 Neutron diffraction
Structural information on the adsorbed H2 phase was obtained by neutron diffraction on the neutron powder diffraction instrument, GEM, at the ISIS neutron source, Rutherford-Appleton Laboratories, UK.25 Results obtained (https://doi.org/10.5286/ISIS.E.RB1910448(2019)) were calibrated and refined against a silicon standard taken prior to the experiment. TE7 and OLC samples were degassed (623 K, 1 × 10−6 mbar) prior to examination to remove adsorbed water and trace gases. Dry samples were loaded into an aluminium sample cell (diameter 0.8 cm; volume 3.5 cm3) inside a He-filled glove box and tamped to reduce the level of interstitial voids between particulates as much as possible. The sample cell was filled and centred to ensure no free-space contribution was included in the beam. Care was taken to ensure the packing between the two samples was similar, so that differences observed in the diffraction patterns could be attributed to differences in the inner micro/mesoporosity of the samples, rather than any hydrogen in the interstitial spaces between particles. Loaded cells were leak tested under vacuum with 2 kbar He, before evacuation at room temperature for at least 1 hour to remove helium from the micropores. To decrease the diffraction signal of the aluminium sample cell, a collimator was used, reducing the beam size to 30 × 5 mm (vertical × horizontal) and removing diffraction banks 4 and 6 from use. Background measurements of the carbon samples were collected at room temperature and 77 K under vacuum for 4 hours (see ESI – Fig. S3–S5†). The background diffraction patterns (recorded under vacuum) indicated an amorphous carbon framework in both cases, with no sharp diffraction peaks and only a broad and diffuse (002) graphitic feature for OLC observed at 3.46 Å (as indicated by powder X-ray diffraction patterns – Fig. S2†).Samples were then dosed with normal-H2 to either 200 or 2000 bar at 77 K before temperature cycling. Neutron diffraction data were collected at 77 K for 3 hours before the samples were cooled to 10 K, after which data was collected for at least 2.5 hours at 10, 15, 25, 52 and 77 K. At 10 K, the pressure inside the cells dropped by ∼3.3% due to thermal contraction. Data was collected from diffraction banks 3 & 5 of the GEM instrument for analysis due to their appropriate d-spacing range for H2 (1.4–5 Å) and resolution (bank 3: ∼2 × 10−2 Δd/d, bank 5: ∼5 × 10−3 Δd/d). Observed diffraction peaks were indexed and treated with full Rietveld refinement using the TOPAS v6-Academic software. Relative percentages of each phase were calculated using all peaks in each diffraction pattern. Clear diffraction peaks from the aluminium sample cell could still be observed throughout the experiment (see Fig. S3– S5†).
2.5 Inelastic neutron scattering
INS spectra collected on IN4 at the Institut Laue Langevin, France used an incident neutron wavelength of 1.6 Å and a Fermi chopper speed of 15000 rpm. This allowed measurement of a large (Q, E) scattering region (energy transfer range E = [−75, 28 meV], momentum transfer range Q = [1, 7 Å−1] at elastic scattering). The energy resolution at 0 meV energy transfer (elastic scattering) is 2.1 meV. Raw data were transformed into the physical observable S(Q, E), often called dynamical structure factor, according to classical time-of-flight procedures, were corrected from the neutron energy dependence efficiency of the detectors and calibrated using a vanadium standard. The measurements allowed quantitative analysis of S(Q, E) as a function of gas pressure. The ∼5 cm3 sample of TE7 beads was acid-washed to remove any residual metal content prior to analysis, then degassed at 623 K for 8 hours under vacuum (1 × 10−6 mbar) and loaded into a cylindrical aluminium sample can inside a dry argon glove box. Sample temperatures were regulated between 4 K and 200 K using an ancillary cryo-furnace. Samples were dosed with normal H2 (Air Liquide, 99.999% purity) using a Hiden Isochema HTP-1 gas sorption analyser and equilibrated at 1 bar and 77 K. Pressure was recorded using a 1 bar baratron and a high-pressure (20 bar) transducer. The TE7 carbon sample was dosed with 1 bar H2 at 77 K and left to equilibrate for 1 hour. The system was then sealed and cooled to 40 K and 4 K. INS data were collected at the appropriate temperatures and pressures over a period of two hours and were background-subtracted with degassed samples under vacuum at the corresponding temperatures and pressures over the same time period, to remove spectral contributions from the carbon support.
Rotational transitions were fitted to a set of Gaussian lines, with the full-width at half-maximum fixed to the resolution of the spectrometer at the corresponding energy transfer. The Gaussian lines in the anti-Stokes side of the spectrum were added to a background composed of a unique elastic contribution (considered as Gaussian) plus a Lorentzian function (to account for a quasielastic like background centred at zero energy transfer). On the Stokes side, the background accounted in addition for a broad recoil-like scattering which was fitted to a large gaussian shape. All Gaussian lines representing the rotational transitions were fitted simultaneously, with their centres and width being tight from one Q value to the other. Their integrated intensity was further fitted to the function:
 | (1) |
Further analysis of the quantum rotations considered a 3D hindered rotational model, the anisotropic potential term being chosen of the form
| W = Vsin2(θ) | (2) |
The total Hamiltonian
| Ĥ = B × J(J + 1) + W | (3) |
2.6 Scanning transmission electron microscopy energy dispersive X-ray (STEM-EDX)
Ground samples were dispersed in ethanol and dropped onto copper TEM grids and left to dry. Grids were analyzed on a JEOL JEM-2100F for STEM-EDX analysis.2.7 Inductively coupled plasma optical emission spectroscopy (ICP-OES)
To analyze the trace metal content, samples were first digested in Piranha acid solution (3H2SO4:H2O2) at 2 mg ml−1. The sample was gently heated up to 80 °C to aid digestion and then to 290 °C to near-complete evaporation. The samples were then diluted in 10 mL of 1% HNO3 solution and filtered through a 0.22 μm filter. Samples were analyzed for trace metals against a calibration standard using an Agilent 710 ICP-OES. Each metal was referenced using at least three optical emission wavelengths.
