
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular electronic refrigeration against parallel phonon heat leakage channels
Fatemeh
Tabatabaei
a,
Samy
Merabia
 a,
Bernd
Gotsmann
b,
Mika
Prunnila
c and
Thomas A.
Niehaus
*a
a,
Bernd
Gotsmann
b,
Mika
Prunnila
c and
Thomas A.
Niehaus
*a
aUniversité Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, Villeurbanne, France. E-mail: thomas.niehaus@univ-lyon1.fr
bIBM Research Europe – Zurich, Rueschlikon, Switzerland
cVTT Technical Research Centre of Finland Ltd., Tietotie 3, FI-02150 Espoo, Finland
First published on 14th July 2022
Abstract
Due to their structured density of states, molecular junctions provide rich resources to filter and control the flow of electrons and phonons. Here we compute the out of equilibrium current–voltage characteristics and dissipated heat of some recently synthesized oligophenylenes (OPE3) using the Density Functional based Tight-Binding (DFTB) method within Non-Equilibrium Green's Function Theory (NEGF). We analyze the Peltier cooling power for these molecular junctions as function of a bias voltage and investigate the parameters that lead to optimal cooling performance. In order to quantify the attainable temperature reduction, an electro-thermal circuit model is presented, in which the key electronic and thermal transport parameters enter. Overall, our results demonstrate that the studied OPE3 devices are compatible with temperature reductions of several K. Based on the results, some strategies to enable high performance devices for cooling applications are briefly discussed.
The advance of experimental techniques to measure the transport properties of molecular junctions1–3 has motivated many theoreticians to investigate these devices.4,5 Most theoretical studies have been done considering the system at equilibrium, that is without an applied bias potential. This is motivated by the fact that key characterizing parameters of the junction like the conductance G, the Seebeck coefficient S and the figure of merit ZT for thermoelectric applications are defined as linear response properties in the limit of vanishing bias.6 In addition, out-of-equilibrium simulations are technically more demanding, as the electro-static potential in the molecular region needs to be determined accurately. On the experimental side, the determination of currents under sizable bias is likewise difficult, given that the electric field can destabilize the delicate metal–molecule bonding. However, to take full advantage of a molecular junction, identifying charge and energy transport out of equilibrium is a necessity.7,8 Studying the I–V characteristics of molecular junctions provides a complete picture of the transport mechanisms at play.
In this field, first principles calculations are a powerful tool to accompany and rationalize experimental data. It has been shown that the Non-equilibrium Green's function (NEGF) method in conjunction with Density Functional Theory (DFT) is well suited to calculate electronic properties of single molecular junctions with or without applied bias.7,9 Besides charge transport, recent efforts in the field of molecular electronics focus also on the question how heat is transported and dissipated in the device (see ref. 11 and 12 for recent reviews). Both electrons and phonons participate in this process, while photon based transfer can typically be neglected. Measurements of the Seebeck coefficient quantify the induced voltage by an applied temperature gradient and are important to investigate the electronic channel.8,13–16 In a recent study, direct Peltier cooling through molecular junctions was demonstrated,17 which is a crucial first step to develop bottom-up cooling devices.18
In this study, we combine parallel electronic and phononic channels of heat transport in a electro-thermal circuit model to provide realistic estimates of the temperature decrease that can be actually reached in prototypical molecular junctions. Note, that we do not evaluate the electron–phonon (e–ph) coupling, which can strongly affect the thermopower in some cases.19–24 As we argue below, this coupling of the channels is not expected to change our main findings.
We study molecular junctions incorporating three oligo (phenyleneethynylene) derivatives (OPE3) (Fig. 1) which have been previously characterized in equilibrium both experimentally and theoretically.10,25 Similar OPE oligoynes were investigated with respect to their electronic transport,26 thermoelectric27,28 and phonon transport properties.29 In a first step, we compute the current–voltage curves using a NEGF formalism based on the approximate DFT method DFTB.30–32 The bias-dependent transmission is then employed to calculate the dissipated heat in the electrodes over a wide range of applied voltages and temperature gradients. Optimal parameter ranges are determined and used in an electro-thermal circuit model that takes into account the phonon heat backflow through the molecule and the parasitic heat leakage of typical experimental setups. It will be shown that under these optimal conditions a cooling power of several nW through a molecule can be reached.
