
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombining confocal microscopy, dSTORM, and mass spectroscopy to unveil the evolution of the protein corona associated with nanostructured lipid carriers during blood–brain barrier crossing†
Matteo
Battaglini
 *a,
Natalia
Feiner
bc,
Christos
Tapeinos
a,
Daniele
De Pasquale
a,
Carlotta
Pucci
a,
Attilio
Marino
a,
Martina
Bartolucci
d,
Andrea
Petretto
d,
Lorenzo
Albertazzi
bc and
Gianni
Ciofani
*a
*a,
Natalia
Feiner
bc,
Christos
Tapeinos
a,
Daniele
De Pasquale
a,
Carlotta
Pucci
a,
Attilio
Marino
a,
Martina
Bartolucci
d,
Andrea
Petretto
d,
Lorenzo
Albertazzi
bc and
Gianni
Ciofani
*a
aIstituto Italiano di Tecnologia, Smart Bio-Interfaces, Viale Rinaldo Piaggio 34, 56025 Pontedera (Pisa), Italy. E-mail: matteo.battaglini@iit.it; gianni.ciofani@iit.it
bEindhoven University of Technology Department of Biomedical Engineering and Institute for Complex Molecular Systems (ICMS), PO Box 513, 5612AZ Eindhoven, The Netherlands
cInstitute for Bioengineering of Catalonia, Nanoscopy for Nanomedicine, Carrer de Baldiri Reixac 10-12, 08028 Barcelona, Spain
dIRCCS Istituto Giannina Gaslini, Core Facilities-Clinical Proteomics and Metabolomics, Via Gerolamo Gaslini 5, 16147 Genova, Italy
First published on 11th August 2022
Abstract
Upon coming into contact with the biological environment, nanostructures are immediately covered by biomolecules, particularly by proteins forming the so-called “protein corona” (PC). The phenomenon of PC formation has gained great attention in recent years due to its implication in the use of nanostructures in biomedicine. In fact, it has been shown that the formation of the PC can impact the performance of nanostructures by reducing their stability, causing aggregation, increasing their toxicity, and providing unexpected and undesired nanostructure–cell interactions. In this work, we decided to study for the first time the formation and the evolution of PC on the surface of nanostructured lipid carriers loaded with superparamagnetic iron oxide nanoparticles, before and after the crossing of an in vitro model of the blood–brain barrier (BBB). Combining confocal microscopy, direct STochastic Optical Reconstruction Microscopy (dSTORM), and proteomic analysis, we were able to carry out a complete analysis of the PC formation and evolution. In particular, we highlighted that PC formation is a fast process, being formed around particles even after just 1 min of exposure to fetal bovine serum. Moreover, PC formed around particles is extremely heterogeneous: while some particles have no associated PC at all, others are completely covered by proteins. Lastly, the interaction with an in vitro BBB model strongly affects the PC composition: in particular, a large amount of the proteins forming the initial PC is lost after the BBB passage and they are partially replaced by new proteins derived from both the brain endothelial cells and the cell culture medium. Altogether, the obtained data could potentially provide new insights into the design and fabrication of lipid nanostructures for the treatment of central nervous system disorders.
Introduction
Nanostructures exposed to a biological environment (e.g., the bloodstream after systemic injection) can be rapidly covered by a large variety of biological molecules, particularly proteins. These molecules, associated with the nanostructure surface, form the so-called “protein corona” (PC).1 PC represents a phenomenon of pivotal importance for understanding and predicting the interaction between nanostructures and living organisms. The PC is usually the first element of a nanostructure to be in contact with biological entities and cells, determining the “biological identity” of the nanostructures themselves. PC can hinder nanostructure properties and alter their performances: it has been shown that the presence of PC can reduce the stability in solution, cause aggregation, increase the nanostructure toxicity, reduce the efficiency of functional molecules associated with the surface of the particles, and lead to an undesired interaction with the biological environment, like an excessive stimulation of the immune system.2–7 On the other hand, it has also been shown that PC formation is not always harmful, yet it can be exploited as a tool to improve nanostructure properties like stability and biocompatibility, or even to engineer ad-hoc designed nanostructure–PC complexes with completely new properties.8–11 PC is usually divided into a “hard corona”, which is characterized by molecules in direct contact with or in proximity to the nanostructure surface, and a “soft corona”, which refers to molecules not directly attached to the nanostructure surface, yet to the hard corona, and is usually characterized by weaker bonds and more loosely organized molecules.12 It has also been shown that PC is both highly heterogeneous, with different nanostructures of the same batch presenting different PCs,13 and highly dynamic, with continuous modification and rearrangements affecting both the PC shape and composition.14,15 It is therefore of pivotal importance to analyze and understand the process of PC formation and its evolution to predict the biological outcome of candidate nanotherapeutics.Concerning nanovectors directed toward the central nervous system (CNS), one of the most relevant biological barriers we have to consider is the blood–brain barrier (BBB), a continuous membrane enveloping the brain and separating the blood flow from the brain environment.16,17 The BBB is mainly composed of brain endothelial cells, astrocyte endfeet, and pericytes, and is characterized by the presence of tight junctions connecting brain endothelial cells and limiting the passive passage of molecules. The selective nature of the BBB plays a vital role in protecting the brain from potentially harmful molecules, yet also hinders therapeutic moieties from reaching the CNS, with more than 98% of small molecule drugs being unable to reach the brain.16
Nanomaterials, owing to their tunability and the possibility to be ad hoc functionalized with molecules able to target and cross the BBB, represent one of the most promising approaches for the treatment of CNS disorders.17,18 Lipid-based nanoparticles, in particular, are one of the main types being investigated as possible therapeutic candidates for CNS treatment. The last generation of lipid-based nanostructures consists of the so-called nanostructured lipid carriers, a class of nanomaterials composed of a matrix of solid lipids containing small droplets of lipids that are liquid at physiological temperature.17–19 We previously reported the preparation and investigation of nanostructured lipid carriers loaded with superparamagnetic iron oxide nanoparticles (lipid magnetic nanovectors, LMNVs) as a multitasking vector for the treatment of glioblastoma multiforme. In our previous work, we showed how LMNVs could cross an in vitro model of the BBB based on the culture of brain endothelial cells on a transwell porous insert.20,21
Here, we aim at investigating for the first time the formation and the evolution of PC on the surface of LMNVs before and after the crossing of an in vitro model of the BBB. To the best of our knowledge, just one work in the literature analyzed the effect of BBB crossing on the composition of PC of drug delivery systems, particularly gold nanostructures.22 Here, direct imaging techniques based on the combination of fluorescently labeled proteins with confocal and direct STochastic Optical Reconstruction Microscopy (dSTORM) and indirect techniques like dynamic light scattering (DLS)/Z-potential measurements and proteomic analysis were combined to provide a complete picture about the formation and the evolution of PC associated with LMNVs. This work presents, to date, a complete analysis of the PC on the surface of lipid nanostructures from the first contact with biological media to the moment when the lipid nanostructures reach their therapeutic target (in our case, the basolateral chamber of the BBB in vitro model). In particular, the main points of our analysis show how PC can greatly vary from particle to particle even among the same nanostructure batch, and how PC composition is affected by both the time of exposure to the protein molecules and the interaction with cell structures. LMNVs that crossed the in vitro BBB model appeared to have partially lost the original PC and to have obtained a new PC derived from the brain endothelial cells. This phenomenon is worth of attention and investigation, since this BBB-derived PC could affect the biological outcome of nanostructures and their interaction with CNS cells.
Results and discussion
LMNV and LMNV-PC characterization
Fig. 1a shows a schematic representation of LMNVs. As shown in Fig. 1b, scanning electron microscopy (SEM) analysis confirmed the spherical morphology of LMNVs; their average hydrodynamic diameter measured through DLS resulted in 351.4 ± 6.1 nm with a polydispersity index (PDI) of 0.226 ± 0.027, while the average Z-potential was −28.9 ± 0.4 mV (green trace in Fig. 1c and d, respectively). The effect of the incubation with FBS for 1 min and 1 h on both the LMNV hydrodynamic size and Z-potential was assessed: the incubation at both time points caused an increment in the average hydrodynamic diameter to 520.0 ± 12.9 nm (PDI of 0.352 ± 0.012, yellow trace in Fig. 1c) after 1 min of incubation, and to 412.0 ± 12.8 nm (PDI of 0.288 ± 0.015, red trace in Fig. 1c) after 1 h of incubation. This size increment suggests the association of protein molecules and the formation of PC around LMNVs. The Z-potential also increased to −16.1 ± 0.4 mV after 1 min of incubation (yellow trace in Fig. 1d) and to −11.8 ± 0.3 mV after 1 h (red trace in Fig. 1d). Overall, the incubation of LMNVs with FBS caused changes in both the average hydrodynamic diameter and Z-potential. The increment observed in size upon incubation with proteins is in line with what has been reported in the literature for other nanostructures, and can be explained by the association of proteins with the surface of LMNVs, resulting in the formation of LMNV-PC complexes that are indeed larger than the plain particles.23 Similarly, the shift of Z-potential towards more positive values upon LMNV interaction with FBS could be explained by a charge shielding effect caused by the association of protein molecules with the surface of LMNVs (it is worth mentioning that this phenomenon has also been observed in the case of other nanostructures24,25). | ||
| Fig. 1 Analysis of LMNV properties. (a) Schematic representation of LMNVs; (b) representative SEM image of LMNVs; (c) and (d) analysis showing respectively the hydrodynamic diameter and Z-potential distributions of plain LMNVs (green trace), LMNVs incubated for 1 min with FBS (yellow trace), and LMNVs incubated for 1 h in FBS (red trace). | ||
The increment in size caused by the association of proteins on the surface of LMNVs could potentially affect the BBB crossing abilities of the nanostructures, as it has been reported that nanostructures with a size below 200 nm have a higher capacity for crossing the BBB.26 It must also be pointed out how the effects of PC formation could be more complex than a “simple” increment in LMNV diameter: the PC associated with LMNVs could be formed by proteins that facilitate the uptake and transcytosis of the nanostructures across the BBB, such as transferrin.27 The shift in charge caused by PC association with LMNVs could also play a role in the interaction of LMNVs with the BBB, as described previously.26 Altogether, the final effect of PC on LMNV BBB crossing ability is the combination of individual effects such as size increment, Z-potential shift, and association of specific protein characterized by different molecular interactions with brain endothelial cells.