3. Results and discussion
3.1 Identification of the crystalline arrangement of the confined H2 phases
To evaluate the effects of nanoscale confinement on the crystal structures and phase diagram of H2, hydrogen sorption in two well-characterized amorphous, activated carbons (a microporous “TE7” activated carbon and a mesoporous onion-like carbon “OLC”) was investigated. Both materials contain slit-shaped pores24,26 exhibiting homogenous graphitic carbon adsorption surfaces with few surface oxygen groups16 and present comparable total pore volumes between 0.8–0.9 cm3 g−1, as determined from gas sorption analysis (see ESI – Table S1†). The different pore diameters of the two activated carbons made them ideal candidates for evaluating the effect of nanoscale confinement of H2 on the stability of the solid phases of confined H2. Highly microporous carbons like TE7 have been shown to cause extreme densification of H2 at temperatures ∼77 K due to their sub-nanometre pores.15,16,22 Mesoporous carbons like OLC show lower levels of H2 densification, owing to larger pore diameters.15,16 The TE7 used in this study exhibits a modal pore size of 0.55 nm with only a small proportion of mesopores (see ESI – Fig. S1†). OLC, the mesoporous activated carbon control, had a modal pore size of 9.5 nm and negligible microporosity in the pore diameter range <1 nm. Examination of the effects of nanoconfinement on the structural arrangement of H2 forming within the pores was conducted at selected pressures and temperatures using “normal-H2” on the GEM neutron diffraction instrument, at the ISIS neutron source, Rutherford-Appleton Laboratories. Neutron diffraction data was collected for both TE7 (microporous) and OLC (mesoporous) carbons under vacuum at 77 K and with adsorbed H2 at 10 K (where bulk solid H2 is known to form14) and at pressures of 200 bar and 2 kbar (where the pore volume has reached saturation) (Fig. 1).Upon dosing both TE7 and OLC porous carbons with H2 at 77 K, cooling to 10 K, and data collection over 2–3 h, sharp diffraction peaks originating from crystalline solid H2 emerged (Fig. 1). To the authors’ knowledge, this represents the first observation of crystalline H2 nanoconfined in the pores of activated carbons.27 Diffraction patterns were indexed (Table S2†), revealing phase I solid H2 (P63/mmc HCP) was present in the pores, as might be expected for bulk H2 at these temperatures and pressures.5,12 However, clear diffraction peaks consistent with a Pa3 FCC H2 crystal packing system were also observed to be present in the microporous TE7, with trace amounts of FCC H2 also seen for the OLC material at 2 kbar. FCC H2 is a metastable and orientationally-ordered phase, requiring the ortho-H2 concentration to be >60% and the alignment of ortho-H2 electric quadrupoles,11,28 indicating a high concentration of ortho-H2 was present in the microporous carbon at low temperatures and that thermodynamic ortho-to-para conversion of confined H2 did not reach the 10 K equilibrium composition ratio (99.8% para-H2).12 Integration of the area under the diffraction peaks revealed the relative proportion of FCC:HCP was significantly higher in microporous carbon than in mesoporous carbons (see Table S3†), indicating that the sub-nanometre pores of TE7 may favour the formation of the FCC phase.
In addition to this, observation of multiple peaks consistent with the FCC ortho-H2 phase at these temperatures is highly significant, as bulk FCC ortho-H2 typically only forms below ∼3 K and 5 K at 200 bar and 2000 bar, respectively.12 FCC H2 has not been previously observed in confined systems, with prior neutron diffraction measurements of H2 confined in mesoporous (7 nm pore diameter) Vycor glasses showing only the HCP phase at 2 K (ref. 29) or only short-range order in mesoporous carbons.27 To gain further insight into the thermal stability of the observed H2 phases confined in the porous carbons, the temperature and pressure dependence of the observed crystalline phases was studied. For comparison, a summary of the confined phases observed in microporous TE7 across the examined temperatures and pressures is contrasted to the bulk H2 phase diagram12 (Fig. 2).
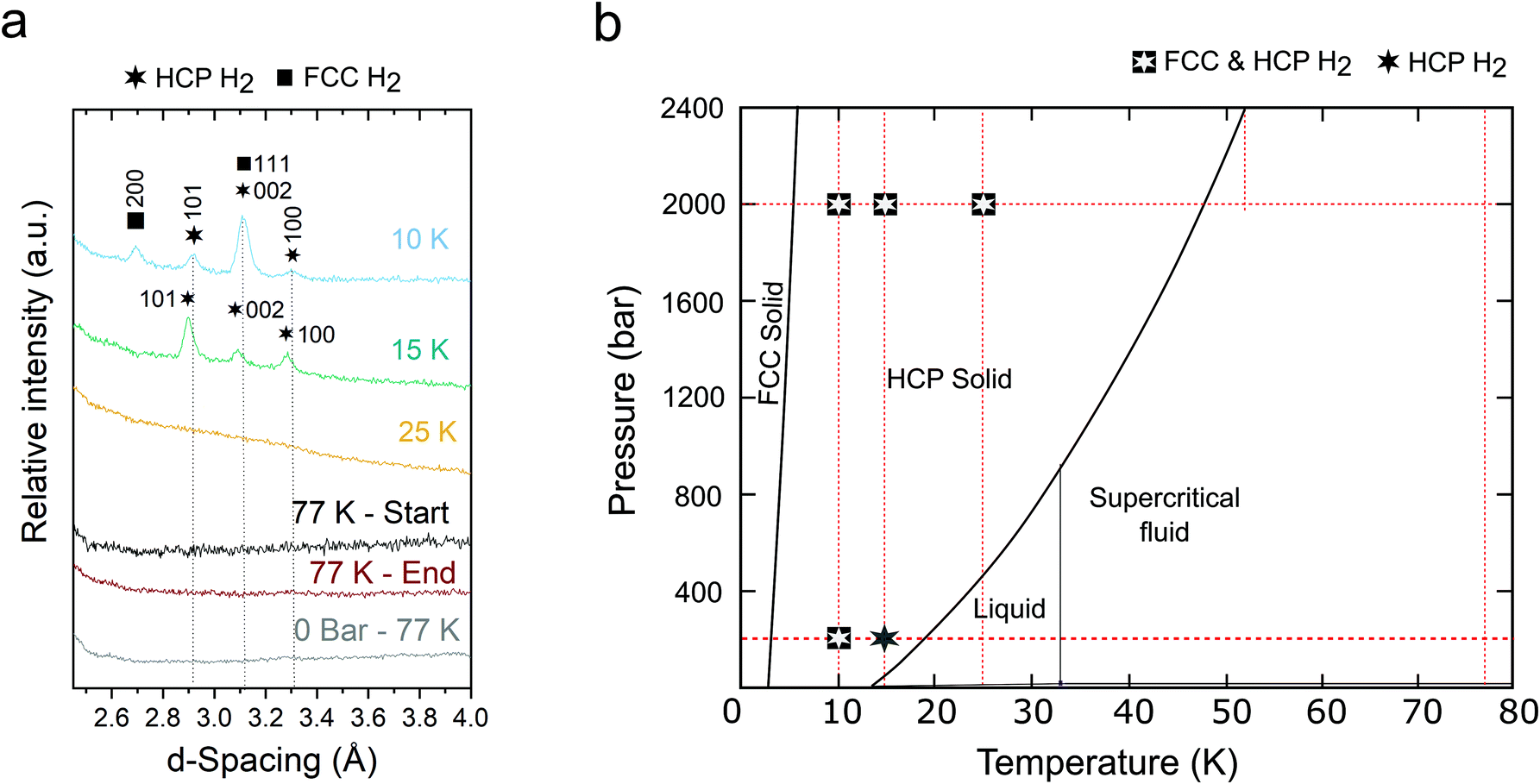 | ||
| Fig. 2 Temperature and pressure dependence of the confined crystalline phases in microporous carbon. (a) Temperature-dependent neutron powder diffraction data of microporous TE7 dosed to 200 bar H2 at 77 K (detector bank 3) showing the disappearance of the FCC phase at 15 K and loss of long-range order at 25 K. Dotted lines are added as a guide to the eye and patterns are offset in the y-direction for clarity. (b) Bulk phase diagram of ortho-H2 at T = 0 as a function of pressure overlaid with the observed crystalline phases of H2 in microporous TE7 in this experiment. Red lines represent the experimental pressures and temperatures observed via neutron diffraction. | ||
At 200 bar, the FCC phase disappeared by 15 K (Fig. 2a & S3†) but at 2 kbar was observed up to a temperature of 25 K (Fig. 2b & S4†), approximately five times higher than achievable in the bulk. However, the ratio of FCC:HCP also decreased with increasing pressure, from 4.7:1 to 1.2:1 (at 10 K). The increased thermal stability of the FCC ortho-H2 phase suggests that ortho-H2 in the subnanometre pores of TE7 may be strongly bound, aiding the molecular ordering of the FCC phase and enhancing the binding energy in the ortho-H2 crystal lattice.30 In contrast, within the range of temperatures and pressures explored, the confined HCP phase appeared to follow the bulk phase diagram, with no crystalline phase observed above the bulk solid/liquid phase boundary (Fig. 2). However, small shifts to this phase boundary may be uncovered with mapping over finer temperature increments.
A broad shoulder appeared in the d-spacing region of 2.6–3.6 Å in detector bank 3, with a corresponding broad peak in detector bank 5, centred at 2.6 Å and persisting for temperatures up to 77 K (Fig. S3 and S4†) suggesting possible short-range order or a glassy state. A glassy solid with the same intermolecular spacing was also observed for H2 confined in a zeolite at very high pressures.31 However, a comparison between changes in momentum transfer (Q) in both diffraction banks 3 & 5 did not show a match in background oscillations (Fig. S7†), therefore, this broad peak is likely due to an incoherent scattering effect than diffuse scattering caused by H2 with short-range order.
