
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multivalent non-covalent interactions lead to strongest polymer adhesion†
Max
Lallemang
 ab,
Leixiao
Yu
c,
Wanhao
Cai
a,
Klaus
Rischka
d,
Andreas
Hartwig
de,
Rainer
Haag
c,
Thorsten
Hugel
*ab and
Bizan N.
Balzer
*abf
ab,
Leixiao
Yu
c,
Wanhao
Cai
a,
Klaus
Rischka
d,
Andreas
Hartwig
de,
Rainer
Haag
c,
Thorsten
Hugel
*ab and
Bizan N.
Balzer
*abf
aInstitute of Physical Chemistry, University of Freiburg, Albertstraße 21, 79104 Freiburg, Germany. E-mail: bizan.balzer@physchem.uni-freiburg.de; thorsten.hugel@physchem.uni-freiburg.de
bCluster of Excellence livMatS @ FIT-Freiburg Center for Interactive Materials and Bioinspired Technologies, University of Freiburg, Georges-Köhler-Allee 105, 79110 Freiburg, Germany
cInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Takusstraße 3, 14195 Berlin, Germany
dFraunhofer Institute for Manufacturing Technology and Advanced Materials IFAM, Wiener Straße 12, 28359 Bremen, Germany
eUniversity of Bremen, Department 2 Biology/Chemistry, Leobener Straße 3, 28359 Bremen, Germany
fFreiburg Materials Research Center (FMF), Albert Ludwig University of Freiburg, 79104 Freiburg, Germany
First published on 16th February 2022
Abstract
Multivalent interactions play a leading role in biological processes such as the inhibition of inflammation or virus internalization. The multivalent interactions show enhanced strength and better selectivity compared to monovalent interactions, but they are much less understood due to their complexity. Here, we detect molecular interactions in the range of a few piconewtons to several nanonewtons and correlate them with the formation and subsequent breaking of one or several bonds and assign these bonds. This becomes possible by performing atomic force microcopy (AFM)-based single molecule force spectroscopy of a multifunctional polymer covalently attached to an AFM cantilever tip on a substrate bound polymer layer of the multifunctional polymer. Varying the pH value and the crosslinking state of the polymer layer, we find that bonds of intermediate strength (non-covalent), like coordination bonds, give the highest multivalent bond strength, even outperforming strong (covalent) bonds. At the same time, covalent bonds enhance the polymer layer density, increasing in particular the number of non-covalent bonds. In summary, we can show that the key for the design of stable and durable polymer coatings is to provide a variety of multivalent interactions and to keep the number of non-covalent interactions at a high level.
Introduction
In nature, the concept of multivalency mediates strong adhesion, in particular for interactions between receptors and ligands, viruses and host cells, or between two cell surfaces. Multivalent bonds are primarily found when a number of covalently linked ligands of certain or various types bind in parallel to an ensemble of acceptors. Due to their high force and selectivity, multivalent bonds have significant advantages in molecular recognition and adhesion. Multivalent bonds may be of identical or different types and strength. However, characterizing individual constituents of multivalent bonds is still challenging because a wide range of forces and interaction lengths are involved.1One important amino acid used in nature to form multivalent bonds is the catecholic 3,4-dihydroxy-L-phenylalanine (DOPA). It is for example used by blue mussels, which can bind in wet and salty conditions to inorganic as well as organic surfaces.2 Catechols can reversibly be oxidized to form quinones, which is highly electrophilic and can irreversibly crosslink with other catechol moieties.3,4 Crosslinking can occur via Michael addition of the free amine with the quinone group, via Schiff Base formation of the amine with the carbonyl group of the quinone or via quinone-quinone coupling.5,6 The oxidation of catechol to semi-quinone and finally to quinone is a crucial step for crosslinking of adhesives based on catechol rich compounds. This evidence can be used to synthesize and tailor mussel-inspired and catechol-based polymers with strong adhesion to surfaces in order to obtain universal/substrate-independent coatings with self-healing or antifouling properties under mild conditions.7–10
Here we used an amphiphilic, catechol-based and multifunctional block copolymer Br/N3-PG-b/r-P(Cat-Ph-A) (Fig. 1a), which has been used before to create well-defined polymer monolayers.11,12 This multifunctional molecule comprises sites for secondary modification and combines the concepts known from blood protein adsorption with those known from mussel adhesion. An azide terminal group allows to perform a copper free type of the Huisgen reaction, which takes advantage of strain-promoted alkyne azide cycloaddition (SPAAC) using dibenzocyclooctine (DBCO) forming 1,2,3-triazoles.13–16 These reaction products are non-toxic, i.e., biocompatible, as well as efficient and practicable under mild conditions.17 Furthermore, the multifunctional molecule offers crosslinking via Schiff base formation or Michael addition.