BBB characterization
The formation of a tight layer of brain endothelial cells in the in vitro model of the BBB was first assessed through transepithelial/transendothelial electrical resistance (TEER) measurements to be 30 Ω cm2, a value in line with the literature for models based on bEnd.3 cells.28 Immunostaining was moreover exploited to assess the expression of zonula occludens-1 (ZO-1) protein (involved in the formation of tight junctions). As shown in Fig. S1a,† after 5 days of growth bEnd.3 cells expressed ZO-1 protein, in particular in the cell–cell contact region. The model was further assessed by evaluating the passage of fluorescent dextran (4 kDa) in the transwell seeded with bEnd.3 cells. As shown in Fig. S1b,† the presence of brain endothelial cells significantly reduced the passage of the fluorescent marker at both 1 and 2 h from its the administration to the apical chamber of the transwell insert. Confocal imaging was performed on bEnd.3 cells treated for 72 h with DiO-stained lipid magnetic nanovectors (DiO-LMNVs), and confirmed the ability of these nanostructures to be internalized by bEnd.3 cells (Fig. S1c†).Confocal analysis of the LMNV-PC complex after blood–brain barrier crossing
Confocal analysis was exploited to assess the effect of BBB crossing on LMNV-PC complexes after the passage of the transwell insert. The passage of LMNV-PC across the BBB model caused a statistically significant reduction in the amount of protein associated with LMNVs both after 24 h and 72 h from the administration of the particles compared to the passage of LMNV-PC complexes across empty transwell inserts (Fig. 2a). In particular, after 24 h the intersection between the LMNV fluorescence signal area and the protein corona fluorescence signal area was reduced from 2.06 ± 0.10 (plain transwell) to 1.54 ± 0.14 (BBB model), and from 2.4 ± 0.12 to 1.72 ± 0.13 after 72 h (Fig. 2b). After 72 h it was possible to observe LMNV-PC associated with relatively huge aggregates of PC in the case of LMNVs passing through control transwells without cells, but these big PC complexes were completely absent in the case of LMNV-PC passing through the BBB model transwell seeded with cells (Fig. 2a). We also determined the combined area between the LMNV fluorescence signal and the protein corona fluorescence signal as more representative data of the soft corona surrounding the nanostructures. Also in this case, we observed a similar trend with respect to the intersection areas, observing a reduction from 4.12 ± 0.2 to 2.75 ± 0.15 (plain transwell vs. the transwell seeded with cells after 24 h from LMNV administration), and a reduction from 3.6 ± 0.15 to 3.09 ± 0.14 (plain transwell vs. the transwell seeded with cells after 72 h from LMNV administration). Confocal microscopy analysis showed that the crossing of the BBB model by LMNVs caused a significant reduction in the amount of associated PC with respect to the situation before BBB crossing. This result suggests that the brain endothelial cells are inducing/promoting some sort of modification of the PC composition of LMNVs, and that part of the PC is lost during the passage of the BBB. The crossing of the BBB seems to drastically reduce the amount of PC associated with LMNVs: this suggests that the PC that remains associated with LMNVs after the crossing is probably mainly the hard corona composed of proteins more tightly attached to the surface of the nanostructures. | ||
| Fig. 2 Analysis of protein corona evolution after in vitro BBB crossing. (a) Confocal images showing the protein corona derived from the stained FBS formed on the surface of LMNVs after the passage of control transwell inserts and of transwell inserts seeded with bEnd.3 cells at two different time points (in green LMNVs, in red the protein corona); (b) analysis of the intersection and union area of LMNVs and PC signals under different conditions (*** p < 0.01). | ||
The effect of FBS incubation time was also evaluated by confocal microscopy (representative confocal images are shown in Fig. S2a†); in particular, we observed a statistically significant increment of PC associated with LMNVs by comparing 1 min of incubation with 1 h (with an increment of intersection area from 0.58 ± 0.16 to 0.76 ± 0.12 and of union area from 1.38 ± 0.26 to 2.39 ± 0.28; Fig. S2b†). Control images obtained on LMNVs not incubated with stained FBS are depicted in Fig. S2c†, and show a complete absence of red signal (Cy3 channel).
dSTORM analysis of LMNV-PC
dSTORM imaging was carried out to quantify the number of proteins associated with single LMNVs after the exposure to three different protein sources (Cy5-stained FBS in Fig. 3, Cy5-stained transferrin in Fig. 4, and Cy5-stained albumin in Fig. 5). Fetal bovine serum (FBS) was chosen as a source of different proteins, albumin was chosen due to its abundance in biological fluids, and transferrin due to its importance in transport mechanisms across the BBB. In all three cases, high heterogeneity in the formation of PC was observed, with few particles being associated with a large number of protein molecules and other particles being completely void of PC. Proteins present in the PC were analyzed in terms of localizations obtained through dSTORM analysis.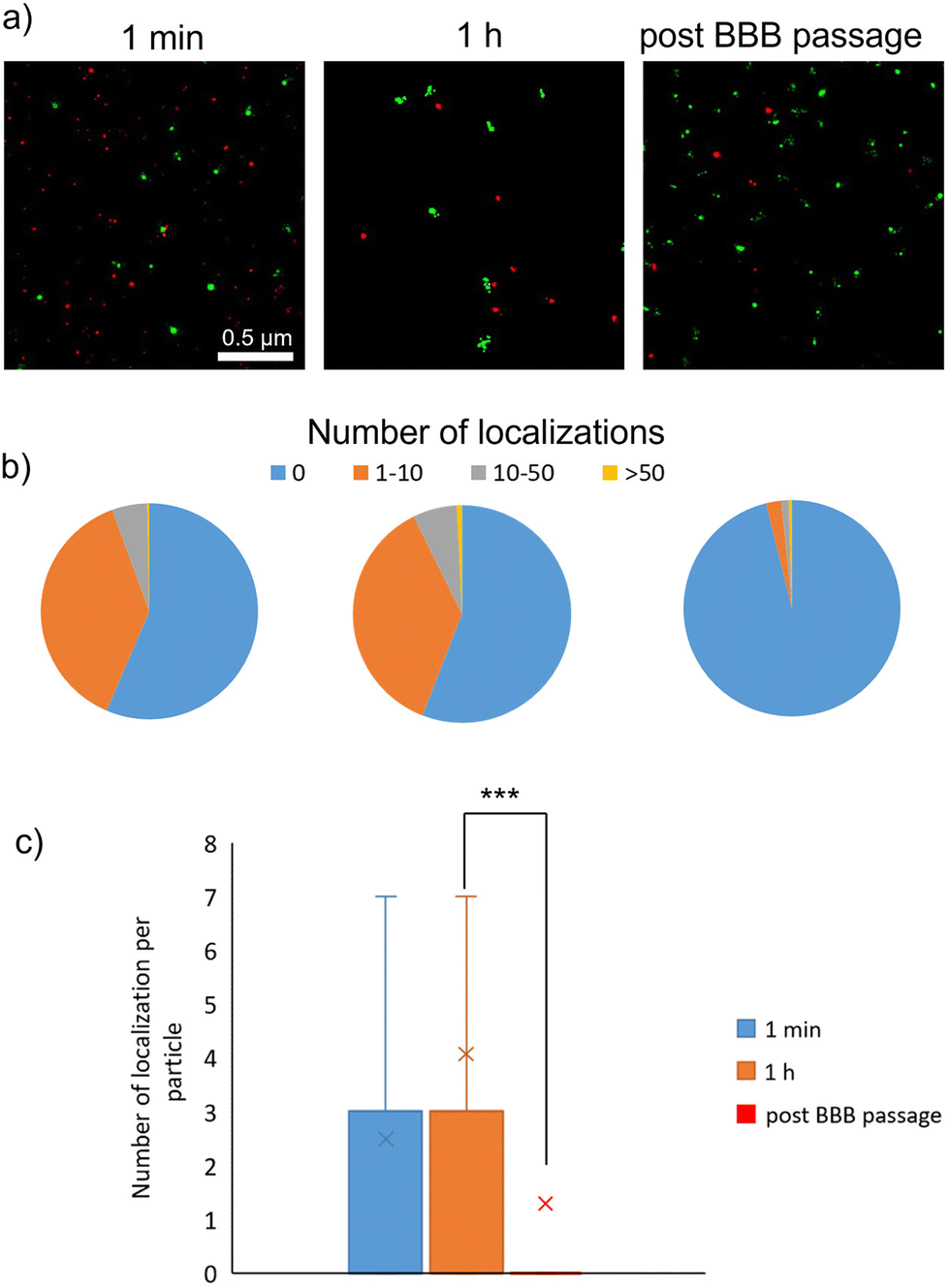 | ||
| Fig. 3 dSTORM analysis of protein corona formation after incubation of LMNVs with stained FBS. (a) dSTORM images showing the protein corona formed on the surface of LMNVs at 1 min of incubation, 1 h, and after the BBB passage (in red the protein corona derived from FBS and in green LMNVs); (b) pie charts showing LMNVs classified based on associated protein signal localizations derived from FBS incubation (in blue LMNVs with no localizations, in orange LMNVs with 1–10 associated protein localizations, in gray LMNVs with 10–50 associated protein localizations, and in yellow LMNVs with more than 50 associated protein localizations); (c) box-plot showing the number of associated protein localization with LMNVs (in blue data of LMNVs incubated with FBS for 1 min, in orange data of LMNVs incubated with FBS for 1 h, and in red data of LMNVs after BBB crossing; ***p < 0.001). | ||
 | ||
| Fig. 4 dSTORM analysis of protein corona formation after incubation of LMNVs with stained transferrin. (a) dSTORM images showing the protein corona formed on the surface of LMNVs at 1 min of incubation, 1 h, and after BBB passage (in red the protein corona derived from transferrin and in green LMNVs); (b) pie charts showing LMNVs classified based on associated protein signal localizations derived from transferrin incubation (in blue LMNVs with no localizations, in orange LMNVs with 1–10 associated protein localizations, in gray LMNVs with 10–50 associated protein localizations, and in yellow LMNVs with more than 50 associated protein localizations); (c) box-plot showing the number of associated protein localization with LMNVs (in blue data of LMNVs incubated with transferrin for 1 min, in orange data of LMNVs incubated with transferrin for 1 h, and in red data of LMNVs after BBB crossing; ***p < 0.001). | ||
 | ||