To determine the effect of confinement on the size of the unit cell, the unit cell parameters were calculated by Rietveld refinement (Table S4†). Notably, the temperature dependence of the unit cell dimensions for HCP H2 was found to differ at the two pressures studied. At the lower H2 pressure, (200 bar) negative thermal expansion was observed (Fig. S3†). As the temperature decreased from 15 to 10 K, the HCP unit cell expanded via elongation of the c-axis, leading to a deviation of the c/a ratio from the ideal value (Table S4†), suggesting that the molecular H2 is becoming anisotropic, as expected for ordered ortho-H2.12 In contrast, at the higher pressure studied (2 kbar), the unit cell exhibited negligible thermal expansion. The negative thermal expansion of the confined HCP phase at 10 K, 200 bar may be related to an incomplete HCP–FCC phase transition occurring at this temperature and pressure, with structural transitions from HCP to FCC typically accompanied by an expansion of volume.32,33 This deviation may be due to strain induced by the nanoconfinement or too low a local concentration of ortho-H2 required for FCC. In comparing the molar volume of the confined phases in the two activated carbons, both HCP and FCC were 7% larger in OLC than in TE7 at comparable pressures indicating that a higher density is achieved in smaller pores, consistent with previous calculations and gas sorption measurements.15,16,20 However, the molar volumes of the nanoconfined crystalline phases appeared to be larger than the bulk solids at similar temperatures and pressures11,13,34 (see Table S5†). Similarly, for H2 confined in a zeolite material at high pressures,31 a larger intermolecular separation than the bulk equivalent was also observed.
With no evidence for a crystalline solid phase at 77 K and larger unit cell volumes of confined crystalline phases, when compared to the bulk, the neutron diffraction suggests that previous reports of high-density solid H2 above the critical temperature15,16,20,21 are likely to be a high-density amorphous/glassy phase. However, previous reports were conducted at lower pressures than used in this study; thus further neutron diffraction experiments across 1–200 bar will be required to understand the confined phase diagram and confirm if the FCC phase can be observed above the critical temperature.22
3.2 Effect of H2 adsorption environments in microporous carbon
To understand how the adsorption environments in TE7 cause the formation of FCC ortho-H2 and increase its thermal stability, the effects of nanoconfinement on the rotational dynamics and distribution of both para and ortho-H2 adsorbed in microporous carbon TE7 was examined by temperature-dependent INS using the IN4C spectrometer at the Institute Laue Langevin, Grenoble. The carbon was dosed with 1 bar high purity (99.999%) normal H2 at 77 K to saturate the pores, subsequently cooled to temperatures between 40 and 4 K, and a detailed analysis of the rotations via INS was conducted (Fig. 3a–d and Table S6†). | ||
| Fig. 3 Nanoconfinement effects on the quantum rotations of adsorbed ortho and para-H2. Temperature-dependent INS of J = 1 → 0 ortho-to-para and J = 0 → 1 para-to-ortho transitions of 1 bar H2 confined in highly microporous TE7 at 4 K (a, b) and 40 K (c, d), Q = 3.4 Å−1. A background spectrum of the carbon under vacuum was recorded at each temperature before dosing H2 and subtracted from the raw data. Peak contributions to the rotational transition are shown in solid lines, freely rotating H2 in pores >1 nm are labelled (1), rotationally hindered H2 in pores <1 nm are labelled (2) and (3). Dashed lines represent contributions to the elastic scattering peak. (e) Quantum rotational levels of H2 in free space and adsorbed in a pore with a strong anisotropic potential causing 1D or 2D rotation. Adsorption into the pore lifts the degeneracy of J = 1 and splits the magnetic rotational sub levels. (f) Distribution of para- and ortho-H2 across the three different adsorption sites in TE7 at 4 K, displaying the dominant molecular rotational orientation. | ||
In bulk H2, the transition energy between the first nuclear rotational levels is 14.7 meV. Because H2 is a quantum solid, this is observed as a singular rotor peak in the INS16,17 and indicative of translationally pinned (hence solid-like) and freely rotating H2 – see peak (1) in Fig. 3a–d. However, upon adsorption, interaction with an anisotropic electrostatic field can hinder the free rotation of the H2 molecule and result in a lowering of rotational energy and the splitting of the rotational transition peak.18,35–37 In porous graphitic carbon, as the pore width narrows to <1 nm,38 the overlap of the anisotropic potential from pore walls is significant enough to hinder the free rotation of H2 and alter the rotational energy levels, lifting the degeneracy of the triplet J = 1 state and causing a 2:1 split in the magnetic rotational sub-levels (m) as shown in Fig. 3e.36,39,40 The ordering of the split states depends upon the negative or positive interaction of electrostatic adsorption potential with the molecular orientation of the rotations of H2. Thus, the rotational energies of a parallel and perpendicular alignment of H2 to the pore surfaces differ.39,41 Furthermore, as the pore width decreases, the overlapping anisotropic potential from the graphitic pore walls increases, further separating the rotational potential energy of each orientation and increasing the magnitude of the energy gap (Δe) between the split states. These relationships have been clearly observed and demonstrated in previous studies of H2 in slit-shaped pores of graphitic carbon.18,36,38 In TE7, the sub-nanometre pore sizes are sufficiently small to fit up to a bilayer of H2,17,22 thus maximising the influence of overlapping anisotropic potential on the solid forming in the pores.