 | ||
| Fig. 1 OPE3 derivatives with dihydrobenzo[b]thiophene (DHBT) anchoring groups and different side chains. Reproduced from ref. 10. | ||
1. Methods
In the past, the approximate DFT method DFTB has been applied successfully to compute transport characteristics of various molecular devices.10,32–37 DFTB is characterized by a second-order expansion of the DFT total energy functional around a suitably chosen reference density. The methods numerical efficiency stems from pre-calculated Hamiltonian matrix elements and the partial neglect and approximation of two-electron integrals.30,38 Here we use DFTB combined with NEGF theory as implemented in the DFTB+ code.39 As described in more detail in ref. 32, the transmission t(E, V) is computed as| t = Tr(ΓLGrΓRGa), | (1) |
 | (2) |
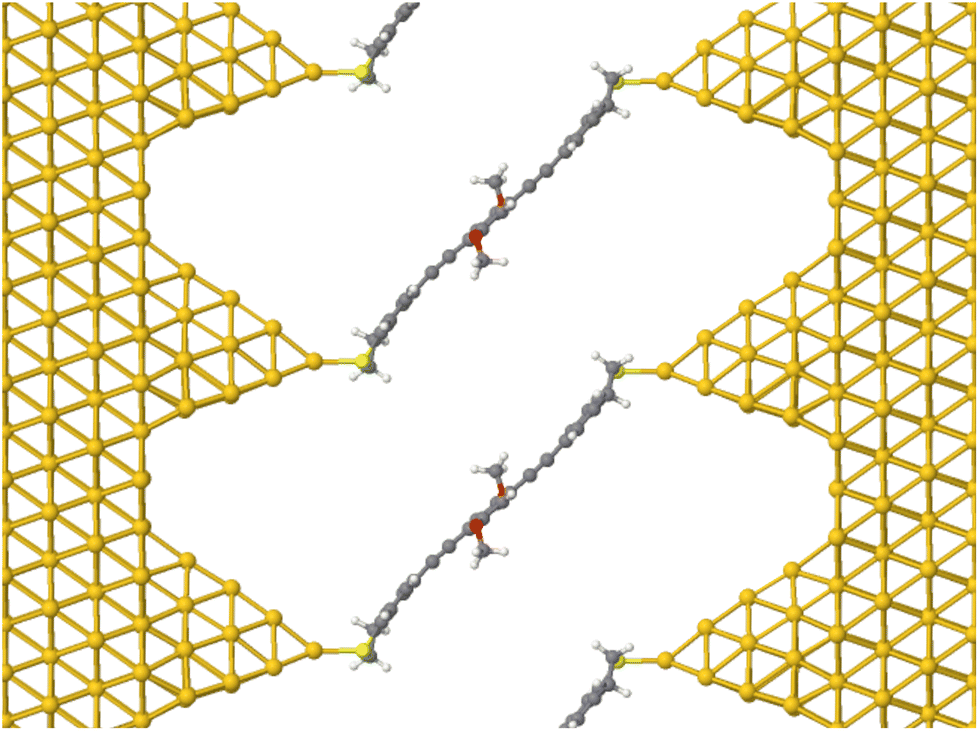 | ||
| Fig. 2 Device geometry for DFTB transport simulations. Shown here is OPE3-Ph(OMe)2 connected to semi-infinite Au(111) leads. The molecules feature a DHBT anchor group (see Fig. 1) that binds to an Au20 cluster, mimicking the tip of a Scanning Tunneling Microscope (STM). Reproduced from ref. 10. | ||
2. Results and discussion
2.1. Current–voltage characteristics
Before discussing the bias dependence of the electronic transport, it seems worthwhile to review the equilibrium properties of the three OPE3 derivatives shortly. Fig. 3 shows the transmission function t(E) taken from ref. 25. The key finding is that all molecules feature a lowest unoccupied molecular orbital (LUMO) that is closer to the Fermi energy than the highest occupied molecular orbital (HOMO). This indicates that for weak bias transport occurs predominantly through the tails of the LUMO resonance. The Seebeck coefficient (also called thermopower) | (3) |
 | ||
| Fig. 3 DFTB transmission for OPE3 derivatives as investigated in ref. 10. For illustrative purposes the DFTB frontier orbital energies of the isolated molecules in the gas phase are given at the top of the figure. Reproduced from ref. 25. | ||
We now go beyond the investigations carried out in ref. 10 and 25. We apply a finite symmetric bias voltage to both leads, such that the chemical potential of the left and the right lead changes to μR = EF − eV/2 and μL = EF + eV/2, respectively, where e = |e| denotes the absolute value of the electron charge. Hence, a current flows from the left lead to the right lead for negative bias. Non-equilibrium transport simulations require a self-consistent cycle in which the Hamiltonian is iteratively updated with the device potential, which in turn leads to a change in the charge density used in the Poisson equation. For large bias values the electronic structure is strongly perturbed which may lead to convergence difficulties. In our simulations we were able to obtain results for bias values up to ±1 V.