| Fig. 5 dSTORM analysis of protein corona formation after incubation of LMNVs with stained albumin. (a) dSTORM images showing the protein corona formed on the surface of LMNVs at 1 min of incubation, 1 h, and after BBB passage (in red the protein corona derived from albumin and in green LMNVs); (b) pie charts showing LMNVs classified based on associated protein signal localizations derived from albumin incubation (in blue LMNVs with no localizations, in orange LMNVs with 1–10 associated protein localizations, in gray LMNVs with 10–50 associated protein localizations, and in yellow LMNVs with more than 50 associated protein localizations); (c) box-plot showing the number of associated protein localization with LMNVs (in blue data of LMNVs incubated with albumin for 1 min, in orange data of LMNVs incubated with albumin for 1 h, and in red data of LMNVs after BBB crossing; ***p < 0.001). | ||
Fig. 3 shows analysis following incubation with Cy5-stained FBS. After 1 min, we observed 56.4% particles without any protein localization, 37.9% particles with a number of localizations between 1 and 10, 5.4% particles with a number of localizations between 10 and 50, and only 0.3% particles with a number of localizations above 50. After 1 h of incubation with stained FBS, no statistically significant differences were observed in terms of PC formation (55.9% particles with no localization, 36.8% particles with 1–10 localizations, 6.5% particles with 10–50 localizations, and 0.8% with over 50 localizations). It is worth mentioning that the number of localization is not equal to the number of protein molecules associated with the nanostructures. This is due to the fact that each protein molecule could be associated with more than one fluorophore molecule, thus providing multiple localization events during dSTORM analysis. However, the number of localizations showed a statistically significant drop after BBB passage (96.1% particles without any localization, 2.3% with 1–10 localizations, 1.1% with 10–50 localizations, and 0.5% with over 50 localizations).
In the case of incubation with transferrin (Fig. 4), similar results were highlighted after 1 min of incubation (65.6% particles without localizations, 29.4% with 1–10 localizations, 4.5% with 10–50 localization, and 0.5% with more than 50 localizations). However, the number of localizations was significantly lower after 1 h of incubation (88.2% particles without localizations, 8.4% particles with 1–10 localizations, 2.7% particles with 10–50 localizations, and 0.7% particles with more than 50 localizations), and after BBB crossing (94.1% particles without localizations, 4.3% particles with 1–10 localizations, 1.1% particles with 10–50 localizations, and 0.4% particles with more than 50 localizations).
The incubation with albumin (Fig. 5) conversely provided a different PC formation process; in particular, after 1 min, the PC was still heterogeneous (54.3% particles without localizations, 38.8% particles with 1–10 localizations, 6.2% particles with 10–50 localizations, and 0.6% particles with more than 50 localizations), but the amount of PC associated with LMNVs significantly increased after 1 h of incubation (21.1% particles with no localizations, 54.5% particles with 1–10 localizations, 21.6% particles with 10–50 localizations, and 2.7% particles with more than 50 localizations). Similarly to the previously analyzed conditions, a significant reduction after BBB crossing was observed (96.3% particles without PC, 1.7% particles with 1–10 localizations, 0.9% particles with 10–50 localizations, and 1.1% with more than 50 localizations). Overall, the modification on LMNV-associated PC observed by confocal microscopy was confirmed by dSTORM analysis, which showed also as different proteins interact differently with LMNVs, with albumin incubation being the experimental condition that leads to a higher number of molecules associated with LMNVs.
Incubation time seems to play a role in the evolution of PC composition with a reduction of transferrin molecules associated to LMNVs over time and an increment of albumin molecules. This could be explained by competition phenomena involving the association of stained and unstained protein molecules with the surface of LMNVs. Time did not show any significant effect on PC composition in the case of incubation with stained FBS. The two main results obtained by dSTORM analysis can be summarized as follows: (i) LMNVs generally show a low amount of associated PC; (ii) PC associated with LMNVs is extremely heterogeneous, with a large portion of particles completely devoid of any PC and some particles instead associated with a large number of protein molecules.13 This heterogeneity in PC formation has already been observed for other nanostructures, such as silica nanoparticles.13 The relatively low amount of PC formed on the surface of LMNVs could be explained by the presence of DSPE-PEG in their composition: PEG, in fact, has repeatedly demonstrated to be able to act as a stealth agent, preventing the formation of PC around nanostructures.24,29 However, it is worth pointing out that PEG is not entirely able to prevent PC formation, and this is in line with the formation of PC that we observed around a small portion of LMNVs.29 Moreover, DSPE-PEG could be heterogeneously present on the surface of LMNVs, contributing to the observed heterogeneity in PC distribution. dSTORM analysis, confirming the confocal microscopy data, showed the effect of BBB model crossing on LMNV-PC complexes, with an almost complete loss of PC associated with LMNVs after the passage of the in vitro BBB model.
Internalization pathways of LMNVs
Internalization pathways and intracellular localization of LMNVs were assessed by confocal microscopy, particularly for evaluating the co-localization with lysosomes, clathrin-positive vesicles, and caveolin-positive vesicles. The confocal microscopy images and Pearson's correlation coefficient of LMNVs for each subcellular compartment are shown in Fig. S3–S5.† In particular, we observed a relatively high co-localization of LMNVs with lysosomes (Pearson's coefficient equal to 0.18 ± 0.12 at 4 h, 0.55 ± 0.04 at 24 h, and 0.53 ± 0.11 at 72 h; Fig. S3†), and a relatively low internalization in both clathrin-positive vesicles (Pearson's coefficient equal to 0.07 ± 0.06 at 4 h, 0.08 ± 0.02 at 24 h, and 0.12 ± 0.02; Fig. S4†) and caveolin-positive vesicles (Pearson's coefficient equal to 0.06 ± 0.03 at 4 h, 0.05 ± 0.02 at 24 h, and 0.08 ± 0.03 at 72 h; Fig. S5†).Through our analysis, we could determine that a relatively high amount of nanostructures is localized at the lysosomal level after 72 h. The entrapment of LMNVs inside of lysosomes could perhaps partially explain the rearrangement of PC observed after BBB crossing. In particular, the exposure to the acidic pH of lysosomes could cause a degradation of the PC associated with LMNVs. However, this process supposes the ability of LMNV-PC complexes to actively escape the lysosomal compartment and to reach the basolateral side of the BBB model. The increment in the lysosomal localization of LMNVs from 24 to 72 h conversely suggests that the nanostructures entrapped inside lysosomes are not able to escape. Usually, lysosome escape has been observed in the early phase of internalization, where nanostructures are able to escape early endosomes:30 we can thus hypothesize that the LMNVs associated with PC that we analyzed after BBB crossing were nanostructures not entrapped inside lysosomes, and that therefore did not undergo the degradative processes typically occurring in the lysosomal compartments.