In the INS, two split peaks in a 2:1 intensity ratio for the J = 0 → 1 excitation (labelled (2) in Fig. 3b & d) were observed, indicating para-H2 confined in a micropore adsorption site <1 nm. For J = 1 → 0, only the return transition from the lowest energy excited state to the ground state should be observed at low temperatures, due to the thermal population of states. Thus, peaks (2) & (3) in Fig. 3a and c appear to be from ortho-H2 in two different micropore adsorption sites <1 nm and not freely rotating ortho-H2. The peak at −13 meV is mirrored in the INS neutron energy loss spectra, hence this peak represents ortho-H2 adsorbed at the same micropore site as para-H2: site (2). The peak at −8 meV likely represents ortho-H2 confined in a much higher anisotropic potential causing an increased Δe, such as an even smaller pore36,38 – designated micropore site (3). Note that it is unlikely that the peak labelled (3) represents a phonon mode, as it is not observed in the neutron energy loss spectra and is too high energy for a quantum translation. Furthermore, peak (3) is unlikely to be the result of a metal impurity phase in the carbon as both EDXA and ICP-OES of samples reveal negligible trace metal content and no residual chlorine after acid washing, confirming the purity of the sample (see ESI – Fig. S8 and Table S7†). Paramagnetic metal centers are also known to rapidly catalyse ortho-to-para conversion.12,42 Hence, the presence of significant metal impurities would preclude the observation of significant proportions of ortho-H2 at this site and hence transitions in the neutron energy gain spectra would not be observed by 4 K.42–44 Finally, if a metal centre were present (magnetic or non-magnetic), an analogous peak in the neutron energy loss spectra would be observed.37,43,45–47 Here no such peak is observed.
The approximate distribution of the adsorption sites and ortho/para-H2 was inferred at 4 K (where all H2 is solid) by comparing the peak areas of the split and unsplit peaks from adsorbed ortho (J = 1 → 0) and para (J = 0 → 1) transitions, allowing percentages of ortho/para-H2 to be calculated at each site (Table S8†). Accounting for the difference in spin-dependent incoherent scattering cross-sections,48 31% of total adsorbed H2 exhibited split rotational peaks (2,3) indicating a barrier to rotation from the strongly anisotropic potential in the pores <1 nm. The remaining contribution came from the single peak at 14.5 meV (1), i.e. freely rotating H2, likely originating from the multiple layers of H2 in pores >1 nm and unaffected by high anisotropic potential.35 The distribution is in good agreement with the total pore volume distribution observed by gas sorption (Fig. S1†). The peaks and splitting observed in the INS are thus consistent with three types of adsorption environment within the carbon affecting H2, one mesopore-type environment >1 nm diameter, site (1), and two different micropore environments <1 nm (sites (2) & (3)), causing substantial perturbation to H2 rotations through nanoconfinement – see Fig. 3f.
3.3 Preferential stabilisation of H2 spin isomers via loss of rotational freedom
As well as the observed differences in the molecular ordering, significant differences in the para:ortho ratio could be found for the different adsorption locations in the carbon, exposing a variation in the thermodynamic conversion of ortho-to-para for the different pore sizes (Fig. S9†). An illustration of the distribution of ortho/para-H2 throughout microporous carbon at 4 K is displayed in Fig. 3f. At 40 K, both freely rotating para and ortho-H2 (87:13) are observed in mesopore adsorption site (1), but by 4 K only freely rotating para-H2 is observed, indicating rapid and complete thermodynamic conversion of ortho-to-para has occurred in pores >1 nm. Such behaviour has also been observed for activated charcoals, used for catalytic conversion of ortho-H2.39 An increased number of nearest neighbour hydrogen molecules (such as would be found in the layers of a filled mesopore) can drive intrinsic thermodynamic ortho-to-para conversion.12 In contrast, for the micropore adsorption sites (2, 3), by 4 K, a 57:43 para:ortho ratio of contributions from adsorbed H2 was observed, signifying the thermodynamic conversion of ortho-to-para in pores <1 nm is significantly lower than in pores >1 nm in activated carbons. Uniquely, at all temperatures, only J = 1 → 0 transitions were observed from site (3), indicating only ortho-H2 is adsorbed at this site. The results indicate preferential adsorption and potential spin trapping of ortho-H2 down to low temperatures at site (3) (lower ortho-to-para thermodynamic conversion rate). This could be caused by the smallest pores in TE7 (∼0.4 nm) or the disordered, amorphous nature of TE7 producing curvature or sites with three to four surrounding walls. Preferential adsorption of ortho-H2 has been experimentally observed in other materials41,49,50 and computationally demonstrated in the pores of carbon nanotubes up to 100 K.51,52 In these studies, the generation of non-equilibrium mixtures of ortho:para H2 was explained by the smaller energy gap between the first excited J = 1 rotational sub-level and ground state caused by the high anisotropic potential in nanoscale pores, observing greater spin selectivity in the smallest pores. As it is unlikely that metallic contaminants are the cause of site (3) due to the careful HCl pre-treatment of the porous carbon, these results demonstrate that subnanometre sized pores in activated carbon can also exhibit this preferential adsorption of ortho-H2. Previous quasi-elastic neutron studies on a very similar highly microporous carbon19 also show extreme confinement of H2 in nanopores hinders the conversion of ortho-to-para H2 as observed and described here.