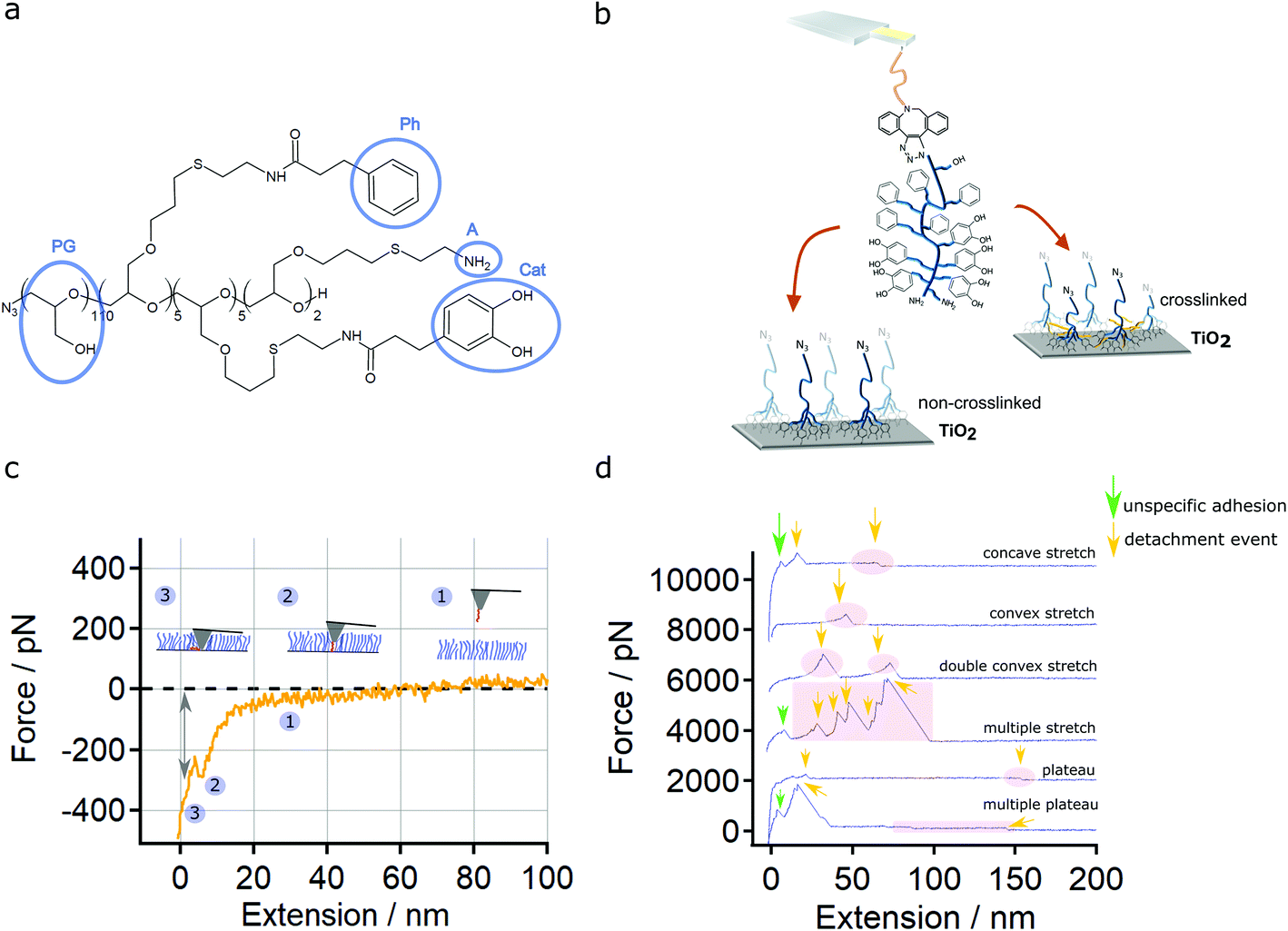 | ||
| Fig. 1 Tools and experiments to quantify multivalent bond strength. (a) Scheme of a multivalent N3-PG110-b-P(Cat5-Ph5-A2) molecule, showing a polyglycol (PG) block and catechol (Cat), phenyl (Ph) and amine (A) groups. (b) Scheme of a PG110-b-P(Cat5-Ph5-A2) molecular force sensor on non-crosslinked (left) and crosslinked (right) N3-PG110-b-P(Cat5-Ph5-A2) layers on TiO2. (c) Approach curves for a PG110-b-P(Cat5-Ph5-A2) molecular force sensor on a N3-PG110-b-P(Cat5-Ph5-A2) brush-like monolayer on TiO2 in H2O revealing a breakthrough event in the range of 100 to 400 pN (indicated by an arrow). (d) Characterization of the retract force-extension curves (offset by 2000 pN) on the N3-PG110-b-P(Cat5-Ph5-A2) layer, showing (from top to bottom) detachment events (marked by yellow arrows): concave stretch, convex stretch, double convex stretch, multiple stretches, plateau of constant force and multiple plateaus of constant force. The adhesion peak (marked by green arrows) at the beginning of the force-extension curves was not taken into account for evaluation, because it results from unspecific interaction of the whole AFM cantilever tip with the underlying sample. | ||
However, the molecular understanding of the interplay between the different and multivalent types of interactions in such a coating, making it so unique, is still lacking because of the variety of different bonds. A better molecular understanding is crucial for a rational design of future polymer coatings.
Single molecule force spectroscopy (SMFS) with an Atomic Force Microscope (AFM) is a powerful method for studying the interactions between single molecules and surfaces or interfaces.18–21 The formation of isolated bonds like, e.g., S–Au,22 Si–O23 and Si–C24 has been studied in detail by SMFS. Many experiments with a big diversity of catechol-based molecules demonstrated the strength of catechols and quinones on different substrates and in different pH and buffer conditions. There, detachment forces varied between approx. 50 and 1000 pN.22,25–35 Additionally, SMFS on systems comprising multivalent interactions have been performed for different biological systems, obtaining detachment forces of up to 1000 pN.36–42 However, the individual contributions to the overall force could not be resolved.
Here, we used covalent attachment of a N3-PG-b-P(Cat-Ph-A) copolymer probe molecule to an AFM cantilever tip to study the multivalent interactions on the molecular level by SMFS. We used polymer layers consisting of the same N3-PG-b-P(Cat-Ph-A) copolymer under different conditions and assign detachment forces to specific types of interaction. This allowed us to directly monitor the respective interactions, namely their multivalent bond strength and rupture force with piconewton resolution. Furthermore, we could show that the formation and rupture of strong (covalent) bonds cannot outperform the presence of interactions of low and intermediate strength (non-covalent). Altogether, this teaches us how to optimize the design of future multifunctional coatings using different types of molecular interactions and multivalency as a concept for creating robust coatings for various applications, especially in the field of chemistry, physics, material science and biomedicine.
Materials and methods
RCA cleaning
The glassware and tweezers were immersed in a RCA solution (Radio Corporation of America) having a volume ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 of H2O (Purelab Chorus 1, Elga LabWater, Celle, Germany, 18.2 MΩ cm), NH3 solution (Roth, Karlsruhe, Germany, 28.0–30.0%), H2O2 (Sigma-Aldrich, St Louis, MO, USA, ≥30%) at 70 °C for 40 min. Afterwards, they were rinsed three times with H2O, dried and stored at 120 °C.
:1:1 of H2O (Purelab Chorus 1, Elga LabWater, Celle, Germany, 18.2 MΩ cm), NH3 solution (Roth, Karlsruhe, Germany, 28.0–30.0%), H2O2 (Sigma-Aldrich, St Louis, MO, USA, ≥30%) at 70 °C for 40 min. Afterwards, they were rinsed three times with H2O, dried and stored at 120 °C.