Proteomic analysis of LMNV-PC before and after BBB crossing
Proteomic analysis was performed to identify the protein associated with LMNVs after the exposure to FBS and after the passage of the BBB in vitro model. The complete lists of proteins identified are listed in Tables S1–S7.† Proteins were classified as derived from FBS or from the brain endothelial cells of the BBB model basing on the animal species of origin (Bos Taurus for FBS and Mus musculus for bEnd.3 cells). Mass spectroscopy (MS) analysis shows that only 110 identified proteins were present both in the PC of LMNVs not exposed to the BBB environment and in the PC of LMNVs after BBB crossing. The PC of LMNVs exposed to only FBS was composed of 261 proteins that were not present in the case of the PC formed on the surface of LMNVs after BBB crossing. These proteins were lost during BBB in vitro model crossing and replaced with other 419 proteins. The proteins associated with LMNVs after BBB passage were derived for 95% from the bEnd.3 cells and for 5% from the cell culture medium.In a further test, LMNVs were administered only to the basolateral side of the transwell inserts seeded with bEnd.3 cells. This experimental condition aimed to discriminate between proteins that associate with LMNVs during BBB passage and proteins that are instead derived from an interaction between LMNVs and the bEnd.3 secretome. In this case, we identified 427 proteins associated with LMNVs, 272 derived from bEnd.3 cells and 155 derived from FBS. 186 of these proteins were exclusively present under this experimental condition, that is in the case of LMNVs exposed to the secretome of bEnd.3 cells (among these, 30 were derived from FBS and 156 from bEnd.3 cells; Table S4†). 115 of these proteins were identified both in the case of LMNVs that crossed the BBB and in the case of LMNVs that were exposed to the bEnd.3 secretome (all derived from bEnd.3 cells; Table S5†). 55 of these proteins were identified as associated with the nanostructures in the case of LMNVs exposed to just FBS and in the case of LMNVs exposed to the bEnd.3 secretome, but not in the case of LMNVs that crossed the BBB (all derived from FBS; Table S6†). Lastly, 70 of these proteins were present in the LMNV-PC of all experimental conditions (Table S7†).
Proteins present in the PC associated with LMNVs both after FBS exposure and after BBB crossing were derived from intracellular organelles (53.33%), from extracellular space (13.33%), from extracellular vesicles (10.00%), from high-density lipoprotein particles (10.00%), from secretory granules (6.67%), from the intrinsic component of the cellular membrane (3.33%), and from lysosomes (3.33%) (Table 1, indicated as “proteins conserved after BBB crossing”). Gene ontology (GO) enrichment analysis performed on protein shared in both groups showed statistically significant enrichment in several terms, including negative regulation of response to the external stimulus, protein activation cascade, cellular macromolecule metabolic process, defense response, regulation of lipid localization, carboxylic acid metabolic process, cofactor metabolic process, wound healing, and coenzyme metabolic process (Fig. 6a).
 | ||
| Fig. 6 GO enrichment for biological function derived from proteomic analysis of proteins present in the PC associated with LMNVs and derived from FBS. (a) GO enrichment for the biological function of proteins associated with LMNVs both after FBS exposure and after BBB crossing. (b) GO enrichment for the biological function of proteins associated with LMNVs after FBS exposure but lost after BBB crossing. Nodes with the label in grey contain a smaller number of associated proteins, while nodes labeled in black contain a larger number of proteins. | ||
| Proteins conserved after BBB crossing | Proteins lost after BBB crossing | Proteins acquired after BBB crossing and derived from bEnd.3 cells | Proteins acquired after BBB crossing and derived from cell culture medium |
|---|---|---|---|
| Intracellular organelle part 53.33%** | Cell surface 59.68%** | Mitochondrion 73.64%** | Endomembrane system 33.33% |
| Extracellular space 13.33%** | Extracellular space 14.52%** | Melanosome 8.18%** | Intracellular organelle 25.00% |
| Extracellular vesicle 10.00%** | Extracellular vesicle 6.45%** | Focal adhesion 7.27%** | Blood microparticles 16.67%** |
| High-density lipoprotein particles 10.00%** | Integral component of the membrane 4.84%** | Sarcomere 0.91% | Extracellular exosome 8.33% |
| Secretory granule 6.67%* | Nucleoplasm 4.84% | Endosome membrane 0.91% | Cytoskeletal part 8.33% |
| Intrinsic component of the membrane 3.33% | Secretory granule 3.23% | Cell cortex 0.91% | Vesicle 8.33% |
| Lysosome 3.33% | Focal adhesion 3.23%* | Caveola 0.91% | |
| Membrane raft 1.61% | Lysosomal membrane 0.91% | ||
| Endoplasmic reticulum lumen 1.61% | Mitochondrial intermembrane space 0.91% | ||
| Mitochondrial outer membrane 0.91% | |||
| Supramolecular fiber 0.91% | |||
| Organelle outer membrane 0.91% | |||
| Extracellular matrix 0.91% | |||
| Secretory granule membrane 0.91% | |||
| Integral component of the endoplasmic reticulum membrane 0.91% |
After exposure to FBS, LMNVs were associated with proteins that were lost after BBB passage (being absent in the PC associated with LMNVs after BBB crossing). These proteins were derived in part from the cell surface (59.68%), from extracellular space (14.52%), from extracellular vesicles (6.45%), and in small percentages from other cellular components (as listed in Table 1 in the column “proteins lost after BBB crossing”). GO analysis showed a significant enrichment in functional terms such as regulation of peptidase activity, negative regulation of phosphorylation, regulation of the RNA metabolic process, the purine nucleotide metabolic process, negative regulation of the cellular protein metabolic process, and transcription (Fig. 6b).
The relative abundance of protein present in the PC of LMNVs both after FBS exposure and BBB crossing was also analyzed and compared (results showed as a heatmap in Fig. 7): 21 proteins were present in low quantities in both groups (cluster 71, 32, and 144), 21 proteins were present in relatively high quantities in both groups (clusters 89, 142, 10, 139), and 68 proteins were present in different quantities between the two groups (cluster 143).
 | ||
| Fig. 7 Heatmap showing the relative abundance of proteins associated with LMNVs exposed to just FBS and with LMNVs after BBB crossing. In blue proteins present in low quantities and in red proteins present in high quantities in the PC associated with LMNVs. | ||
LMNVs that crossed the BBB in vitro model developed a partially new PC composed of protein originating from the bEnd.3 cells and derived from mitochondria (73.64%), from melanosomes (8.18%), from focal adhesion (7.27%), and in small percentage from other cellular compartments (as listed in Table 1 in the column “proteins acquired after BBB crossing and derived from bEnd.3 cells”). GO analysis performed on these proteins showed functional enrichments in several functional terms including protein transport, the fatty acid metabolic process, the nucleotide metabolic process, transcription by RNA polymerase II, blood vessel morphogenesis, regulation of angiogenesis, and vasculature development (Fig. 8a).
 | ||
| Fig. 8 GO enrichment for biological function derived from proteomic analysis of proteins present in the PC associated with LMNVs after BBB crossing. (a) GO enrichment for the biological function of proteins associated with LMNVs after BBB crossing derived from bEnd.3 cells. (b) GO enrichment for the biological function of proteins associated with LMNVs after BBB crossing derived from cell culture medium. Nodes with the label in grey contain a lower number of associated proteins, while nodes with the label in black contain a larger number of proteins. | ||
A small amount of the proteins present exclusively in the PC associated with LMNVs after BBB crossing was also derived from the cell culture medium; in particular, these proteins were derived from the endomembrane system (33.33%), from intracellular organelles (25.00%), from blood microparticles (16.67%), from extracellular exosomes (8.33%), from cytoskeletal parts (8.33%), and from vesicles (8.33%), as listed in Table 1 in the column “proteins acquired after BBB crossing and derived from cell culture medium”. GO enrichment performed on the cell culture medium-derived protein present in the PC associated with LMNVs formed after BBB crossing showed enrichment in functional terms such as the hydrogen peroxide metabolic process, removal of superoxide radicals, response to wounding, and negative regulation of catalytic activity (Fig. 8b).
The complete list of proteins identified with their reference cluster is shown in supplementary materials in Tables S1–S3.† Proteomics provided relevant insights about the observed PC evolution phenomenon; in particular, it confirmed that LMNVs after BBB crossing partially lose the proteins forming their initial PC, and they develop a newly formed PC partially derived from both proteins present in the brain endothelial cells and in FBS. All these observations provide evidence on the dynamic state of the protein corona which has been already discussed previously.22 The original PC associated with LMNVs presents proteins involved in key biological functions (in particular metabolic processes involving nucleotides) that are only partially preserved after the BBB passage. The PC associated with LMNVs after BBB crossing is mainly constituted by proteins involved in metabolic processes and in transport systems. The presence of proteins involved in the transport mechanism (such as caveolin-2) in the LMNVs that underwent BBB crossing suggests that the nanostructures are actively transported across the BBB model, and the proteins involved in this transport are partially retained in the PC. It is worth mentioning that the PC associated with LMNVs after BBB crossing could be the result of protein interchange phenomena during transcytosis due to the dynamic nature of PC. However, the “final” composition of PC associated with LMNVs after BBB crossing could also be made of proteins that interacted with LMNVs after crossing the brain endothelial cell layer. As demonstrated by our control experiment, where LMNVs were exposed to the bEnd.3 conditioned medium (LMNVs administered to the basolateral side of the BBB in vitro model), LMNVs showed a PC composed of proteins derived from both bEnd.3 cells and FBS. In particular, the PC derived from conditioned medium was partially overlapped with the PC associated with particles that crossed the BBB and with the PC associated with LMNVs exposed to just FBS. Therefore, the PC associated with LMNVs after BBB crossing is suggested to be derived from a combination of phenomena: (i) a rearrangement caused by the BBB crossing itself, where previously adsorbed proteins are lost and substituted with new proteins derived from brain endothelial cells; (ii) the interaction with BBB-derived proteins triggered by bEnd.3 cells and present on the basolateral side of the transwell insert.