To understand why particular sites seemed to favour higher concentrations of ortho-H2, the potential barrier (V) to out-of-plane rotation from the adsorption sites and rotational constant (B) of confined H2 was calculated by fitting the rotational energy levels to a double-well hindering adsorption potential18 (see Experimental section). Micropore site (2) was consistent with a perturbed 3D rotator model53 and exhibited a Δe of ∼4 meV, corresponding to the estimated values V = −10 meV and B = 7.1 meV, thus promoting adsorption of H2 molecules with their molecular axis oriented parallel to the surface. The potential barrier to rotation V is approximately two times larger than observed for other microporous carbons18,54 and three times larger than that observed in carbon nanotubes35,55 indicating that the H2 is strongly confined in the anisotropic environment of these micropore sites. For micropore site (3), two scenarios are possible: (i) almost free 2D rotations originating from a (J, m) = (1, ±1) → (0, 0) transition, which can occur for strongly attractive potential surfaces with V ∼ −150 meV and B = 7.1 meV or (ii) severely restricted rotations fitting the ‘1D vibrator’ model with the H2 molecular axis oriented perpendicular to the surface41originating from a (J, m) = (1, 0) → (0, 0) transition, with a repulsive anisotropic potential of V ∼ 30 meV and B = 7.35 meV. Due to the low thermal population of states, the higher energy transition of the split states is not expected to be observed for adsorbed ortho-H2, however, these are projected to occur at −55 meV and −21 meV, for scenarios (i) and (ii), respectively. Examining the neutron energy gain spectrum (see ESI – Fig. S10†), a small contribution is observed at −23 meV, very close to that calculated for scenario (ii), indicating a 1D vibrator model at site (3) out of thermal equilibrium. Given both the extremely high interaction potential barrier for scenario (i), and the possible observation of the higher energy split state of scenario (ii), scenario (ii) seems much more probable in the framework of H2 physisorption in a system out of thermodynamic equilibrium. This type of anisotropic potential has not been observed in carbons previously and is more commonly observed for ortho-enrichment surfaces.41 However, in all previous INS studies of H2 in porous carbons the neutron energy gain spectrum has not been analysed in detail16–18,22,35,36,38,42,48,55–57 and therefore may have overlooked the existence of preferentially adsorbing sites with high anisotropic potential caused by extreme confinement. These data are also consistent with previous computational studies that indicate increased potential barrier in nanoscales pores generate greater spin selectivity and produce non-equilibrium mixtures of ortho:para H2 in carbonaceous materials.51,52 Additionally, the calculated mean squared displacement 〈U2〉 of H2 (Table S6 and Fig. S6†) is significantly lower at site (3) than site (2) and (1) by ∼17% and 29% respectively, indicating a smaller delocalization in space – validating stronger confinement, adsorption and perturbed rotation at site (3).
3.4 Effects of nanoscale confinement on stabilization of H2 phases
Considering the relationship of these observed confinement effects to the observed changes to phase behaviour, a greater understanding of why the observed crystalline phases form can be obtained. The decreased thermodynamic conversion of ortho-to-para in pores <1 nm, preferential adsorption, and spin trapping of ortho-H2 at site (3) observed via INS correlate well with the increased proportion of molecularly ordered FCC ortho-H2 found in microporous carbons via neutron diffraction. Likewise, the high level of thermodynamic conversion of ortho-to-para observed in pores >1 nm observed via INS correlates with the lower proportion of the FCC phase and majority HCP phase found in mesoporous carbons. Additionally, the increase in unit cell volume could be related to a small increase in H–H bond length. In site (2), the most widespread micropore site, the calculated rotational constant of H2 deviated from normal, suggesting an elongation to the bond length by ∼+2%. In turn, this could cause an increase in molar volume by ∼6%, similar to what was observed via diffraction (Table S5†). Previous INS studies have also estimated elongation up to 7% for H2 confined in single-walled carbon nanotubes (10, 10).35 However, as bond elongation is dependent on the polarization of the H2 molecule (for example, from an increased level of anisotropic potential such as that found in subnanometre pores) this explanation does not account for the greater increase in molar volume observed in mesoporous carbon, and thus requires further study.Regarding the increase in thermal stability of the FCC ortho-H2 phase confined in microporous carbon, similar changes to phase behaviour has been observed for other small molecules confined in nanoscale pores and have been shown to be related to both the pore size and the ratio of absorbate–substrate:absorbate–absorbate interactions.58–60 In these cases, stronger adsorbate–substrate interactions in nanoscale pores increase the thermal stability of the confined solid61–64 and stronger adsorbate–adsorbate interactions in nanoscale pores reduce the phase transition temperature.64,65 Here we posit that the same mechanism is in play, where substrate–absorbate interactions are greater than absorbate–adsorbate interactions, lowering the total internal energy of the confined crystal with respect to the bulk phase and inducing molecular order in the confined phase at higher temperatures. This can be understood by first comparing the bulk to confined phase FCC ortho-H2 and observing changes to the rotational spectrum. For bulk H2, the lowest energy crystal confirmation for high ortho-H2 concentrations is the FCC phase.12,66 In the HCP phase, the molecules are free to rotate in free space (Fig. 1C), making it a higher energy, molecularly disordered quantum solid. In the FCC phase, the electric quadrupoles (EQQ) of the ortho-H2 molecules orient themselves to lower interaction energy (Fig. 1C), reducing the rotational degrees of freedom of the molecules and reducing the total internal energy of the solid12,67,68 resulting in a molecularly ordered solid. The reduction in energy from EQQ coupling between molecules can be considered the ‘binding energy’ of the FCC phase. This binding energy can be correlated to a drop in the rotational energy across the bulk HCP–FCC transition for various concentrations of ortho-H2.28,30 As the concentration of ortho-H2 increases in the solid, the observed binding energy in the solid also increases. Consequently, increasing EQQ coupling in the solid raises the HCP–FCC transition temperature in tandem.69 However, even for a 100% ortho-H2 solid, the average increase in the binding energy of an ortho molecule to all other ortho molecules in bulk FCC solid is particularly low (∼0.3 meV (ref. 28 and 30)) and explains why there is such a low transition temperature in the bulk (temperature easily overcomes the binding energy and disorders the molecules). Thus, the adsorbate–adsorbate interaction is very weak for solid H2 compared to the interaction energy between adsorbate–substrate of H2 adsorbed to a surface or confined in a pore. Interestingly, changes to rotations and internal energy upon the bulk HCP–FCC transition, are strikingly similar to the changes observed for ortho-H2 upon adsorption/confinement in highly anisotropic potential. As described previously, the binding strength to the interface can be strong enough that the free rotation of H2, which usually persists even in the solid-state, is hindered, accompanied by loss of degrees of rotational freedom and a loss of rotational energy in the process. The stronger the binding, the more significant a perturbation to the rotational energy.70 It is known that ortho-H2 typically binds more strongly to the substrate than para-H252,70–74 due to the electrostatic interaction of the substrate with the electric quadrupoles of ortho-H2 causing preferred orientation or imposed molecular ordering of ortho-H2 on the surface, lowering its rotational energy and increasing its overall binding strength to the substrate. This is demonstrated by higher desorption temperatures/enthalpies of adsorption for the ortho species,70,72,74 and loss of rotational degrees of freedom upon adsorption or confinement.