In Fig. 4 we show exemplarily the potential in the device region for OPE3-Ph coupled perpendicularly to the leads at V = −1 V and V = 0 V. At equilibrium, the potential is nearly symmetrical with respect to the center of the molecule, which is expected since the device is symmetrical. For the junction under bias, the potential drops linearly along the molecular part with some fluctuations on the atomic scale. The potential in the metallic Au20 pyramids is similar to the equilibrium and just shifted by ±0.5 V as a whole, indicating a rather efficient screening of the field in this small cluster.
 | ||
| Fig. 4 (Top panel) 2D color map of the electrostatic potential (xy-plane crossing the apexes of the gold pyramids) in the device region for OPE3-Ph and sketch of the device region. The scale bar on the top right is in units of V. (bottom panel) Potential along the line indicated in the top panel. Small circles with the same color show the same coordinate in transport direction. (a) OPE3-Ph under bias 0 V, (b) OPE3-Ph under bias −1 V. | ||
Given the smooth density of states for the gold electrodes, as well as the nearly symmetrical metal–molecule coupling, we do not expect major changes in the transport characteristics compared to the equilibrium. The applied bias has however a non-negligible effect on the transmission of the junction as shown in Fig. 5a exemplarily for OPE3-Ph. For V in the range [0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.8] V, the LUMO resonance (around 0.4 eV) does not shift strongly in energy but exhibits a reduced transmission at higher bias. Large changes are also found around E − EF = −0.5 V, where the weakly transmitting resonance diminishes as the bias increases and a new feature directly at the Fermi energy arises. In ref. 42, the corresponding state at E − EF = −0.5 V was tentatively assigned to a localized state at the molecule–Au20 interface, which has no gas phase counterpart in contrast to the HOMO/LUMO frontier orbitals that keep their spatial form also in the metal–molecule–metal complex. Such localized features have been coined chemisorption-induced gap states (CIGS)43 and have been investigated theoretically44,45 and experimentally46 for a variety of metal–molecule interfaces. For oligomer junctions, the presence of CIGS can be detected by the length dependence of the thermopower.47
:0.8] V, the LUMO resonance (around 0.4 eV) does not shift strongly in energy but exhibits a reduced transmission at higher bias. Large changes are also found around E − EF = −0.5 V, where the weakly transmitting resonance diminishes as the bias increases and a new feature directly at the Fermi energy arises. In ref. 42, the corresponding state at E − EF = −0.5 V was tentatively assigned to a localized state at the molecule–Au20 interface, which has no gas phase counterpart in contrast to the HOMO/LUMO frontier orbitals that keep their spatial form also in the metal–molecule–metal complex. Such localized features have been coined chemisorption-induced gap states (CIGS)43 and have been investigated theoretically44,45 and experimentally46 for a variety of metal–molecule interfaces. For oligomer junctions, the presence of CIGS can be detected by the length dependence of the thermopower.47
 | ||
| Fig. 5 (a) Evolution of the electronic transmission under bias for OPE3-Ph, (b) the current as a function of bias for OPE3 derivatives. | ||
The current–voltage curves for the different OPE3 derivatives are shown in Fig. 5b. In general, the I–V characteristics are nonlinear, as reported for other organic molecular junctions.48,49 Additionally, the symmetry of I–V curves proves that the coupling to the left and right leads is very similar in our transport device.49,50 As the bias value increases, OPE3-An and OPE3-Ph(OMe)2 show a higher value of current compared to OPE3-Ph. This is due to the larger transmission around the Fermi energy (cf. Fig. 3) for these two molecules. Around V = 0.5 V all systems show a steeper increase of the current, since the LUMO resonance centered at ≈0.4 eV starts to enter the bias window which extends from −V/2 to V/2. Given the larger transmission of OPE3-An at the LUMO resonance, the highest current values are obtained for this compound.