Substrates
Ti-wafers, named TiO2 in the following, were prepared by using 3 inches N/Phosphor doped Si wafers with a thickness of 380 μm, a 100 orientation and a resistance of 7.5 Ω cm (4P219/90, Silchem, Germany). A 50 nm thick Ti-layer was applied by a sputter process using a high-vacuum coater (Leica EM ACE600, Leica Microsystems GmbH, Wetzlar, Germany). The thickness was controlled during the sputter process (current 100 mA, Ar pressure 0.01 mbar, working distance 50 mm, tilt angle 20°) by a quartz crystal thickness measurement.Polymer
N3-PG110-b-P(Cat5-Ph5-A2) with a molecular weight of 12 kDa is an amphiphilic block copolymer used in this work (Fig. 1a). The polyglycol (PG) acts as a hydrophilic domain. Poly (allyl glycidyl ether) is functionalized by amine groups (A, 2 units), catechol groups (Cat, 5 units) and phenyl groups (Ph, 5 units). An azide group serves as a functional site for secondary modification. The contour length (43 nm) of the polymer was calculated by multiplying the number of polyethylene glycol (PEG) and PG groups N by the length l of one PEG or PG-repeat unit (0.356 nm).43,44 Together with the triethoxysilane-PEG-DBCO (silane-PEG-DBCO, NANOCS, Boston, MA, USA) linker having a length of about 41 nm, the final PG110-b-P(Cat5-Ph5-A2) molecular force sensor used in the SMFS experiments has a total contour length of about 84 nm.Substrate and polymer layer preparation
The TiO2-wafers were cleaned ultrasonically for 5 min first with H2O followed by MeOH. Afterwards, the TiO2-wafers were cleaned with an oxygen plasma (15% power, 0.1 mbar, 20 min, Diener Electronics, Germany). To prepare polymer layers, the cleaned slides were incubated for 2 h in a solution of N3-PG110-b-P(Cat5-Ph5-A2) (1 mg mL−1) in a pH 6 3-(N-morpholino)propanesulfonic acid (MOPS, 0.1 M, Applichem GmbH, Darmstadt, Germany) buffer at 22 °C.11 The pH was adjusted by using a pH meter (LLG pH meter 7, Meckenheim, Germany) and solutions of NaOH (1.0 M) for the pH 8.6 buffer and HCl (1.0 M) for the pH 6.0 buffer. Afterwards, the slides were rinsed with H2O, followed by MeOH and finally dried by a N2 stream. To induce the crosslinking reaction, the slides were stored for 1 h in a solution of K2S2O8 (10 mg mL−1) in pH 8.6 MOPS (0.1 M) buffer at 22 °C.11 After that, the slides were rinsed with H2O, followed by MeOH and finally dried by a N2 stream. For control experiments with SiO2, SiO2-wafers were cleaned in ethanol for 5 min at 22 °C using a sonicator (Elmasonic S15, Elma, Singen, Germany).AFM cantilever tip functionalization
The following processes have been optimized to ensure a proper passivation of the cantilever tip surface with 5 kDa silane-PEG and the attachment of PG110-b-P(Cat5-Ph5-A2) to finally obtain detachment events in the SMFS experiment.PG110-b-P(Cat5-Ph5-A2) molecular force sensor
For functionalization of cantilever tips with N3-PG110-b-P(Cat5-Ph5-A2) which is schemed in Fig. S1,† Si3N4 MLCT-BIO-DC cantilevers (Bruker AFM probes, Camarillo, CA, USA) were used. First, the cantilevers were activated with oxygen plasma (Diener Electronics, Germany) to gain hydroxyl groups on the surface of the cantilever tips. The MLCT-BIO-DC probes were treated for 2 min with 40% power at a pressure of 0.1 mbar. As a next step, a 5 kDa silane-PEG-DBCO (NANOCS, Boston, MA, USA) linker was bound to the cantilever tip. Due to the sensitivity of DBCO towards light, the following steps were performed in the dark. The cantilevers were incubated in a solution of silane-PEG-DBCO (PEG-DBCO) in toluene (1.25 mg mL−1, 2 h) at 22 °C. Then, the PEGylated cantilevers were rinsed in toluene, EtOH, H2O and were finally incubated in a solution of N3-PG110-b-P(Cat5-Ph5-A2) in a 1:1 solution of PBS buffer (Dulbecco's Phosphate Buffered Saline, pH 7.2, Sigma-Aldrich, USA) and DMSO (1.25 mg mL−1, 24 h) at 22 °C to induce the copper-free click chemistry of the azide group of N3-PG110-b-P(Cat5-Ph5-A2) and DBCO. After final rinsing, the cantilevers were stored in HEPES (Sigma-Aldrich, St Louis, MO, USA) buffer (pH 7.0, 50 mM NaCl) at 4 °C until use in the AFM experiment. For each functionalization, control cantilevers (carrying the 5 kDa silane-PEG-DBCO only) were additionally prepared by the same procedure incubating the cantilevers in the pure solvent (DMSO) instead of N3-PG110-b-P(Cat5-Ph5-A2).
AFM-based force spectroscopy
All SMFS experiments were performed with a Cypher ES (Asylum Research, an Oxford Instruments company, Santa Barbara, CA, USA). Before each SMFS experiment, the inverse optical lever sensitivity (InvOLS) was determined by fitting a linear function to the repulsive region of a force–extension curve. In order to reduce errors, the determination of the InvOLS was performed by using an average of at least five individual InvOLS values. The spring constant of the cantilever was determined by the thermal noise method.45PG110-b-P(Cat5-Ph5-A2) on the N3-PG110-b-P(Cat5-Ph5-A2) layer on TiO2 took place in ultrapure H2O, MOPS buffer pH 6.0 (0.1 M) and MOPS buffer pH 8.6 (0.1 M). PG110-b-P(Cat5-Ph5-A2) and silane-PEG-DBCO were probed on SiO2 in ultrapure water.
SMFS parameters were defined as follows: force distance, 1 μm; velocity, 1 μm s−1; contact force, 50 to 3000 pN; sampling rate, 5 kHz; contact time towards the surface, 0–10 s and temperature, 25 °C. To reduce the influence of local sample variations, force-extension curves were recorded in a grid-like manner with 10·10 points covering 20·20 μm2 (force maps). At least two force maps were obtained per cantilever. Prior to a series of measurements, at least one control cantilever (same procedure but the incubation took place in pure solvent instead of the target molecule) was measured on different surface spots to verify a contamination-free functionalization. Once the control cantilevers (silane-PEG-DBCO on SiO2) showed an absence of stretching events or plateaus of constant force, the PG110-b-P(Cat5-Ph5-A2) functionalized cantilevers were measured, respectively. For data evaluation, a self-programmed evaluation software based on Igor Pro (Wavemetrics, Portland, OR, USA) was used. For the analysis of detachment forces, all detachment events such as stretches and plateaus were taken into account, except the adhesion peak at the beginning of the force-extension curves (Fig. 1d), resulting from unspecific interactions of the whole AFM cantilever tip and the surface.