For a nanopore of dimensions allowing up to 2 layers of H2, the effect on the confined phase is maximised. Here the strong adsorbate–substrate interactions facilitate the molecular ordering required for the low energy FCC phase even at high temperatures by lifting the degeneracy of the J = 1 state and altering the rotational degrees of freedom (Fig. 3c). The strong binding to the surface lowers the rotational energy of all the molecules (Fig. 3e) and hence total internal energy of the confined, 2-layer solid is reduced compared to the bulk equivalent, thus increasing the thermal stability of the FCC phase when confined. This can be semi-quantitatively observed in the changes to the rotational energy spectrum. In the micropores of TE7, the rotational energy of confined ortho-H2 was observed at 13.3 meV and 8 meV as compared to 13.7 meV for bulk FCC ortho-H228,30 indicating the confined ortho-H2 is in a lower rotational energy state than the bulk. Likewise, the calculated potential barriers to rotation in the confined phase are much higher than the EQQ coupling binding energy observed and calculated in the bulk phase.28,30,68,75
With this understanding of how confinement can affect the phase behaviour of H2, the thermal stability of the confined phase could theoretically be increased further. If other materials that exhibit preferential adsorption, high enthalpies of adsorption and significant perturbations to the rotational degrees of freedom of H2, can be engineered to have subnanometre pores similar to those shown in activated carbons, the overlap of anisotropic potential will be much greater and enhance the confinement effects, increasing the proportion and thermal stability of confined FCC ortho-H2 higher than observed here. Temperature-dependent structural studies and rotational dynamics studies of H2 confined in sub-nanometre pores of materials such as Al2O3 or BN would be recommended to test the theory.
The pore geometry may also have an influence on the thermal stability of the confined phase. In porous carbons, the degree of rotational splitting is greater for slit-like pores when compared to cylindrical,18,35,54,55 likewise, calculated densities of confined H2 are also higher17 indicating slit-shaped pores may exhibit a greater enhancement to the thermal stability of FCC ortho-H2. However, the effects could differ in other material compositions with surface inhomogeneities and require further study.
4. Conclusions
Through judicious evaluation of the structural and dynamical properties of H2 confined in porous activated carbons, we have demonstrated that the thermal stability of known crystalline phases of H2 can be enhanced by nanoconfinement to enable departures from the phase diagram of bulk H2. We show adsorbed H2 is distributed across three adsorption environments in the TE7 activated carbon, indicating two micropore sites <1 nm with high potential barriers perturbing the rotations of H2 and a third mesopore site >1 nm with close-to-free rotation, correlating well with the differential pore volume distribution. Uniquely, microporous site (3), believed to correspond to the smallest pores found in the TE7 activated carbon, causes preferential adsorption of ortho-H2 and perpendicular orientation of molecules to the pore surface, which is best described by the 1D vibrator model. The severe restriction of rotation at site (3) also results in a reduced thermodynamic conversion rate from ortho-to-para, resulting in spin trapping down to low temperatures.For the first time, neutron diffraction also revealed that both HCP solid H2 and FCC solid ortho-H2 were present inside both microporous and mesoporous carbon, with a higher FCC:HCP ratio found in microporous carbon and a much lower ratio in mesoporous carbon. The increased proportion of metastable orientationally ordered FCC correlates to the preferentially adsorbed and spin-trapped ortho-H2 found in micropore site (3) as well as the reduced thermodynamic conversion found in pores <1 nm found by INS. Importantly, we discovered that nanoconfinement of H2 significantly increases the structural transition temperature of FCC to HCP H2 up to five times higher than is seen in the bulk at 2 kbar. The increased thermal stability of the orientationally ordered FCC ortho-H2 phase is linked to the high potential barrier to rotation observed in micropore sites, aiding molecular ordering of the molecules, increasing the binding energy of the crystal lattice and lowering the total internal energy of the confined solid with respect to the bulk equivalent. In contrast, the HCP phase followed the expected bulk liquid/solid transition. Calculated unit cells of confined H2 revealed that the unit cell volumes of the adsorbed H2 are smaller in microporous carbon than in mesoporous carbon, but larger than that of bulk H2 at comparable temperatures and pressures. We also determine that above the bulk HCP solid/liquid phase boundary, the H2 may display solid-like properties, but no crystalline phases are observed at the pressures and temperature examined. Further neutron diffraction experiments across 1–200 bar would be useful to understand the confined phase diagram and confirm if the FCC phase can be observed above the critical temperature.
The findings reported here demonstrate how the unique interactions of nanoconstrained spaces with para and ortho-H2 can allow manipulation of the phase diagram. The results present new insight into how materials could be engineered for interactions with individual nuclear spin isomers of H2 to improve temperature stability in hydrogen sorbent systems and applications that require compact and condensed phases of H2. Further alteration of hydrogen's rotational energy levels by an increasingly anisotropic potential in nanoconfined spaces may allow us to manipulate the phase diagram further or achieve other exotic non-equilibrium solid phases of interest to condensed matter physics or hydrogen storage research at more amenable temperatures and pressures.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the US Army Research Office for funding this project (W911NF-19-1-0321-P00001) and the UK Engineering and Physical Sciences Research Council (EPSRC) for an EPSRC Research Fellowship for VPT (EP/R01650X/1). The authors thank the Science and Technology Facilities Council (STFC) for funding and allocation of ISIS beamtime (Proposal number RB1910448) and the Institut Laue Langevin for beamtime (Proposal number IN4C 7-05-468). Thanks also goes to Prof Steve Tennison and Tom Avery (MAST Carbon International) for providing the TE7 carbon beads and Volker Presser (INM, University of Saarbruken, Germany) for providing the OLC carbons, and to Rafael Balderas Xicohtencatl and Anibal J. Ramirez-Cuesta (Oak Ridge National Laboratories) for useful discussions. VPT designed and managed the project and obtained the funding. LT and MT performed the materials characterization and testing. LT, VPT, and MT carried out the neutron experiments with the support of IdS, SR and SB. LT led the data analysis, with all authors contributing to data interpretation and the drafting of the manuscript.References
- S. van Renssen, Nat. Clim. Change, 2020, 10, 799 CrossRef.