Intriguingly, the major part of BBB-derived protein present in the PC associated with LMNVs after BBB crossing is of mitochondrial origin: this phenomenon could also be explained by a process of “corona interactome” as previously reported for other nanostructures.31 In particular, LMNVs exposed to a protein source develop a first layer of PC composed of protein molecules directly interacting with the surface of the particles; when exposed to other protein sources (e.g., brain endothelial cells during BBB crossing), other protein molecules may indirectly associate with LMNVs by interacting with the protein already present on the surface of LMNV-PC complexes.
Our analysis demonstrates how the study of PC evolution after LMNV BBB crossing represents a pivotal point to fully understand the interaction between nanostructures and CNS cells. In particular, as previously discussed, PC is commonly the first “layer” of the nanomaterial to come in contact with biological structures, dictating the biological identity and the ultimate interaction of the nanostructures with cells. In our analysis, we highlighted how LMNVs that reached the basolateral side of a BBB in vitro model were associated with protein derived from brain endothelial cells. Moreover, the newly developed BBB-derived PC was characterized by proteins involved in several biological functions as highlighted by the GO enrichment analysis. The BBB-derived PC could potentially lead to an unexpected interaction between nanostructures and CNS cells in terms of internalization rates, uptake pathways, intracellular localization, biocompatibility, and efficiency as drug carriers. For example, the GO analysis (Fig. 8) highlighted an enrichment in proteins involved in fatty acid and lipid metabolism in the PC of LMNVs after BBB crossing. These newly associated proteins could potentially lead to effects in terms of overall lipid metabolism in CNS cells exposed to LMNVs. In view of this, it is clear how a complete characterization of the PC phenomenon, including its potential evolution due to the exposure of the nanomaterials to different biological environments, should become a standard to predict and characterize the interaction between nanomaterials and biological structures.
Conclusion
Overall, this work provides new insights into the phenomenon of PC formation associated with lipid nanostructures. In particular, LMNVs exposed to biological media are rapidly covered by protein molecules in a highly heterogeneous way, and the PC associated with LMNVs undergoes a series of rearrangements and modifications, both in terms of morphology and composition. The LMNVs that could cross an in vitro BBB model resulted in being covered by various proteins that are just partially originated by their first exposure to biological media, and that are largely derived from the subsequent internalization and transcytosis through brain endothelial cells. Eventually, the final PC associated with LMNVs is the first component that will come into contact with CNS cells, dictating the nanoparticle biological outcome: it is therefore of pivotal importance for the potential exploitation of LMNVs and of other lipid nanostructures in clinical practice to deeply investigate the phenomena of PC formation and evolution in complex in vitro and in vivo systems before nanovectors reach their therapeutic target.Materials and methods
LMNV preparation
The protocol for the fabrication of lipid magnetic nanovectors (LMNVs) was adapted from previous works of our group combining hot ultra-sonication and high-pressure homogenization (HPH) methods.20 Briefly, we mixed different lipids including 2.5 mg of oleic acid (Sigma-Aldrich), 25 mg of 1-stearoyl-rac-glycerol (Sigma-Aldrich), 2.5 mg of oleic acid (Sigma-Aldrich), 2.5 mg of 1,2-dipalmitoyl-rac-glycero-3-phosphocholine (Sigma-Aldrich), and 4 mg of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine with conjugated methoxyl poly(ethylene glycol) (mPEG-DSPE) (5000 Da, Nanocs) with 84.5 μl of an ethanol solution of superparamagnetic iron oxide nanoparticles (SPIONs) (3 nm diameter, 15 wt%; US Research Nanomaterials Inc.) into a 6 ml glass vial. 3 ml of pre-warmed (70 °C) Tween® 80 (Sigma-Aldrich) solution (1.0 wt%) were added to the lipid/SPION dispersion and sonicated using an ultrasonic tip (Fisherbrand™ Q125 Sonicator) for 15 min (amplitude 30%, 120 W). After the sonication, the mixture underwent high-pressure homogenization with a homogenizer at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psi (5 passages of high-pressure homogenization were performed). The nanovectors were purified by centrifugation at 16000g for 90 min at 4 °C (three passages) and then re-dispersed in water. For confocal imaging, LMNVs were labeled with the fluorescent Vybrant DiO cell-labeling dye (Invitrogen) by incubating 5 mg of nanovectors with 20 μM of DiO for 2 h at 37 °C and then washing them by centrifugation at 16000g for 90 min at 4 °C (three passages). For Direct STochastic Optical Reconstruction Microscopy (dSTORM) analysis, the staining was obtained by mixing 3 mg of mPEG-DSPE (5000 Da, Nanocs) with 1 mg of DSPE-PEG-Cy3 (5000 Da) during the preparation procedure.
000 psi (5 passages of high-pressure homogenization were performed). The nanovectors were purified by centrifugation at 16000g for 90 min at 4 °C (three passages) and then re-dispersed in water. For confocal imaging, LMNVs were labeled with the fluorescent Vybrant DiO cell-labeling dye (Invitrogen) by incubating 5 mg of nanovectors with 20 μM of DiO for 2 h at 37 °C and then washing them by centrifugation at 16000g for 90 min at 4 °C (three passages). For Direct STochastic Optical Reconstruction Microscopy (dSTORM) analysis, the staining was obtained by mixing 3 mg of mPEG-DSPE (5000 Da, Nanocs) with 1 mg of DSPE-PEG-Cy3 (5000 Da) during the preparation procedure.
Dynamic light scattering measurements
DLS (Malvern-Zetasizer Nano ZS90) was used to measure the average hydrodynamic diameter and the Z-potential of LMNVs and LMNVs associated with the protein corona (LMNV-PC). Briefly, 100 μg ml−1 of LMNVs were used to measure the average hydrodynamic diameter of the nanostructures using polystyrene cuvettes (Malvern Zetasizer Nano series), and to measure the Z-potential using disposable folded capillary cells (Malvern Zetasizer Nano series). To assess how PC affects the average hydrodynamic diameter and the Z-potential of the nanostructures, 100 μg ml−1 of LMNVs were dispersed in full serum for 1 min or 1 h and then washed three times by centrifugation at 16000g for 5 min at RT (three passages). At the end of the washing steps, LMNV-PC complexes were re-suspended in Milli-Q water and the average hydrodynamic diameter and Z-potential were measured as previously described.
Electron microscopy
The LMNV morphology was assessed using scanning electron microscopy (SEM). Briefly, 5 μl of a suspension of 10 μg ml−1 of LMNVs were drop-cast on a small piece of a silicon wafer and left to dry. After the drop dried, the sample was gold-sputtered using a Quorum Tech Q150RES Gold Sputter Coater at 30 mA for 60 s and imaged using an SEM system, Helios NanoLab 600i FIB/SEM, FEI.Cell culturing and the BBB in vitro model
To simulate the crossing of the BBB, an in vitro model based on a transwell insert was exploited. Mouse-derived brain endothelial cells (bEnd.3, ATCC CRL-2299) were cultured in high-glucose Dulbecco's Modified Eagle's Medium (DMEM; Sigma-Aldrich) supplemented with 10% of heat-inactivated Fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 IU ml−1 of penicillin, and 100 μg ml−1 of streptomycin (all from Gibco). For the characterization of the BBB model, confocal imaging, and dSTORM analysis, bEnd.3 cells were seeded at a density of 100000 cells per cm2 on transparent poly(ethylene terephthalate) membranes inserted in 24-well plates (inserts provided by Falcon, pores size 3 μm). For proteomic analysis, bigger inserts (a 6-well size transwell insert with 3 μm pore size; Falcon) were used. Cells were grown for 5 days, and then the model was characterized in terms of electrical properties and permeability to dextran passage. Bioelectrical properties of the BBB model were assessed by measuring the transepithelial/transendothelial electrical resistance (TEER) with a Millipore Millicell ERS-2 Volt-Ohmmeter, while the BBB integrity was assessed by measuring the passage of fluorescein-5-isothiocyanate (FITC)-dextran (4 kDa; Sigma) at different time points (1 and 2 h). Briefly, fresh medium (500 μl) was added in the abluminal compartments and 300 μl of 500 μg ml−1 FITC-dextran solution were added in the apical compartments (solutions were prepared in phenol red-free complete medium). Membranes without cells were considered as a control. Analyses were conducted by measuring the fluorescence (excitation 485 nm, emission 535 nm) of the medium recovered in the abluminal compartment at 1 and 2 h with a Victor X3 Plate Reader (PerkinElmer). To assess the formation of tight junctions, a protocol of immunostaining against ZO-1 protein was carried out as described in the previous publications from our group.32 Briefly, bEnd.3 after 5 days of culture were fixed with 4% paraformaldehyde (PFA) for 20 min at 4 °C and permeabilized with Triton 0.1% X-100 for 15 min; afterward, cultures were blocked with 10% of goat serum (GS) for 1 h and incubated with the primary antibody anti-ZO-1 (2.5 μg ml−1; Abcam) for 3 h at room temperature. Cells were washed three times in Dulbecco's phosphate-buffered saline (DPBS) and incubated with goat serum 10% supplemented with 10 μg ml−1 of F(ab′)2-goat-anti-Rabbit IgG (H + L) Alexa Fluor 488 conjugate (Invitrogen), 2.5 μg ml−1 of tetramethylrhodamine (TRITC) phalloidin (Sigma), and 5 μg ml−1 of Hoechst (Invitrogen) at 37 °C for 45 min. After three DPBS rinsing steps, images were acquired by confocal microscopy (C2s, Nikon) using a 60× oil immersion objective.