OriginPro 2019 (OriginLab, Northampton, MA, USA) was used for data presentation and to test the significance. The latter was performed by an unpaired two-tailed Mann–Whitney U test was performed with for the force values. The null hypothesis in the Mann–Whitney U test is that X and Y are two random selected values from two populations, the probability of Y being greater than X is equal to the probability of X being greater than Y. The level of significance was set to 0.05.
AFM-based scratching
The thickness of the N3-PG110-b-P(Cat5-Ph5-A2) polymer layer was determined by AFM scratching experiments on a Cypher ES (Asylum Research, an Oxford Instruments company, Santa Barbara, CA, USA) using AC240TS cantilevers (Olympus, Japan). AFM imaging of those polymer layers was performed in pH 6.0 MOPS buffer (0.1 M) at a temperature of 25 °C. The first image of each sample was taken in the intermittent-contact mode with a scan size of 15·15 μm2. Then, the surface was scratched in contact mode with a scan size of 1·1 μm2 and an applied force (trigger force) of 648 nN. The imaging mode was switched back again to intermittent-contact mode for further imaging. All images were taken at a scan rate of 2 Hz. The depth of the hole, created by the cantilever tip in contact mode, was used to determine the presence and thickness of the polymer layer on the respective substrate.Results
Characterization of multivalent interactions by SMFS
We used a PG110-b-P(Cat5-Ph5-A2) molecule (Fig. 1a) covalently bound to the AFM cantilever tip (Fig. 1b and S1†) as a molecular force sensor. The molecular force sensor was measured on N3-PG110-b-P(Cat5-Ph5-A2) layers on TiO2 (Fig. 1b) to understand bond formation and rupture. The presence and thickness of this layers on TiO2 have been tested by contact angle and AFM imaging experiments (Fig. S2 and S3†).The covalent attachment of one single polymer to a cantilever is crucial for single molecule force experiments. It enables a high reproducibility when performing force spectroscopy with a certain polymer on a specific cantilever tip. In our case, the successful attachment of one single polymer has been verified by measuring the PG110-b-P(Cat5-Ph5-A2) molecular force sensor on SiO2 showing single molecule detachment events (Fig. S4†). Control measurements with silane-PEG-DBCO (5 kDa) show that these detachment events are indeed caused by the PG110-b-P(Cat5-Ph5-A2) and not by unspecific adhesion between AFM cantilever tip and the SiO2 surface (Fig. S5†).
Finally, a variation of the contact force and contact time (Fig. S6 and S7†) revealed that a contact force of 500 pN and a contact time of 1 s was ideal to obtain reproducible indentation into the layer (breakthrough) and detachment events as depicted in Fig. 1c and d.
The variety of the possible interactions between the PG110-b-P(Cat5-Ph5-A2) molecular force sensor and the N3-PG110-b-P(Cat5-Ph5-A2) layer is reflected by the obtained force-extension traces. We classified them into concave stretch, convex stretch, double (multiple) convex stretches, single plateau of constant force and multiple plateaus of constant force (Fig. 1d).
In order to assign the detachment events to specific interactions, we performed SMFS measurements under different conditions. First in MOPS buffer at pH 6.0 (0.1 M). Then, the buffer was exchanged to MOPS buffer pH 8.6 (0.1 M) for the next SMFS experiments. Second, a crosslinked N3-PG110-b-P(Cat5-Ph5-A2) layer was prepared using the oxidant (K2S2O8) in MOPS buffer pH 8.6 (0.1 M) according to the protocol of Yu et al.11 Then, the crosslinked polymer layer was taken for SMFS experiments in MOPS buffer pH 8.6 (0.1 M).
Fig. 2a illustrates a representative force-extension curve (condition: pH 8.6) that comprises several detachment events. Further force-extension curves of that type are shown in Fig. S8† (condition: pH 8.6 crosslinked). Fig. 2b represents the different measured detachment forces, resulting mostly from convex stretches (see Table S1† for a quantification). The median and average forces increased from pH 6.0 to pH 8.6 crosslinked. An unpaired two-tailed Mann–Whitney U test (with a level of significance of 0.05, see Materials and Methods) showed that the difference between pH 6.0 and pH 8.6 and between pH 8.6 crosslinked and the other two conditions (pH 6.0 and pH 8.6) was significant, respectively. In general, the forces of PG110-b-P(Cat5-Ph5-A2) on N3-PG110-b-P(Cat5-Ph5-A2) exceed the forces on SiO2 (Fig. S4†) by a factor of more than 10, demonstrating the strength of multivalent interactions.