- M. I. Eremets and I. a. Troyan, Nat. Mater., 2011, 10, 927 CrossRef CAS PubMed.
- R. P. Dias and I. F. Silvera, Science, 2017, 355, 715 CrossRef CAS PubMed.
- P. Loubeyre, F. Occelli and P. Dumas, Nature, 2020, 577, 631 CrossRef CAS PubMed.
- W. H. Keesom, D. E. Smedt and H. H. Mooy, Commun. Phys. Libr. Leiden Univ., 1930, 19, 814 Search PubMed.
- C. J. Pickard and R. J. Needs, Nat. Phys., 2007, 3, 473 Search PubMed.
- R. T. Howie, C. L. Guillaume, T. Scheler, A. F. Goncharov and E. Gregoryanz, Phys. Rev. Lett., 2012, 108, 125501 CrossRef PubMed.
- P. Dalladay-Simpson, R. T. Howie and E. Gregoryanz, Nature, 2016, 529, 63 CrossRef CAS PubMed.
- R. P. Dias, O. Noked and I. F. Silvera, Phys. Rev. B, 2019, 100, 184112 CrossRef CAS.
- I. Goncharenko and P. Loubeyre, Nature, 2005, 435, 1206 CrossRef CAS PubMed.
- A. F. Schuch, R. L. Mills and D. A. Depatie, Phys. Rev., 1968, 165, 1032 CrossRef CAS.
- I. Silvera, Rev. Mod. Phys., 1980, 52, 393 CrossRef CAS.
- R. L. Mills and A. F. Schuch, Phys. Rev. Lett., 1965, 15, 722 CrossRef CAS.
- B. A. Younglove, J. Phys. Chem. Ref. Data, 1982, 14, 1 Search PubMed.
- N. C. Gallego, L. He, D. Saha, C. I. Contescu and Y. B. Melnichenko, J. Am. Chem. Soc., 2011, 133, 13794 CrossRef CAS PubMed.
- V. P. Ting, A. J. Ramirez-Cuesta, N. Bimbo, J. E. Sharpe, A. Noguera-Diaz, V. Presser, S. Rudic and T. J. Mays, ACS Nano, 2015, 9, 8249 CrossRef CAS PubMed.
- M. Tian, M. J. Lennox, A. J. O'Malley, A. J. Porter, B. Krüner, S. Rudić, T. J. Mays, T. Düren, V. Presser, L. R. Terry, S. Rols, Y. Fang, Z. Dong, S. Rochat and V. P. Ting, Carbon, 2021, 173, 968 CrossRef CAS.
- J. Bahadur, C. I. Contescu, A. J. Ramirez-Cuesta, E. Mamontov, N. C. Gallego, Y. Cheng, L. L. Daemen and Y. B. Melnichenko, Carbon, 2017, 117, 383 CrossRef CAS.
- C. I. Contescu, D. Saha, N. C. Gallego, E. Mamontov, A. I. Kolesnikov and V. V. Bhat, Carbon, 2012, 50, 1071 CrossRef CAS.
- J. E. Sharpe, N. Bimbo, V. P. Ting, A. D. Burrows, D. Jiang and T. J. Mays, Adsorption, 2013, 19, 643 CrossRef CAS.
- L. He, Y. B. Melnichenko, N. C. Gallego, C. I. Contescu, J. Guo and J. Bahadur, Carbon, 2014, 80, 82 CrossRef CAS.
- R. J. Olsen, A. K. Gillespie, C. I. Contescu, J. W. Taylor, P. Pfeifer and J. R. Morris, ACS Nano, 2017, 11, 11617 CrossRef CAS PubMed.
- A. J. Ramirez-Cuesta, M. O. Jones and W. I. F. David, Mater. Today, 2009, 12, 54 CrossRef CAS.
- M. Zeiger, N. Jäckel, M. Aslan, D. Weingarth and V. Presser, Carbon, 2015, 84, 584 CrossRef CAS.
- W. G. Williams, R. M. Ibberson, P. Day and J. E. Enderby, Physica B: Condens. Matter, 1997, 241–243, 234 CrossRef CAS.
- A. Hruzewicz-Kołodziejczyk, V. P. Ting, N. Bimbo and T. J. Mays, Int. J. Hydrogen Energy, 2012, 37, 2728 CrossRef.
- T. Roussel, R. J. M. Pellenq, M. Bienfait, C. Vix-Guterl, R. Gadiou, F. Béguin and M. Johnson, Langmuir, 2006, 22, 4614 CrossRef CAS PubMed.
- H. Stein, H. Stiller and R. Stockmeyer, J. Chem. Phys., 1972, 57, 1726 CrossRef CAS.
- P. E. Sokol, R. T. Azuah, R. M. Gibbs and S. M. Bennington, J. Low Temp. Phys., 1996, 103, 23 CrossRef CAS.
- A. Bickermann, H. Spitzer, H. Stiller and R. Scherm, Z. Phys. B: Condens. Matter Quanta, 1978, 31, 339 CrossRef CAS.
- W. Xu, X. Di Liu, M. Peña-Alvarez, H. C. Jiang, P. Dalladay-Simpson, B. Coasne, J. Haines, E. Gregoryanz and M. Santoro, J. Phys. Chem. C, 2021, 125, 7511 CrossRef CAS.
- J. X. Yang, H. L. Zhao, H. R. Gong, M. Song and Q. Q. Ren, Sci. Rep., 2018, 8, 1 Search PubMed.
- B. Li, G. Qian, A. R. Oganov, S. E. Boulfelfel and R. Faller, J. Chem. Phys., 2017, 146, 214502 CrossRef PubMed.
- A. Driessen and I. F. Silvera, J. Low Temp. Phys., 1984, 54, 361 CrossRef CAS.
- P. A. Georgiev, D. K. Ross, A. De Monte, U. Montaretto-Marullo, R. A. H. Edwards, A. J. Ramirez-Cuesta, M. A. Adams and D. Colognesi, Carbon, 2005, 43, 895 CrossRef CAS.
- P. A. Georgiev, D. K. Ross, P. Albers and A. J. Ramirez-Cuesta, Carbon, 2006, 44, 2724 CrossRef CAS.
- A. J. Ramirez-Cuesta, P. C. H. Mitchell, D. K. Ross, P. A. Georgiev, P. A. Anderson, H. W. Langmi and D. Book, J. Mater. Chem., 2007, 17, 2533 RSC.
- R. J. Olsen, L. Firlej, B. Kuchta, H. Taub, P. Pfeifer and C. Wexler, Carbon, 2011, 49, 1663 CrossRef CAS.