2.2. Heat dissipation out of equilibrium
Next, we used the bias dependent transmission to estimate the dissipated heat in the leads. The power PL associated with the total heat dissipated in the left lead is given within the Landauer–Büttiker formalism as:17 | (4) |
Note that the total dissipated heat includes both Joule heating and the Peltier effect, consequently, net refrigeration happens when PL < 0. Fig. 6 depicts the results for the different OPE3 derivatives. Shown in the first column is a 2D plot of PL as a function of bias V and the shift Δ of the Fermi energy towards more positive values. Experimentally such a shift can be realized by gating in a three-terminal device51 or by means of electro-chemical gating.52 Our motivation to investigate the effect of modifying the Fermi energy (or equivalently, the energetical position of the molecular levels) is twofold. First, as will we explained below, the attainable cooling power increases strongly when the Fermi energy approaches a molecular resonance. Second, like for most DFT based methods, the calculation of the precise Fermi level alignment between molecule and metal is quite delicate. Our approach predicted LUMO based transport in full agreement with the experimental results, but the thermopower was overestimated in absolute terms, which speaks for a slight underestimation of the ELUMO − EF difference. By modifying EF we arrive at more general conclusions.
 | ||
| Fig. 6 (left column) Bias dependent net heating/cooling in OPE3 derivatives. Shown is the power dissipated in the left lead PL for TL = TR = 300 K as a function of bias voltage and the shift of the Fermi energy (Δ) towards the LUMO. Red color indicates positive values (heating) and blue color negative values (cooling). (a) OPE3-Ph, (c) OPE3-Ph(OMe)2, (e) OPE3-An (right column) PL as function of bias and ΔT = TL − TR, with TR = 300 K and Δ = 0 eV. (b) OPE3-Ph, (d) OPE3-Ph(OMe)2, (f) OPE3-An. | ||
If we consider first the results for OPE3-An in Fig. 6e, three different transport scenarios may be distinguished that are schematically presented in Fig. 7. For Δ < 0.2 eV (corresponding to Fig. 7a), the Fermi level is relatively far away from the LUMO. For V > 0, hot charge carriers in the left lead may only transfer to the right through the tails of the LUMO resonance. The factor μL − E in eqn (4) is negative in this case, but the cooling effect in the left lead is weak. For 0.2 eV < Δ < 0.4 eV (corresponding to Fig. 7b), hot electrons from the left lead are close to the LUMO resonance and have a high probability for transmission. The resulting current leads to cooling of the left lead. Note that this Peltier cooling is compensated by a heating of the right lead such that total dissipated heat is always given by PL + PR = IV, which corresponds to Joule heating.17 Finally, for Δ > 0.4 eV, the LUMO resonance is placed in the bias window [μL, μR] or below and electrons with energy E < μL are predominantly transmitted. This results in heating of the left lead. Changing the bias polarity inverts the roles of left and right lead as can be seen in Fig. 6e. From the foregoing discussion it might appear beneficial to increase the bias potential, instead of – or in addition to – gating the device, in order to achieve maximal cooling. Also in this case the chemical potential μL approaches the LUMO resonance and electrons transmitted at E > μL effectively shuffle heat from L to R. This is however more than offset by the transport in the increased bias window which always leads to (Joule) heating. Consequently, in Fig. 6a, c and e, cooling is only observed in a narrow region around vanishing bias.
 | ||
| Fig. 7 Schematic representation of Peltier cooling in molecular devices. The right lead is at lower chemical potential (V > 0), electrons move from left to right. The occupation probability of the electrons in the two leads (T > 0) is shown in blue color versus energy and the dotted line corresponds to the Fermi energy of the device. Gating leads to a shift of the molecular density of states, shown in grey color, which is represented here by HOMO and LUMO levels. (a) μL far away from LUMO, moderate cooling of the left lead, (b) μL close to resonance, strong cooling of left lead, (c) μL above resonance, heating of left lead. | ||
Comparing the different OPE3 derivatives, we see that the dissipated power reflects the different transmission characteristics shown in Fig. 3. OPE3-An features ideal LUMO transmission (t = 1) at E − EF = 0.4 eV which translates into the simple power spectrum Fig. 6e discussed above. Both OPE3-Ph and OPE3-(OMe)2 exhibit weaker LUMO transmission (t = 0.01 at E − EF = 0.4 eV) and have near perfect transmission only at higher energies, namely at 0.7 V and 0.8 V, respectively. From this, the broader region of cooling (0.4 eV < Δ < 0.8 eV) observed in Fig. 6a and c can be understood. Interpolation of the bias dependent transmission allows us to quantify the maximum cooling power (PoptL) for each of the OPE3 derivatives used in this study. The results are presented in Table 1 and show that nW cooling may be reached at moderate bias and gating for OPE3-An.