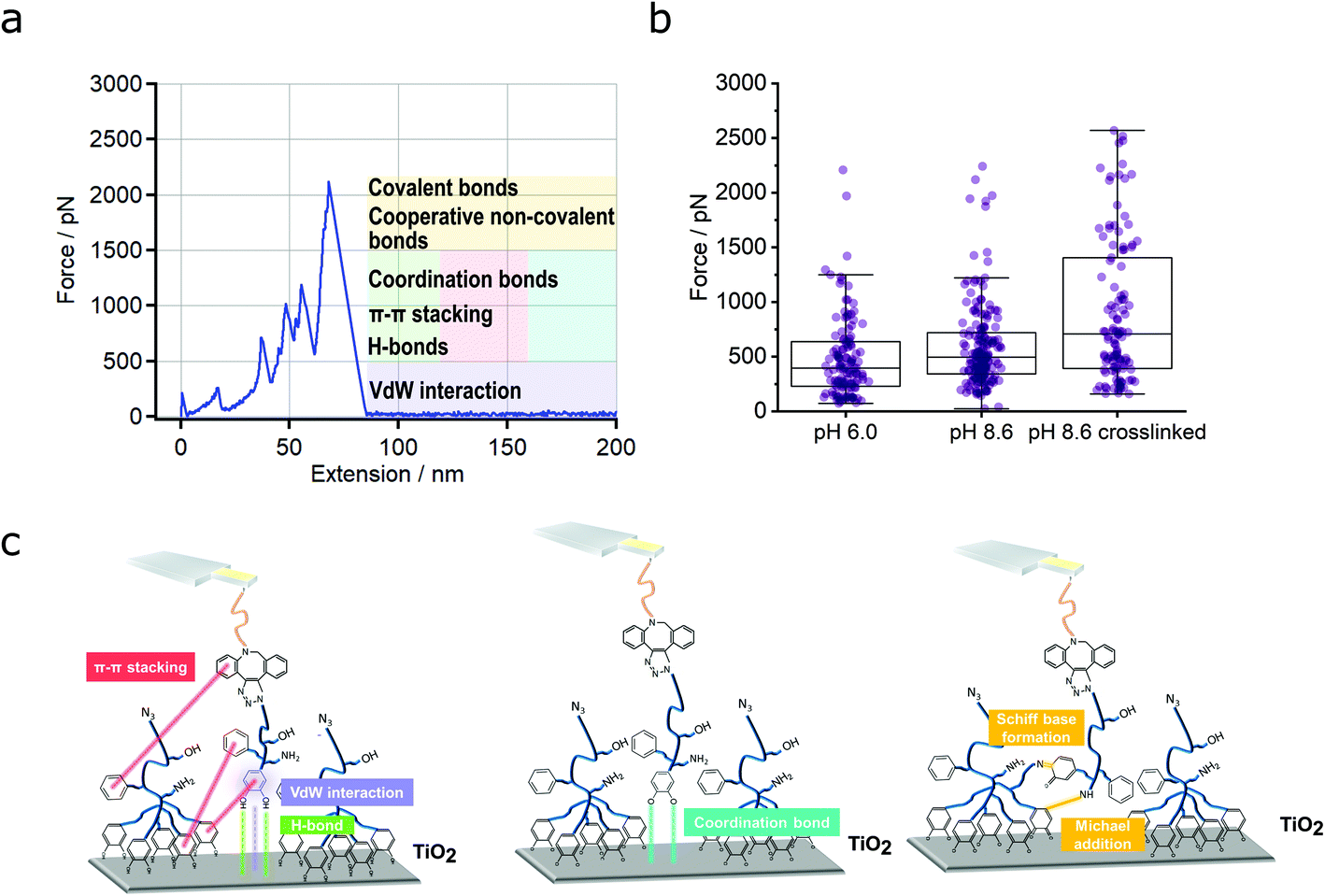 | ||
| Fig. 2 Dependence of multivalent interactions on pH value and crosslinking. (a) An exemplary force-extension curve for a PG110-b-P(Cat5-Ph5-A2) molecular force sensor on a non-crosslinked N3-PG110-b-P(Cat5-Ph5-A2) layer on TiO2 (condition: pH 8.6). The first peak at zero extension was due to unspecific adhesion of the whole AFM cantilever tip and the polymer layer and has been omitted for evaluation. (b) The force distributions given by box plots are shown for pH 6.0, pH 8.6 and pH 8.6 crosslinked, revealing an increase of the median force with increasing pH and crosslinking of the polymer layer. The whiskers correspond to the lower quartile Q1 − 1.5·interquartile range (minimum) and the upper quartile Q3 + 1.5·interquartile range (maximum) or the last value within that range, respectively. 119 detachment events were obtained for pH 6.0, 171 for pH 8.6 and 110 for pH 8.6 crosslinked. For pH 6.0, the median value corresponds to 396 pN, the lower quartile Q1 and the upper quartile Q3 have values of 228 pN and 638 pN, respectively. For pH 8.6, the median value corresponds to 495 pN, the lower quartile Q1 and the upper quartile Q3 have values of 343 pN and 719 pN, respectively. For pH 8.6 crosslinked, the median has a value of 707 pN, the lower quartile Q1 and the upper quartile Q3 correspond to 391 pN and 1406 pN, respectively. The average detachment force corresponds to (491 ± 373) pN for pH 6.0, (598 ± 389) pN for pH 8.6 and (903 ± 655) pN for pH 8.6 crosslinked. The errors correspond to the standard deviation. (c) Scheme of the different types of possible interactions. | ||
Assignment of different interactions of the multivalent system
Fig. 2c shows the possible types of interactions between the PG110-b-P(Cat5-Ph5-A2) molecular force sensor and the N3-PG110-b-P(Cat5-Ph5-A2) layer. These can be van der Waals (vdW) interaction (depicted in purple), hydrogen bonds44 (depicted in green) between hydrogen atoms of the catechol (5 groups) and oxygen of TiO2,46,47 π–π stacking (depicted in red) between the aromatic rings of the catechols (5 groups), phenyls (5 groups) and DBCO (Fig. 2c, left).21,48 Coordination bonds (depicted in blue) can be formed between oxygen atoms of quinone groups (5 groups) and Ti atoms (Fig. 2c, middle). Finally, strong interaction between amines (2 groups) and quinone groups (5 groups), accounting for the formation of a covalent N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (Schiff base reaction) or NH–C (Michael addition) bond with a subsequent rupture event (Fig. 2c, right) are also possible.
C (Schiff base reaction) or NH–C (Michael addition) bond with a subsequent rupture event (Fig. 2c, right) are also possible.
In principle, the detachment events in the force-extension curves could directly be assigned to the respective interactions, but the multitude of possible interactions makes this difficult. Therefore, we also evaluated the frequency of the detachment forces and their positions. Fig. 3a shows a summary of the results, which guided us in assigning the different interactions. We subdivided the distribution of the forces into three different regions (R1, R2 and R3). Forces in region 1 (R1, up to 500 pN) probably originate from low force interactions like vdW interaction (in purple, Fig. 2c). But also π–π stacking (in red, Fig. 2c), hydrogen bonds (in green, Fig. 2c) and coordination bonds (depicted in blue) will already contribute here. This is based on literature values for the single bond types: forces of vdW interactions, range between 21 to 100 pN.48–50 The forces of hydrogen bonds range from a few to several tens of pN depending on the exact geometry of the molecules and direction of force application.51,52 Altogether, 232 hydrogen bonds (PEG backbone and OH side groups) could be formed at maximum, which then would exceed 500 pN by far (see regions 2 and 3). Additionally, phenyl and catechol groups could contribute via π–π stacking where the forces are reported to be between 2 and 8 pN for DNA base pair unstacking and approx. 70 to 80 pN for strong donor–acceptor π-interactions in H2O, respectively.53,54 In our multivalent system, there are 12 possibilities for π–π stacking (5·phenyl, 5·catechol, 2·phenyl from DBCO) leading to forces in the range of several 100 pN. In case of cooperative effects (non-linear increase of force with number of bonds) a further increase of the observed force is possible.55–57 Finally, the interaction between a single catechol and TiO2 is 300 to 1000 pN, according to the literature.25,26,29 For more basic pH values (e.g., pH = 8.6), more and more quinone groups are formed,58,59 which interact with TiO2via coordination bonds with forces of 50 to 300 pN.25,26,29 Therefore, also a catechol–TiO2 bond can be found in region 1, although rarely.