To assess the ability of bEnd.3 cells to internalize LMNVs, DiO-stained nanostructures were administered to bEnd.3 cells grown for 5 days as previously described at 100 μg ml−1 for 72 h. After 72 h, the cells were washed in DPBS, fixed in PFA at 4% in DPBS at 4 °C, and washed twice in DPBS. After the fixation procedure, the cells were incubated for 40 min in GS 10% in DPBS at RT, and then stained for 1 h with a solution of GS 10% in DPBS supplemented with 2.5 μg ml−1 of TRITC-phalloidin (Sigma) and 5 μg ml−1 of Hoechst (Invitrogen). After the staining, the cells were washed twice in DPBS and imaged with a confocal microscope (C2s, Nikon) using a 60× oil immersion objective.
Protein staining for protein corona analysis
To image the association of protein to the surface of LMNVs and the subsequent formation of PC, FBS, transferrin, and albumin were stained with a fluorescent marker (Cy5). In particular, for dSTORM analysis and confocal analysis of intracellular localization of LMNV-PC, full serum, 3 mg ml−1 of transferrin, and 10 mg ml−1 of albumin were dispersed in sodium bicarbonate (pH 8.5, Sigma-Aldrich) with the addition of 2 eq. of Cy5-NHS (Lumiprobe). The reaction was then put under agitation for 4 h at RT and then dialyzed using a Pur-A-Lyzer™ Midi 3500 (Sigma Aldrich, cut-off 3.5 kDa) against MilliQ water. For confocal analysis of LMNV-PC after BBB crossing, full FBS was stained using the same procedure but exploiting 2 eq. of Cy3-NHS (Lumiprobe).Confocal imaging of LMNV-PC after BBB crossing
Confocal imaging was carried out to assess the effect of BBB crossing on the composition of PC associated with LMNVs. DiO-stained LMNVs (400 μg ml−1) were incubated for 1 h with 90% FBS and 10% Cy3-stained FBS under agitation at 37 °C. After the incubation, LMNV-PC complexes were purified through centrifugation (16000g for 5 min at RT) and re-suspended in cell medium at 100 μg ml−1. Cell culture medium containing LMNV-PC complexes was then added to both a 24-well plate transwell insert without cells and to a transwell insert seeded with bEnd.3 cells grown for 5 days as previously described; the transwell inserts were then placed inside a μ-Plate 24-Well Black (Ibidi®). LMNV-PC complexes present in the basolateral compartment were imaged at 24 and 72 h of incubation by confocal microscopy (C2s, Nikon) using a 60× oil immersion objective. To assess the effects of incubation time on PC formation, DiO-stained LMNVs (8 mg ml−1) were incubated for either 1 min or 1 h with 90% FBS and 10% Cy3-stained FBS under agitation. After incubation, LMNV-PC complexes were purified through centrifugation (16000g for 5 min at RT) and re-suspended in PBS at 100 μg ml−1 in a μ-Plate 24-well Black (Ibidi®). DiO-stained LMNVs were also incubated with unstained FBS for 1 h, as a control. Control LMNVs and LMNVs incubated with stained FBS for either 1 min or 1 h were imaged with a confocal microscope (C2s, Nikon) using a 60× oil immersion objective. The signal threshold was used to select LMNV and PC region of interests (ROIs), and PC ROIs were filtered through the fluorescence intensity to select only ROIs associated with LMNVs. Both the intersection and the union (combined) areas of LMNV and PC ROIs were measured and used to compare the abundance of PC associated with LMNVs under the different conditions.
Intracellular localization of LMNVs
To assess the intracellular localization of LMNV-PC complexes in bEnd.3 cells, DiO-stained LMNVs were incubated for 1 h with 90% unstained FBS and 10% Cy5-stained FBS at 37 °C under agitation. After the incubation, LMNV-PC were recovered through centrifugation as previously described, re-suspended in phenol red-free medium at 100 μg ml−1, and administered to bEnd.3 cells previously seeded and grown for 5 days in μ-Plate 24-Well Black (Ibidi®). For the analysis of intracellular localization into lysosomes, cells treated for 4, 24, and 72 h were rinsed with DPBS and incubated with phenol red-free medium containing 5 μg ml−1 of Hoechst (Invitrogen) and 1 μM LysoTracker Red (Thermo Fisher) for lysosome staining. After the incubation, the cells were washed twice with DPBS, incubated in phenol red-free medium complemented with HEPES, and imaged using a confocal microscope (C2s system, Nikon) with a 60× oil immersion objective. The intracellular localization was analyzed through the NIS elements software by measuring Pearson's correlation coefficient between the LMNV-PC signal and the lysosome signal.To assess the localization of LMNV-PC inside caveolae or clathrin-positive vesicles, bEnd.3 cells treated for 4, 24, and 72 h with LMNV-PC were washed twice in DPBS and then fixed using 4% PFA for 20 min at 4 °C. After fixation, the cells were incubated for 40 min with a blocking solution of 10% GS in DPBS, and thereafter with a solution of 10% GS in DPBS supplemented with either 6.7 μg ml−1 of anti-caveolin-1 primary antibody (Abcam) or 6.7 μg ml−1 of clathrin primary antibody (Abcam) for 3 h at RT. After the incubation with the primary antibody, the cells were washed three times with a solution of 10% GS in DPBS and stained for 1 h with a DPBS solution containing 5 μg ml−1 of Hoechst (Invitrogen) and 10 μg ml−1 of either TRITC-labeled goat anti-rabbit or goat anti-mouse IgG (H + L) secondary antibody (Invitrogen). After the staining, the cells were washed three times in DPBS and imaged using a confocal microscope (C2s system, Nikon) with a 60× oil immersion objective. The intracellular localization was again analyzed through the NIS elements software by measuring Pearson's correlation coefficient between the LMNV signal and the caveolin-1/clathrin signal.
dSTORM imaging
For dSTORM imaging, 400 μg ml−1 of Cy3-labeled LMNVs were incubated with three different solutions of FBS containing respectively 1% of Cy5-stained FBS, 1% of Cy5-stained albumin, and 1% of Cy5-stained transferrin for either 1 min or 1 h at 37 °C under agitation. Thereafter, particles were recovered through centrifugation, washed twice in PBS, and re-suspended in a dSTORM buffer composed of an oxygen scavenging system (0.5 mg ml−1 glucose oxidase, 40 μg ml−1 catalase), glucose (5% w/v), and cysteamine (100 mM). To perform imaging of LMNV-PC, nanostructures were immobilized on a flow chamber composed of a glass slide and a coverslip bound together through a double-face adhesive tape (24 mm × 24 mm with 0.15 mm depth).For assessing the effect of BBB crossing on PC, LMNV-PC complexes obtained by 1 h of incubation with various stained proteins as previously mentioned were administered at 100 μg ml−1 to the transwell insert seeded with bEnd.3 cells at 150000 cells per cm2 and grown for 5 days. After 72 h of incubation, LMNV-PC complexes were recovered from the basolateral chamber of the transwell inserts through centrifugation and re-suspended in dSTORM buffer. Once again, particles recovered after BBB crossing were immobilized on a glass chamber prepared as previously described. dSTORM images were acquired using a Nikon N-dSTORM system configured for total internal reflection fluorescence imaging with a 100× oil immersion objective. Cy3-labeled LMNVs were imaged using a 561 nm laser (80 mW) and Cy5-labeled PC was imaged using a 647 nm laser (140 mW) without the use of UV activation. 10000 frames were acquired for both channels, and the obtained dSTORM images were analyzed using the NIS element Nikon software. Analysis of dSTORM images was carried out by generating a localization list using a Gaussian fit of blinking dyes in the acquired time-lapse of the microscopy images. The first 100 frames of each acquisition were discharged from the processing to avoid interference due to the initial activation of labeled molecules. The localization list was filtered by applying a density filter of 20 localizations in a radius of 100 nm on the 561 nm channel. The list of filtered localization was further analyzed using a MATLAB script described by Feiner et al.13 to determine the number of localizations for each particle.