| Molecule | V opt (V) | Δ opt (eV) | P optL (nW) |
|---|---|---|---|
| OPE3-Ph | −0.04 | 0.8 | −8.25 |
| OPE3-Ph(OMe) 2 | −0.03 | 0.9 | −9.29 |
| OPE3-An | 0.06 | 0.3 | −11.29 |
We also shortly discuss the dependence of PL on the temperature difference (ΔT = TL − TR) between the two leads, keeping TR fixed at 300 K. Fig. 6b, d, and f indicate a rather weak dependence on this parameter. At zero bias, cooling occurs if the left lead is at higher temperature as the left lead, in line with the schematic representations given in Fig. 7a.
2.3. Electro-thermal circuit for OPE3 derivatives
In this section we provide estimates for the temperature reduction that can be reached in real devices based on a simple electro-thermal circuit. To do so, we base our discussion on the experimental setup which was used to measure the charge and heat transport for one of the molecular junctions (OPE3-Ph) used in this study.53,54 In the experiment, a micro-electro-mechanical system (MEMS) was suspended using four silicon nitride beams. The characteristic thermal conductance of this support can be reduced to below κPhsupp = 10–8 W K−1. In the central membrane of the MEMS device, a thermometer and a gold surface are located on which the molecules are deposited. Heat and electronic transport measurements are then carried out upon contact with a scanning tunnelling microscope (STM) tip.We model this setup with the electro-thermal circuit as shown in Fig. 8, which was inspired by a similar model for a semiconductor–superconductor thermionic junction,54 where the quasiparticle gap gives similar energy filtering as the HOMO–LUMO gap in this work. Here the left lead is identified with the gold surface on the central MEMS platform, while the right lead corresponds to the STM tip. The temperature of the tip stays constant due to the good thermal contact with the environment. When the net heating power is negative, the left lead cools down due to the Peltier effect which is of electronic origin. In the phonon channel, the thermal conductance of both molecule and the support need to be considered. At equilibrium, one has:
| PelL + Pphsupp + Pphmol = 0, | (5) |
 | ||
| Fig. 8 Illustration of the electro-thermal circuit used to model the temperature gradient in molecular junctions. | ||
As mentioned above, we also neglect effects of direct e–ph coupling, which leads to significant changes of the thermopower if small variations in the molecular geometry entail large changes in the electronic conductance. As an example, Sergueev and co-workers report on a biphenyl junction that exhibits two major configurations.24 The low energy configuration features a small relative torsion of the phenyl rings and has high conductance, while the other configuration is characterized by orthogonal phenyl rings. Due to the breaking of the conducting π-system, the conductance of the latter is very sensitive to torsional vibrations. Consequently there is a strong increase of |S| under appropriate gating, if e–ph interactions are taken into account. In contrast, the backbone of the OPE3 derivatives studied here is rigid. We performed additional MD simulations† for OPE-An showing that the molecule remains planar during a 20 ns trajectory with angles between the phenyl groups of maximal 10°. Our experimental and theoretical studies also indicate low conformational flexibility with respect to the metal–molecule binding due to the nature of the DHBT anchor group.10 We therefore expect only modest effects of e–ph coupling even under gating.