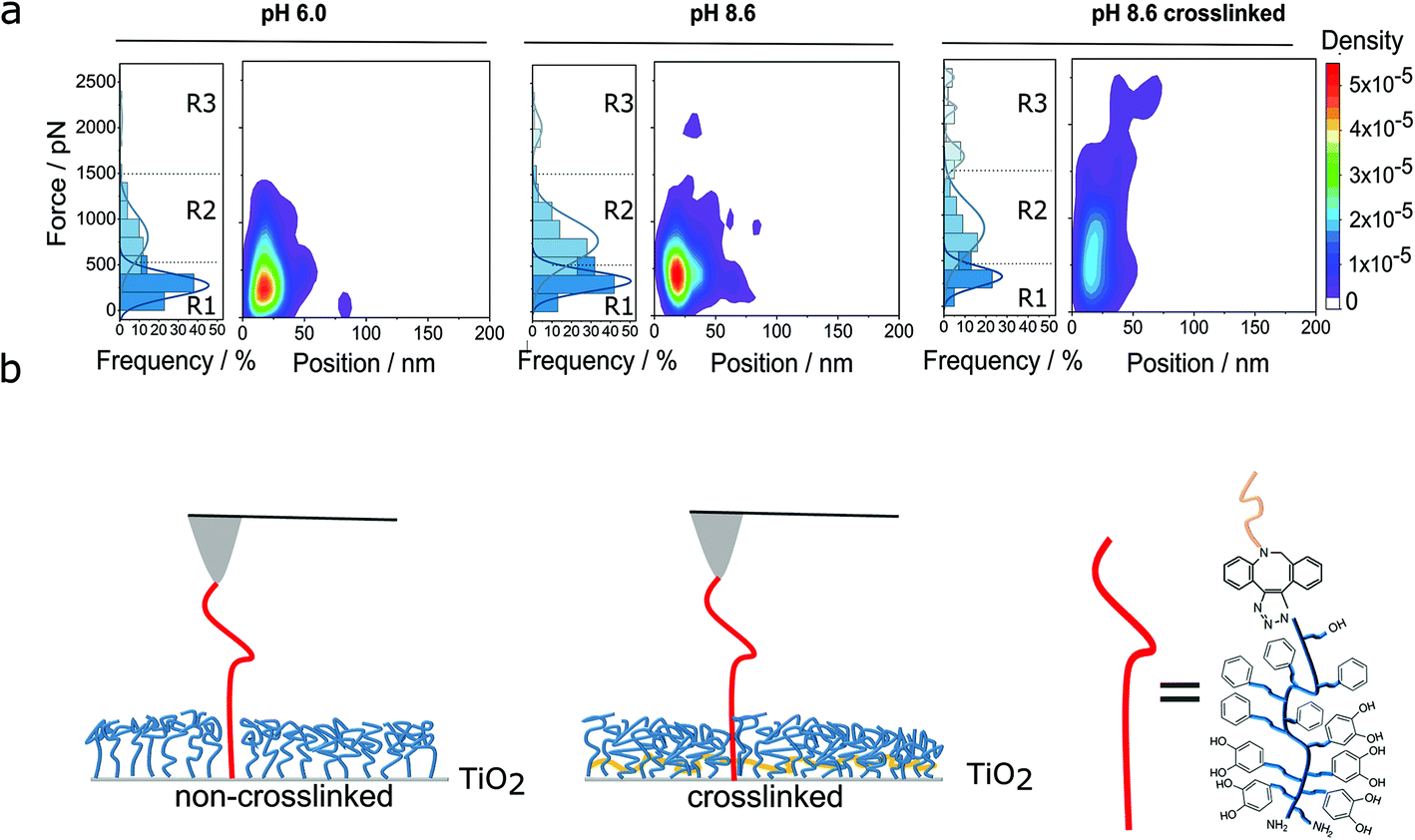 | ||
| Fig. 3 Classification of multivalent interactions. (a) Heat maps of detachment events for the PG110-b-P(Cat5-Ph5-A2) molecular force sensor on the non-crosslinked and crosslinked N3-PG110-b-P(Cat5-Ph5-A2) layers on TiO2 under the different preparation conditions of the polymer layer, as given in Fig. 2b. The frequency of the detachment events as well as the heat maps of the detachment force vs. the detachment position are shown. There the density corresponds to the probability of the detected force and position values. The detachment forces are subdivided into different regions R1, R2 and R3 (see main text for details). (b) Scheme of non-crosslinked and crosslinked (in yellow) polymer layers for incorporation of the molecular force sensor into the polymer film. | ||
In force region 2 (R2, 500 to 1500 pN), previous studies showed that multiple bonds between catechol and TiO2 are likely to be formed and show forces of above 500 pN, depending on the polymer architecture and the time in contact between the polymer and the underlying surface.25,33,35 Therefore, catechol, quinone, amine and phenyl groups might mostly contribute to the forces in that region. In other words, several catechol groups of the PG110-b-P(Cat5-Ph5-A2) molecular force sensor could interact simultaneously with the TiO2 surface and the N3-PG110-b-P(Cat5-Ph5-A2) layer on TiO2. Once several of the five catechol/quinone groups of the molecular force sensor form coordination bonds with TiO2, they could even deliver forces above 1000 pN. Thus, force region 2 most likely originates from multivalent interaction of catechols/quinones and TiO2 in addition to interactions from R1. In particular, a multitude of hydrogen bonds resulting from the PEG backbone and OH side groups likely contribute significantly to the forces observed in the region R2.
Finally, force region 3 (R3), corresponding to the third populations in the histograms, shows forces higher than 1500 pN. This force region (R3) is only significantly populated in the case of the crosslinked polymer layer. There, the detachment events could either result from the breakage of several non-covalent bonds, such as given above, in a possibly cooperative fashion, or due to covalent bonds.