- K. Fukutani and T. Sugimoto, Prog. Surf. Sci., 2013, 88, 279 CrossRef CAS.
- D. White and E. N. Lassettre, J. Chem. Phys., 1960, 32, 72 CrossRef CAS.
- I. F. Silvera and M. Nielsen, Phys. Rev. Lett., 1976, 37, 1275 CrossRef CAS.
- C. M. Brown, T. Yildirim, D. A. Neumann, M. J. Heben, T. Gennett, A. C. Dillon, J. L. Alleman and J. E. Fischer, Chem. Phys. Lett., 2000, 329, 311 CrossRef CAS.
- P. D. C. Dietzel, P. A. Georgiev, J. Eckert, R. Blom, T. Strässle and T. Unruh, Chem. Commun., 2010, 46, 4962 RSC.
- C. M. Brown, A. J. Ramirez-Cuesta, J. H. Her, P. S. Wheatley and R. E. Morris, Chem. Phys., 2013, 427, 3 CrossRef CAS.
- J. Z. Larese, T. Arnold, L. Frazier, R. J. Hinde and A. J. Ramirez-Cuesta, Phys. Rev. Lett., 2008, 101, 165302 CrossRef CAS PubMed.
- S. K. Callear, A. J. Ramirez-Cuesta, W. I. F. David, F. Millange and R. I. Walton, Chem. Phys., 2013, 427, 9 CrossRef CAS.
- A. J. Ramirez-Cuesta, P. C. H. Mitchell, D. K. Ross, P. A. Georgiev, P. A. Anderson, H. W. Langmi and D. Book, J. Alloys Compd., 2007, 446–447, 393 CrossRef CAS.
- S. Mamone, M. Jiménez-Ruiz, M. R. Johnson, S. Rols and A. J. Horsewill, Phys. Chem. Chem. Phys., 2016, 18, 29369 RSC.
- T. R. Prisk, M. S. Bryan and P. E. Sokol, Phys. Chem. Chem. Phys., 2014, 16, 17560 RSC.
- D. W. Brown, P. E. Sokol and S. A. Fitzgerald, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 258 Search PubMed.
- T. Lu, E. M. Goldfield and S. K. Gray, J. Phys. Chem. B, 2006, 110, 1742 CrossRef CAS PubMed.
- R. A. Trasca, M. K. Kostov and M. W. Cole, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 1 CrossRef.
- P. C. H. Mitchell, S. F. Parker, A. J. Ramirez-Cuesta and J. Tomkinson, Vibrational Spectroscopy with Neutrons: With Applications in Chemistry, Biology, Materials Science and Catalysis, World Scientific, Hackensack, NJ, 2005 Search PubMed.
- R. J. Olsen, M. Beckner, M. B. Stone, P. Pfeifer, C. Wexler and H. Taub, Carbon, 2013, 58, 46 CrossRef CAS.
- F. Fernandez-Alonso, F. J. Bermejo, C. Cabrillo, R. O. Loutfy, V. Leon and M. L. Saboungi, Phys. Rev. Lett., 2007, 98, 2 Search PubMed.
- Y. Ren and D. L. Price, Appl. Phys. Lett., 2001, 79, 3684 CrossRef CAS.
- P. A. Georgiev, D. K. Ross, A. De Monte, U. Montaretto-Marullo, R. A. H. Edwards, A. J. Ramirez-Cuesta and D. Colognesi, J. Phys.: Condens. Matter, 2004, 16, L73 CrossRef CAS.
- R. Radhakrishnan, K. E. Gubbins and M. Sliwinska-Bartkowiak, J. Chem. Phys., 2002, 116, 1147 CrossRef CAS.
- C. Alba-Simionesco, B. Coasne, G. Dosseh, G. Dudziak, K. E. Gubbins, R. Radhakrishnan and M. Sliwinska-Bartkowiak, J. Phys.: Condens. Matter, 2006, 18, R15 CrossRef CAS PubMed.
- B. K. Peterson, J. P. R. B. Walton and K. E. Gubbins, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 1789 RSC.
- K. Kaneko, A. Watanabe, T. Iiyama, R. Radhakrishan and K. E. Gubbins, J. Phys. Chem. B, 1999, 103, 7062 CrossRef.
- R. Radhakrishnan, K. E. Gubbins, A. Watanabe and K. Kaneko, J. Chem. Phys., 1999, 111, 9058 CrossRef CAS.
- A. Watanabe, T. Iiyama and K. Kaneko, Chem. Phys. Lett., 1999, 305, 71 CrossRef CAS.
- M. Sliwinska-Bartkowiak, G. Dudziak, R. Sikorski, R. Gras, K. E. Gubbins and R. Radhakrishnan, Phys. Chem. Chem. Phys., 2001, 3, 1179 RSC.
- S. Gautam, A. I. Kolesnikov, G. Rother, S. Dai, Z.-A. Qiao and D. Cole, J. Phys. Chem. A, 2018, 122, 6736 CrossRef CAS PubMed.
- N. S. Sullivan, Can. J. Chem., 1988, 66, 908 CrossRef CAS.
- N. S. Sullivan and R. V. Pound, Phys. Rev. A, 1972, 6, 1102 CrossRef CAS.
- H. M. James and J. C. Raich, Phys. Rev., 1967, 162, 649 CrossRef CAS.
- R. L. Mills, A. F. Schuch and D. A. Depatie, Phys. Rev. Lett., 1966, 17, 1131 CrossRef CAS.
- A. Van Itterbeek, R. Hellemans and W. Van Dael, Physica, 1964, 30, 324 CrossRef CAS.
- T. Sugimoto and K. Fukutani, Phys. Rev. Lett., 2014, 112, 1 CrossRef PubMed.
- J. Heidberg, N. Gushanskaya and R. Schwarte, Surf. Sci., 1995, 333, 1473 CrossRef.
- T. Magome, K. Fukutani and T. Okano, J. Vac. Soc. Jpn., 1999, 42, 286 CrossRef CAS.
- L. Amiaud, A. Momeni, F. Dulieu, J. H. Fillion, E. Matar and J. L. Lemaire, Phys. Rev. Lett., 2008, 100, 1 CrossRef PubMed.
- A. B. Harris and A. J. Berlinsky, Theoretical Analysis of Inelastic Neutron Scattering in Solid Hydrogen, 1977 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: EDX, ICP-OES and pore characteristics of the activated carbons, neutron and X-ray powder diffraction data, unit cell dimensions, inelastic neutron scattering peak parameters and ortho/para-H2 distributions. See DOI: https://doi.org/10.1039/d2nr00587e |
| This journal is © The Royal Society of Chemistry 2022 |