As additional approximation we will assume that the thermal conductances are not temperature dependent. This is well justified, because the temperatures considered here are well above the Debye temperature of the gold contacts. We do, however, account for the temperature dependence of PL according to eqn (4) and solve eqn (5) in the form
 | (6) |
 | ||
| Fig. 9 Temperature difference between both leads ΔT versus thermal conductance of the support (TR = 300 K, κphmol = 22 pW K−1). | ||
3. Conclusion
In summary, we performed NEGF transport simulations at the DFTB level for three OPE3 derivatives that were recently synthesized and experimentally characterized. Full I–V curves were computed and show a strong current increase at around 0.4 eV when the LUMO resonance enters the window of conduction. OPE3-Au exhibits the largest currents in the studied bias range, which can be explained by its enhanced LUMO transmission compared to the other two molecules. The voltage bias dependent transmission was then used to quantify the heat transport through the molecules under non-equilibrium conditions. It was found that junction gating has a profound beneficial impact on the cooling power which can reach several nW under optimal conditions. Cooling is observed at small bias voltages and large bias leads to simple Ohmic heating. Given that good electrostatic control of molecular junctions is still difficult to achieve, a viable strategy might consist of pushing the frontier orbitals closer to the Fermi energy. This could be achieved by appropriate electron-withdrawing or donating functional groups or using quantum interference effects as recently discussed by several groups.62,63 Finally, we used a combination of experimental data and the theoretical results of this study to set up an electro-thermal circuit model that combines the electronic and phononic transport channels. As a result, we obtained promising values for the attainable temperature reduction of several K in these bottom-up molecular devices. Further improvements are expected for cross-linked molecular architectures. Simulations of these more complex devices are currently under way.Author contributions
F. Tabatabaei: data curation; formal analysis; investigation; software; writing original draft. S. Merabia, B. Gotsmann, M. Prunnila: conceptualization; methodology; validation; writing – review and editing. T.A. Niehaus: conceptualization; methodology; formal analysis; project administration; supervision; writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the European Commission H2020 projects ‘EFINED’ http://www.efined-h2020.eu Grant Agreement No. 766853 for providing financial resources to our project. We thank the GENCI for computational resources (under project DARI A0050810637 and A0070810637). MP acknowledges financial support of the Academy of Finland through project HyPhEN (No. 342586) and Centre of Excellence Program No. 336817. TAN would like to thank Alessandro Pecchia for advice and access to the gdftb code. We thank the EFINED team for inspiring discussions.References
- J. C. Cuevas and E. Scheer, Molecular Electronics: An Introduction to Theory and Experiment, World Scientific, 2010 Search PubMed.
- A. Nitzan and M. A. Ratner, Science, 2003, 300, 1384–1389 CrossRef CAS PubMed.
- H. Song, M. A. Reed and T. Lee, Adv. Mater., 2011, 23, 1583–1608 CrossRef CAS PubMed.
- M. Koentopp, C. Chang, K. Burke and R. Car, J. Phys.: Condens. Matter, 2008, 20, 083203 CrossRef.
- F. Evers, R. Korytár, S. Tewari and J. M. van Ruitenbeek, Rev. Mod. Phys., 2020, 92, 035001 CrossRef CAS.
- B. K. Nikolić, K. K. Saha, T. Markussen and K. S. Thygesen, J. Comput. Electron., 2012, 11, 78–92 CrossRef.
- P. Darancet, J. R. Widawsky, H. J. Choi, L. Venkataraman and J. B. Neaton, Nano Lett., 2012, 12, 6250–6254 CrossRef CAS PubMed.
- A. Tan, S. Sadat and P. Reddy, Appl. Phys. Lett., 2010, 96, 013110 CrossRef.
- M. Brandbyge, J.-L. Mozos, P. Ordejón, J. Taylor and K. Stokbro, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 165401 CrossRef.
- H. Dekkiche, A. Gemma, F. Tabatabaei, A. S. Batsanov, T. Niehaus, B. Gotsmann and M. R. Bryce, Nanoscale, 2020, 12, 18908–18917 RSC.
- K. Wang, E. Meyhofer and P. Reddy, Adv. Funct. Mater., 2020, 30, 1904534 CrossRef CAS.
- S. Park, J. Jang, H. Kim, D. I. Park, K. Kim and H. J. Yoon, J. Mater. Chem. A, 2020, 8, 19746–19767 RSC.
- P. Reddy, S.-Y. Jang, R. A. Segalman and A. Majumdar, Science, 2007, 315, 1568–1571 CrossRef CAS PubMed.
- J. R. Widawsky, P. Darancet, J. B. Neaton and L. Venkataraman, Nano Lett., 2012, 12, 354–358 CrossRef CAS PubMed.