Fig. 3b shows the schematic comparison between the non-crosslinked (Fig. 3b, left) and crosslinked (Fig. 3b, right) polymer layer.11 In particular, crosslinking of the N3-PG110-b-P(Cat5-Ph5-A2) layer could enhance the density of chains11 (which is consistent with the measured decrease in layer thickness, see Fig. S3†). Thus, the PG110-b-P(Cat5-Ph5-A2) molecular force sensor can interact with more neighboring chains of the polymer layer possibly leading to more multivalent interactions and more covalent bonds. The higher density of the crosslinked polymer layer might support the formation of up to five catechol/quinone coordination bonds in a cooperative way and also lead to forces higher than 1500 pN.
Furthermore, with increasing pH, quinone groups are increasingly formed both, in the molecular force sensor and the polymer layer on TiO2. Quinone groups have a higher reactivity and are prone to attacks by nucleophiles and therefore, the formation of a covalent NC (Schiff base reaction) or NH–C (Michael addition) bond with a subsequent rupture is likely.60 Finally, by contacting the PG110-b-P(Cat5-Ph5-A2) molecular force sensor and the N3-PG110-b-P(Cat5-Ph5-A2) layer, a high pressure of approx. 0.4 MPa is applied (taking an indentation force of 500 pN and a nominal tip radius of 20 nm). This is sufficient to form pressure induced covalent bonds.61
Also, the highest forces (R3) had the highest position values (Fig. 3a, right). Thus, the highest interactions resulted from the very end of the polymer, namely the quinone and amine groups which are possibly involved in the formation of covalent bonds. In theory, the maximum number of covalent bonds per PG110-b-P(Cat5-Ph5-A2) polymer amounts to 7 (2·NH2, 5·catechol, which could also form coordination bonds). This would lead to a maximum force of more than 7 nN, when just assuming additivity of the respective interaction without cooperative effects. This has not been observed here and underlines that the simultaneous formation of multiple covalent bonds between molecular force sensor and polymer layer within seconds is unlikely.
Additionally, the increase of possible bonding positions of the molecular force sensor to the polymer layer may cause a misalignment between pulling direction and bond rupture direction. As discussed by Schoeler et al.,62 this misalignment could also lead to a higher bond rupture force.
The difference between pH 8.6 and pH 8.6 crosslinked is the crosslinking of the polymer layer, resulting in a higher density. Therefore, most likely, more bonds of any kind are formed, resulting in the observed increase of molecular rupture events in R3, likely in a cooperative manner.
Discussion
Multivalent bond strength and high density as key factor for stable and durable coatings
After identifying and quantifying all the different interactions that we could possibly observe, we can draw a conclusion on the question which type of interaction is mostly responsible for the robustness of a N3-PG110-b-P(Cat5-Ph5-A2) layer on TiO2. To that aim, we determine a new quantity, namely the multivalent bond strength, which is the product of the force and its relative frequency of occurrence. Fig. 4 shows the highest level of multivalent bond strength for region R2 for every condition. Therefore, the region R2, which does not contain covalent bonds, contributes mostly to the stability and durability of the coating. The multivalent bond strength of non-covalent interactions, in particular hydrogen and coordination bonds, on average outperformed the bond strength possibly assigned to covalent bonds, because many more non-covalent interactions were formed. The dominance of these non-covalent interactions can be even enhanced, once the respective interactions act in a cooperative manner.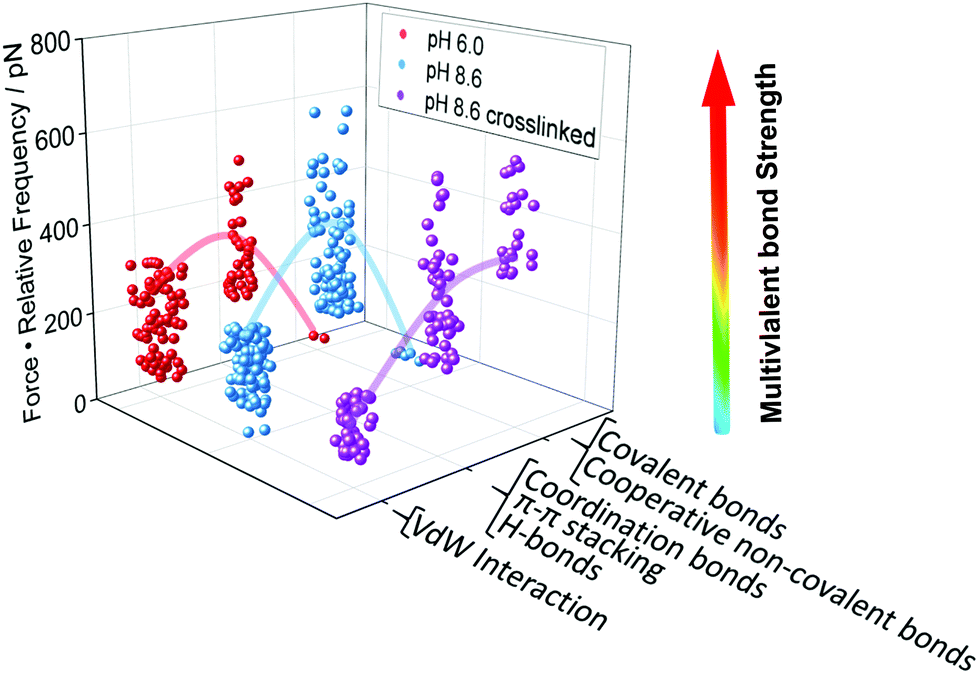 | ||
| Fig. 4 Multivalent bond strength as basis for the design of polymer coatings. We have quantified the multivalent bond strength, which is the product of the force and the relative frequency of its occurrence, for the PG110-b-P(Cat5-Ph5-A2) molecular force sensor on the N3-PG110-b-P(Cat5-Ph5-A2) layer on TiO2, as given in Fig. 3a. Bonds of intermediate strength showed the highest contribution to the overall strength of the interaction, even in the case of possible covalent bond formation. The lines are a guide to the eye. | ||
A quantification of the energy (area under the force extension curve) for multivalent bonds further supports the finding that interactions of intermediate strength (R2) are most advantageous for stable coatings. For the interaction of the single PG110-b-P(Cat5-Ph5-A2) molecule and the N3-PG110-b-P(Cat5-Ph5-A2) layer on TiO2, we found an average detachment energy of (608 ± 569) kBT at pH 6.0, (676 ± 414) kBT at pH 8.6 and (814 ± 868) kBT at pH 8.6 crosslinked. These values were higher than the energy necessary to break a covalent bond.63 In addition, the number of detachment events found in a single force-extension trace reached up to 11 upon crosslinking of the N3-PG110-b-P(Cat5-Ph5-A2) layer (Fig. S9†). Furthermore, the average energy per detachment event increases with the number of detachment events per curve n. In particular, this increase is strong for n ≥ 4 for pH 8.6 crosslinked (Fig. S10†). This result underlines the increased presence of cooperative non-covalent bonds and also covalent bonds for the crosslinked polymer layer.