Sample preparation for proteomics analysis
LMNVs were pre-incubated with FBS as previously described to form LMNV-PC, and then recovered through centrifugation. 6-well transwell inserts (3 μm pore size) seeded with bEnd.3 grown for 5 days and cultured as previously mentioned were treated with 400 μg ml−1 of LMNV-PC, while wells without cells containing only medium doped with LMNV-PC were used as a control. To assess the presence of proteins derived from the bEnd.3 secretome and not due to the direct crossing of the BBB, a third group of 6-well transwell inserts was seeded with bEnd.3 cells grown for 5 days, but in this case LMNV-PC were administered at 400 μg ml−1 just on the basolateral side of the inserts. After 72 h from the addition of LMNV-PC, the nanostructures present on the basolateral side were isolated using centrifugation (16000g for 5 minutes at RT, three times in PBS). After the last centrifugation passage, the obtained supernatant was discharged, and the LMNV-PC pellet was frozen at −80 °C. After thawing, the samples were solubilized in 30 μl of 2% sodium deoxycholate (SDC), 40 mM chloroacetamide, 10 mM tris(2-carboxyethyl)phosphine (TCEP), and 100 mM Tris HCl pH 8 at 95 °C for 10 min and sonicated with an Ultrasonic Processor UP200St (Hielscher), 3 cycles of 30 s. Lysate samples were digested with trypsin and LysC, at a 1:50 and 1:100 ratio of enzyme to sample protein, respectively, overnight at 37 °C. Then nanoparticles were separated from the samples using a DynaMag-2 magnetic particle concentrator (Invitrogen) and treated with 30 μl of 5% NH4OH. The supernatant separated from the nanoparticles was concentrated and joined with the rest of the sample and processed by the iST protocol.33
NanoLC and the mass spectrometer setup
The resulting peptides were analyzed using a nano-UHPLC-MS/MS system with an Ultimate 3000 RSLC coupled to an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific Instrument). Elution was performed with an EASY spray column (75 μm × 50 cm, 2 μm particle size, Thermo Scientific) maintained at 55 °C at a flow rate of 250 nl min−1 with a 140 min non-linear gradient of 7–45% solution B (80% acetonitrile, 20% H2O, 5% DMSO and 0.1% formic acid). Orbitrap detection was used for MS1 measurements at a resolving power of 120 K in the range between 375 and 1500 m/z and with a standard AGC target. MS/MS spectra were acquired in the linear ion trap (rapid scan mode) after higher-energy C-trap dissociation (HCD) at a collision energy of 28% with a standard AGC target and a maximum injection time of 50 ms. Quadrupole isolation with a 1.6 m/z isolation window was used, and dynamic exclusion was enabled for 30 s.Proteomics data analysis
MaxQuant software,34 version 1.6.8.0, was used to process the raw data. The false discovery rate (FDR) for the identification of proteins, peptides, and PSM (peptide-spectrum match) was set to 0.01. A minimum length of 6 amino acids was required for peptide identification. Andromeda engine, incorporated into MaxQuant software, was used to search MS/MS spectra against Uniprot Mouse and Bos taurus database (release Mouse_UP000000589 and Bos taurus_UP000009136 June 2019). In the processing, the variable modifications were Acetyl (Protein N-term), Oxidation (M), and Deamidation (NQ). Carbamidomethyl (C) was selected as a fixed modification. Algorithm MaxLFQ was chosen for the protein quantification with the activated option “match between runs” to reduce the number of the missing proteins. The intensity values were extracted and statistically evaluated using the ProteinGroup Table and Perseus software35 version 1.6.10.43. Gene ontology (GO) enrichment analysis was performed with the app ClueGO of Cytoscape software.36Statistical analysis
The normal distribution of data was assessed through the Shapiro–Wilk normality test. Normally distributed data were compared using the ANOVA test followed by the LSD post-hoc test with Bonferroni's correction, and expressed as mean ± standard deviation. Data non-normally distributed were compared using the Kruskal–Wallis test followed by a pairwise-Wilcox post-hoc test with Holm correction, and expressed in a box plot as median ± 95% confidence interval. Mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE37 partner repository, with the dataset identifier PXD031034Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 709613, SLaMM).References
- T. Lima, K. Bernfur, M. Vilanova and T. Cedervall, Understanding the Lipid and Protein Corona Formation on Different Sized Polymeric Nanoparticles, Sci. Rep., 2020, 10(1), 1129, DOI:10.1038/s41598-020-57943-6.
- S. Dominguez-Medina, L. Kisley, L. J. Tauzin, A. Hoggard, B. Shuang, A. S. D. S. Indrasekara, S. Chen, L. Y. Wang, P. J. Derry, A. Liopo, E. R. Zubarev, C. F. Landes and S. Link, Adsorption and Unfolding of a Single Protein Triggers Nanoparticle Aggregation, ACS Nano, 2016, 10(2), 2103–2112, DOI:10.1021/acsnano.5b06439.
- Z. Wang, C. Wang, S. Liu, W. He, L. Wang, J. Gan, Z. Huang, Z. Wang, H. Wei, J. Zhang and L. Dong, Specifically Formed Corona on Silica Nanoparticles Enhances Transforming Growth Factor B1 Activity in Triggering Lung Fibrosis, ACS Nano, 2017, 11(2), 1659–1672, DOI:10.1021/acsnano.6b07461.
- G. Su, H. Jiang, B. Xu, Y. Yu and X. Chen, Effects of Protein Corona on Active and Passive Targeting of Cyclic RGD Peptide-Functionalized PEGylation Nanoparticles, Mol. Pharm., 2018, 15(11), 5019–5030, DOI:10.1021/acs.molpharmaceut.8b00612.
- R. del Caño, L. Mateus, G. Sánchez-Obrero, J. M. Sevilla, R. Madueño, M. Blázquez and T. Pineda, Hemoglobin Bioconjugates with Surface-Protected Gold Nanoparticles in Aqueous Media: The Stability Depends on Solution PH and Protein Properties, J. Colloid Interface Sci., 2017, 505, 1165–1171, DOI:10.1016/j.jcis.2017.07.011.
- J. H. Shannahan, R. Podila and J. M. Brown, A Hyperspectral and Toxicological Analysis of Protein Corona Impact on Silver Nanoparticle Properties, Intracellular Modifications, and Macrophage Activation, Int. J. Nanomed., 2015, 10(1), 6509–6521, DOI:10.2147/IJN.S92570.
- J. Ren, R. Cai, J. Wang, M. Daniyal, D. Baimanov, Y. Liu, D. Yin, Y. Liu, Q. Miao, Y. Zhao and C. Chen, Precision Nanomedicine Development Based on Specific Opsonization of Human Cancer Patient-Personalized Protein Coronas, Nano Lett., 2019, 19(7), 4692–4701, DOI:10.1021/acs.nanolett.9b01774.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. F. Puntes, Hardening of the Nanoparticle–Protein Corona in Metal (Au, Ag) and Oxide (Fe3O4, CoO, and CeO2) Nanoparticles, Small, 2011, 7(24), 3479–3486, DOI:10.1002/smll.201101511.
- M. Garvas, A. Testen, P. Umek, A. Gloter, T. Koklic and J. Strancar, Protein Corona Prevents TiO2 Phototoxicity, PLoS One, 2015, 10(6), e0129577 CrossRef PubMed.
- F. Giulimondi, L. Digiacomo, D. Pozzi, S. Palchetti, E. Vulpis, A. L. Capriotti, R. Z. Chiozzi, A. Laganà, H. Amenitsch, L. Masuelli, G. Peruzzi, M. Mahmoudi, I. Screpanti, A. Zingoni and G. Caracciolo, Interplay of Protein Corona and Immune Cells Controls Blood Residency of Liposomes, Nat. Commun., 2019, 10(1), 3686, DOI:10.1038/s41467-019-11642-7.
- X. Zeng, J. Sun, S. Li, J. Shi, H. Gao, W. S. Leong, Y. Wu, M. Li, C. Liu, P. Li, J. Kong, Y.-Z. Wu, G. Nie, Y. Fu and G. Zhang, Blood-Triggered Generation of Platinum Nanoparticle Functions as an Anti-Cancer Agent, Nat. Commun., 2020, 11(1), 567, DOI:10.1038/s41467-019-14131-z.
- S. Kihara, N. J. van der Heijden, C. K. Seal, J. P. Mata, A. E. Whitten, I. Köper and D. J. McGillivray, Soft and Hard Interactions between Polystyrene Nanoplastics and Human Serum Albumin Protein Corona, Bioconjugate Chem., 2019, 30(4), 1067–1076, DOI:10.1021/acs.bioconjchem.9b00015.
- N. Feiner-Gracia, M. Beck, S. Pujals, S. Tosi, T. Mandal, C. Buske, M. Linden and L. Albertazzi, Super-Resolution Microscopy Unveils Dynamic Heterogeneities in Nanoparticle Protein Corona, Small, 2017, 13(41), 1701631, DOI:10.1002/smll.201701631.
- T. Miclăuş, C. Beer, J. Chevallier, C. Scavenius, V. E. Bochenkov, J. J. Enghild and D. S. Sutherland, Dynamic Protein Coronas Revealed as a Modulator of Silver Nanoparticle Sulphidation in Vitro, Nat. Commun., 2016, 7(1), 11770, DOI:10.1038/ncomms11770.