- S. Guo, G. Zhou and N. Tao, Nano Lett., 2013, 13, 4326–4332 CrossRef CAS PubMed.
- P. Gehring, J. K. Sowa, C. Hsu, J. de Bruijckere, M. van der Star, J. J. Le Roy, L. Bogani, E. M. Gauger and H. S. van der Zant, Nat. Nanotechnol., 2021, 16, 426–430 CrossRef CAS PubMed.
- L. Cui, R. Miao, K. Wang, D. Thompson, L. A. Zotti, J. C. Cuevas, E. Meyhofer and P. Reddy, Nat. Nanotechnol., 2018, 13, 122–127 CrossRef CAS PubMed.
- J. Ding, W. Zhao, W. Jin, C.-a. Di and D. Zhu, Adv. Funct. Mater., 2021, 31, 2010695 CrossRef CAS.
- O. Entin-Wohlman, Y. Imry and A. Aharony, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 115314 CrossRef.
- M. Leijnse, M. Wegewijs and K. Flensberg, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 045412 CrossRef.
- D. M.-T. Kuo, Jpn. J. Appl. Phys., 2010, 49, 095205 CrossRef.
- M. Galperin, A. Nitzan and M. A. Ratner, Mol. Phys., 2008, 106, 397–404 CrossRef CAS.
- D. Segal, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 165426 CrossRef.
- N. Sergueev, S. Shin, M. Kaviany and B. Dunietz, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195415 CrossRef.
- H. Dekkiche, A. Gemma, F. Tabatabaei, A. S. Batsanov, T. Niehaus, B. Gotsmann and M. R. Bryce, Nanoscale, 2021, 13, 4685–4686 RSC.
- P. Moreno-García, M. Gulcur, D. Z. Manrique, T. Pope, W. Hong, V. Kaliginedi, C. Huang, A. S. Batsanov, M. R. Bryce and C. Lambert, et al. , J. Am. Chem. Soc., 2013, 135, 12228–12240 CrossRef PubMed.
- B. Huang, X. Liu, Y. Yuan, Z.-W. Hong, J.-F. Zheng, L.-Q. Pei, Y. Shao, J.-F. Li, X.-S. Zhou and J.-Z. Chen, et al. , J. Am. Chem. Soc., 2018, 140, 17685–17690 CrossRef CAS PubMed.
- R. Miao, H. Xu, M. Skripnik, L. Cui, K. Wang, K. G. Pedersen, M. Leijnse, F. Pauly, K. Warnmark and E. Meyhofer, et al. , Nano Lett., 2018, 18, 5666–5672 CrossRef CAS PubMed.
- M. D. Noori, S. Sangtarash and H. Sadeghi, Appl. Sci., 2021, 11, 1066 CrossRef CAS.
- G. Seifert, D. Porezag and T. Frauenheim, Int. J. Quantum Chem., 1996, 58, 185–192 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260 CrossRef CAS.
- A. Pecchia, G. Penazzi, L. Salvucci and A. Di Carlo, New J. Phys., 2008, 10, 065022 CrossRef.
- X.-M. Wang and S.-S. Lu, J. Phys. Chem. C, 2013, 117, 19740–19745 CrossRef CAS.
- C. Yam, L. Meng, G. Chen, Q. Chen and N. Wong, Phys. Chem. Chem. Phys., 2011, 13, 14365–14369 RSC.
- S. Shenogin, L. Ferguson and A. K. Roy, Polymer, 2020, 198, 122502 CrossRef CAS.
- Y. Zhang, L. Meng, C. Yam and G. Chen, J. Phys. Chem. Lett., 2014, 5, 1272–1277 CrossRef CAS PubMed.
- M. Ghorbani-Asl, S. Borini, A. Kuc and T. Heine, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 235434 CrossRef.
- T. Frauenheim, G. Seifert, M. Elstner, T. Niehaus, C. Köhler, M. Amkreutz, M. Sternberg, Z. Hajnal, A. Di Carlo and S. Suhai, J. Phys.: Condens. Matter, 2002, 14, 3015 CrossRef CAS.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafé, A. Buccheri, C. Camacho, C. Cevallos, M. Deshaye, T. Dumitrică and A. Dominguez, et al. , J. Chem. Phys., 2020, 152, 124101 CrossRef CAS PubMed.