- V. Mirshafiee, R. Kim, S. Park, M. Mahmoudi and M. L. Kraft, Impact of Protein Pre-Coating on the Protein Corona Composition and Nanoparticle Cellular Uptake, Biomaterials, 2016, 75, 295–304, DOI:10.1016/j.biomaterials.2015.10.019.
- W. M. Pardridge, The Blood-Brain Barrier: Bottleneck in Brain Drug Development, NeuroRx, 2005, 2(1), 3–14, DOI:10.1602/neurorx.2.1.3.
- C. Martinelli, C. Pucci, M. Battaglini, A. Marino and G. Ciofani, Antioxidants and Nanotechnology: Promises and Limits of Potentially Disruptive Approaches in the Treatment of Central Nervous System Diseases, Adv. Healthcare Mater., 2020, 9(3), 1901589, DOI:10.1002/adhm.201901589.
- C. Tapeinos, M. Battaglini and G. Ciofani, Advances in the Design of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Targeting Brain Diseases, J. Controlled Release, 2017, 264(August), 306–332, DOI:10.1016/j.jconrel.2017.08.033.
- M. Battaglini, C. Tapeinos, I. Cavaliere, A. Marino, A. Ancona, N. Garino, V. Cauda, F. Palazon, D. Debellis and G. Ciofani, Design, Fabrication, and In Vitro Evaluation of Nanoceria-Loaded Nanostructured Lipid Carriers for the Treatment of Neurological Diseases, ACS Biomater. Sci. Eng., 2019, 5(2), 670–682, DOI:10.1021/acsbiomaterials.8b01033.
- A. Marino, A. Camponovo, A. Degl'Innocenti, M. Bartolucci, C. Tapeinos, C. Martinelli, D. De Pasquale, F. Santoro, V. Mollo, S. Arai, M. Suzuki, Y. Harada, A. Petretto and G. Ciofani, Multifunctional Temozolomide-Loaded Lipid Superparamagnetic Nanovectors: Dual Targeting and Disintegration of Glioblastoma Spheroids by Synergic Chemotherapy and Hyperthermia Treatment, Nanoscale, 2019, 11(44), 21227–21248, 10.1039/c9nr07976a.
- C. Tapeinos, A. Marino, M. Battaglini, S. Migliorin, R. Brescia, A. Scarpellini, C. De Julián Fernández, M. Prato, F. Drago and G. Ciofani, Stimuli-Responsive Lipid-Based Magnetic Nanovectors Increase Apoptosis in Glioblastoma Cells through Synergic Intracellular Hyperthermia and Chemotherapy, Nanoscale, 2018, 11(1), 72–88, 10.1039/c8nr05520c.
- A. Cox, P. Andreozzi, R. Dal Magro, F. Fiordaliso, A. Corbelli, L. Talamini, C. Chinello, F. Raimondo, F. Magni, M. Tringali, S. Krol, P. J. Silva, F. Stellacci, M. Masserini and F. Re, Evolution of Nanoparticle Protein Corona across the Blood–Brain Barrier, ACS Nano, 2018, 12(7), 7292–7300, DOI:10.1021/acsnano.8b03500.
- C. Rodriguez-Quijada, H. de Puig, M. Sánchez-Purrà, C. Yelleswarapu, J. J. Evans, J. P. Celli and K. Hamad-Schifferli, Protease Degradation of Protein Coronas and Its Impact on Cancer Cells and Drug Payload Release, ACS Appl. Mater. Interfaces, 2019, 11(16), 14588–14596, DOI:10.1021/acsami.9b00928.
- K. Natte, J. F. Friedrich, S. Wohlrab, J. Lutzki, R. von Klitzing, W. Österle and G. Orts-Gil, Impact of Polymer Shell on the Formation and Time Evolution of Nanoparticle-Protein Corona, Colloids Surf., B, 2013, 104, 213–220, DOI:10.1016/j.colsurfb.2012.11.019.
- U. Martens, D. Böttcher, D. Talbot, U. Bornscheuer, A. Abou-Hassan and M. Delcea, Maghemite Nanoparticles Stabilize the Protein Corona Formed with Transferrin Presenting Different Iron-Saturation Levels, Nanoscale, 2019, 11(34), 16063–16070, 10.1039/C9NR04967C.
- V. Ceña and P. Játiva, Nanoparticle Crossing of Blood–Brain Barrier: A Road to New Therapeutic Approaches to Central Nervous System Diseases, Nanomedicine, 2018, 13(13), 1513–1516, DOI:10.2217/nnm-2018-0139.
- S. Li, Z. Peng, J. Dallman, J. Baker, A. M. Othman, P. L. Blackwelder and R. M. Leblanc, Crossing the Blood-Brain-Barrier with Transferrin Conjugated Carbon Dots: A Zebrafish Model Study, Colloids Surf., B, 2016, 145, 251–256, DOI:10.1016/j.colsurfb.2016.05.007.
- J. Sun, W. Ou, D. Han, A. Paganini-Hill, M. J. Fisher and R. K. Sumbria, Comparative Studies between the Murine Immortalized Brain Endothelial Cell Line (BEnd.3) and Induced Pluripotent Stem Cell-Derived Human Brain Endothelial Cells for Paracellular Transport, PLoS One, 2022, 17(5), e0268860 CrossRef CAS PubMed.
- M. Papi, D. Caputo, V. Palmieri, R. Coppola, S. Palchetti, F. Bugli, C. Martini, L. Digiacomo, D. Pozzi and G. Caracciolo, Clinically Approved PEGylated Nanoparticles Are Covered by a Protein Corona That Boosts the Uptake by Cancer Cells, Nanoscale, 2017, 9(29), 10327–10334, 10.1039/C7NR03042H.
- B. Rathore, K. Sunwoo, P. Jangili, J. Kim, J. H. Kim, M. Huang, J. Xiong, A. Sharma, Z. Yang, J. Qu and J. S. Kim, Nanomaterial Designing Strategies Related to Cell Lysosome and Their Biomedical Applications: A Review, Biomaterials, 2019, 211, 25–47, DOI:10.1016/j.biomaterials.2019.05.002.
- C. Pisani, J.-C. Gaillard, M. Odorico, J. L. Nyalosaso, C. Charnay, Y. Guari, J. Chopineau, J.-M. Devoisselle, J. Armengaud and O. Prat, The Timeline of Corona Formation around Silica Nanocarriers Highlights the Role of the Protein Interactome, Nanoscale, 2017, 9(5), 1840–1851, 10.1039/C6NR04765C.
- M. Battaglini, A. Marino, A. Carmignani, C. Tapeinos, V. Cauda, A. Ancona, N. Garino, V. Vighetto, G. La Rosa, E. Sinibaldi and G. Ciofani, Polydopamine Nanoparticles as an Organic and Biodegradable Multitasking Tool for Neuroprotection and Remote Neuronal Stimulation, ACS Appl. Mater. Interfaces, 2020, 12(32), 35782–35798, DOI:10.1021/acsami.0c05497.
- N. A. Kulak, G. Pichler, I. Paron, N. Nagaraj and M. Mann, Minimal, Encapsulated Proteomic-Sample Processing Applied to Copy-Number Estimation in Eukaryotic Cells, Nat. Methods, 2014, 11(3), 319–324, DOI:10.1038/nmeth.2834.
- J. Cox and M. Mann, MaxQuant Enables High Peptide Identification Rates, Individualized ppb-Range Mass Accuracies and Proteome-Wide Protein Quantification, Nat. Biotechnol., 2008, 26(12), 1367–1372, DOI:10.1038/nbt.1511.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data, Nat. Methods, 2016, 13(9), 731–740, DOI:10.1038/nmeth.3901.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks, Genome Res., 2003, 13(11), 2498–2504, DOI:10.1101/gr.1239303.
- Y. Perez-Riverol, J. Bai, C. Bandla, D. García-Seisdedos, S. Hewapathirana, S. Kamatchinathan, D. J. Kundu, A. Prakash, A. Frericks-Zipper, M. Eisenacher, M. Walzer, S. Wang, A. Brazma and J. A. Vizcaíno, The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences, Nucleic Acids Res., 2022, 50(D1), D543–D552, DOI:10.1093/nar/gkab1038.
Footnote |
| † Electronic supplementary information (ESI) available: Five supplementary figures depicting the BBB characterization, the time-dependent evolution of PC associated with LMNVs analyzed by confocal microscopy, and the co-localization analysis between LMNVs and, respectively, lysosomes, clathrin-positive vesicles, and caveolin-positive vesicles. Seven supplementary tables reporting the lists of proteins present in LMNV-PC after the BBB passage, of proteins present in LMNV-PC exposed just to FBS without the passage through the BBB, of proteins conserved before and after the BBB passage, of proteins associated with LMNVs exposed to the secretome of bEnd.3 cells, of proteins associated both with LMNVs that crossed the BBB and with LMNVs that were exposed to the bEnd.3 secretome, of proteins associated both with LMNVs exposed to only FBS and LMNVs exposed to the bEnd.3 secretome, and finally of proteins present under all experimental conditions. See DOI: https://doi.org/10.1039/d2nr00484d |
| This journal is © The Royal Society of Chemistry 2022 |