- T. A. Niehaus, M. Elstner, T. Frauenheim and S. Suhai, J. Mol. Struct.: THEOCHEM, 2001, 541, 185–194 CrossRef CAS.
- A. Fihey, C. Hettich, J. Touzeau, F. Maurel, A. Perrier, C. Köhler, B. Aradi and T. Frauenheim, J. Comput. Chem., 2015, 36, 2075–2087 CrossRef CAS PubMed.
- F. Tabatabaei, Ph.D. thesis, 2022.
- S. Masuda, T. Kamada, K. Sasaki, M. Aokia and Y. Morikawa, Phys. Chem. Chem. Phys., 2010, 12, 10914–10918 RSC.
- C. Zeng, B. Li, B. Wang, H. Wang, K. Wang, J. Yang, J. Hou and Q. Zhu, J. Chem. Phys., 2002, 117, 851–856 CrossRef CAS.
- C.-C. Kaun and H. Guo, Nano Lett., 2003, 3, 1521–1525 CrossRef CAS.
- S. Masuda, Y. Koide, M. Aoki and Y. Morikawa, J. Phys. Chem. C, 2007, 111, 11747–11750 CrossRef CAS.
- S. Park, N. Cho and H. J. Yoon, Chem. Mater., 2019, 31, 5973–5980 CrossRef CAS.
- W. Lee, K. Kim, W. Jeong, L. A. Zotti, F. Pauly, J. C. Cuevas and P. Reddy, Nature, 2013, 498, 209–212 CrossRef CAS PubMed.
- Y. Selzer, M. A. Cabassi, T. S. Mayer and D. L. Allara, J. Am. Chem. Soc., 2004, 126, 4052–4053 CrossRef CAS PubMed.
- M. L. Perrin, R. Eelkema, J. Thijssen, F. C. Grozema and H. S. van Der Zant, Phys. Chem. Chem. Phys., 2020, 22, 12849–12866 RSC.
- H. Song, Y. Kim, Y. H. Jang, H. Jeong, M. A. Reed and T. Lee, Nature, 2009, 462, 1039–1043 CrossRef CAS PubMed.
- F. Zhang, X.-H. Wu, Y.-F. Zhou, Y.-H. Wang, X.-S. Zhou, Y. Shao, J.-F. Li, S. Jin and J.-F. Zheng, ChemElectroChem, 2020, 7, 1337–1341 CrossRef CAS.
- N. Mosso, H. Sadeghi, A. Gemma, S. Sangtarash, U. Drechsler, C. Lambert and B. Gotsmann, Nano Lett., 2019, 19, 7614–7622 CrossRef CAS PubMed.
- E. Mykkänen, J. S. Lehtinen, L. Grönberg, A. Shchepetov, A. V. Timofeev, D. Gunnarsson, A. Kemppinen, A. J. Manninen and M. Prunnila, Sci. Adv., 2020, 6, eaax9191 CrossRef PubMed.
- A. Gemma, Ph.D. thesis, 2021.
- J.-C. Klöckner, J. C. Cuevas and F. Pauly, Phys. Rev. B, 2017, 96, 245419 CrossRef.
- T. Markussen, J. Chem. Phys., 2013, 139, 244101 CrossRef PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- D. Kony, W. Damm, S. Stoll and W. F. Van Gunsteren, J. Comput. Chem., 2002, 23, 1416–1429 CrossRef CAS PubMed.
- B. J. Henz, P. W. Chung, J. W. Andzelm, T. L. Chantawansri, J. L. Lenhart and F. L. Beyer, Langmuir, 2011, 27, 7836–7842 CrossRef CAS PubMed.
- H. Heinz, R. Vaia, B. Farmer and R. Naik, J. Phys. Chem. C, 2008, 112, 17281–17290 CrossRef CAS.
- H. Sadeghi, J. Phys. Chem. C, 2019, 123, 12556–12562 CrossRef CAS PubMed.
- C. Jia, M. Famili, M. Carlotti, Y. Liu, P. Wang, I. M. Grace, Z. Feng, Y. Wang, Z. Zhao and M. Ding, et al. , Sci. Adv., 2018, 4, eaat8237 CrossRef CAS PubMed.
Footnote |
| † We used the OPLS force field for the molecule,58,59 the potential by Henz et al. for the gold–molecule interaction60 and the Heinz potential for Au.61 |
| This journal is © The Royal Society of Chemistry 2022 |