Finally, the robustness of a coating comprising a complex multifunctional molecule such as N3-PG110-b-P(Cat5-Ph5-A2) towards environmental changes is a result of the different interactions present. Once the catechol interaction with TiO2 decreases with increasing pH value, the possibility of crosslinking of quinone groups to form covalent bonds with amine moieties increases, enhancing the density of the polymer layer, which in turn increases the number of possibly cooperative non-covalent bonds. Thus, in this system, the formation of one type of bond compensates the reduction of another type of bond again demonstrating the power of the interplay between various bond types in a multivalent system.
Conclusion
Multivalent bonds are important in a wide range of biological systems for, e.g., virus–cell interactions, antigen–antibody interactions or cell–cell signalling. Here, we have performed AFM-based SMFS for the molecular investigation of catecholic, multivalent and heterofunctional polymer layers in order to identify the multivalent bond strengths and their interplay for technical applications. We were able to quantify the force range of a variety of interactions. The formation of vdW interactions, hydrogen bonds, π–π stacking and coordination bonds could be assigned to detachment events with forces of several 10 to 500 pN. A simultaneous occurrence of these non-covalent interactions leads to forces of up to 1500 pN. In addition, forces greater than 1500 pN could be attributed to the formation of non-covalent bonds in a cooperative way as well as covalent bonds. The formation of these bonds, which result in highest forces, is enhanced by crosslinking of the polymer layer beforehand. Crosslinking leads to a denser layer, which favours a higher number of multivalent interactions per polymer and area.Surprisingly, the multivalent bond strength was not highest for covalent bonds but for non-covalent bonds, like hydrogen and coordination bonds, because more bonds of these types could be formed and possibly act in a cooperative manner.
Thus, the combination of non-covalent and covalent interactions, i.e., the multivalency of the presented system, explain the interaction strength obtained by SMFS experiments. Interactions of intermediate strength (non-covalent) take a prominent role among the molecular interactions, even outperforming strong interactions (covalent bonds) under many environmental conditions. In robust and durable coatings, non-covalent bonds could therefore serve as sacrificial bonds that can be reformed upon breakage enhancing the longevity of polymer-based coatings. Our findings underline the need of complex molecules for coatings for future applications under varying conditions and provide quantitative values for their design.
Author contributions
BNB, TH and RH conceived and designed the study. LY synthesized the multifunctional block copolymers. ML and WC performed the experiments and the evaluation of the data with the support of LY, KR, AH, RH, TH and BNB. All authors contributed to the writing of the manuscript and approved its final submitted version. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC-2193/1 – 390951807 (ML, TH and BNB) and the collaborative research center SFB 765 (LY and RH) and the grant HU 997/13-1 (WC and TH) for financial support.References
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- Q. Lin, D. Gourdon, C. Sun, N. Holten-Andersen, T. H. Anderson, J. H. Waite and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3782–3786 CrossRef CAS PubMed.
- J. Yang, M. A. C. Stuart and M. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- W. Zhang, R. Wang, Z. Sun, X. Zhu, Q. Zhao, T. Zhang, A. Cholewinski, F. K. Yang, B. Zhao and R. Pinnaratip, Chem. Soc. Rev., 2020, 49, 433–464 RSC.
- J. Yang, V. Saggiomo, A. H. Velders, M. A. Cohen Stuart and M. Kamperman, PLoS One, 2016, 11, e0166490 CrossRef PubMed.
- A. H. Hofman, I. A. van Hees, J. Yang and M. Kamperman, Adv. Mater., 2018, 30, 1704640 CrossRef PubMed.
- M. Shin, S.-G. Park, B.-C. Oh, K. Kim, S. Jo, M. S. Lee, S. S. Oh, S.-H. Hong, E.-C. Shin and K.-S. Kim, Nat. Mater., 2017, 16, 147–152 CrossRef CAS PubMed.
- Y. Wang, J. P. Park, S. H. Hong and H. Lee, Adv. Mater., 2016, 28, 9961–9968 CrossRef CAS PubMed.
- G. Zhu, T. Gao, S. Si, Z. Zhang, Q. Liu and G. Zhou, Appl. Surf. Sci., 2020, 507, 145080 CrossRef CAS.
- Q. Wei, T. Becherer, S. Angioletti-Uberti, J. Dzubiella, C. Wischke, A. T. Neffe, A. Lendlein, M. Ballauff and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 8004–8031 CrossRef CAS PubMed.
- L. Yu, C. Cheng, Q. Ran, C. Schlaich, P.-L. M. Noeske, W. Li, Q. Wei and R. Haag, ACS Appl. Mater. Interfaces, 2017, 9, 6624–6633 CrossRef CAS PubMed.
- L. Yu, Y. Hou, C. Cheng, C. Schlaich, P.-L. M. Noeske, Q. Wei and R. Haag, ACS Appl. Mater. Interfaces, 2017, 9, 44281–44292 CrossRef CAS PubMed.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. F. Debets, S. S. Van Berkel, J. Dommerholt, A. J. Dirks, F. P. Rutjes and F. L. Van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed.
- T. Yamada, C. G. Peng, S. Matsuda, H. Addepalli, K. N. Jayaprakash, M. R. Alam, K. Mills, M. A. Maier, K. Charisse and M. Sekine, J. Org. Chem., 2011, 76, 1198–1211 CrossRef CAS PubMed.
- G. Pourceau, A. Meyer, J.-J. Vasseur and F. Morvan, J. Org. Chem., 2009, 74, 6837–6842 CrossRef CAS PubMed.
- T. A. Schaub, J. T. Margraf, L. Zakharov, K. Reuter and R. Jasti, Angew. Chem., Int. Ed., 2018, 57, 16348–16353 CrossRef CAS PubMed.
- M. Rief, F. Oesterhelt, B. Heymann and H. E. Gaub, Science, 1997, 275, 1295–1297 CrossRef CAS PubMed.
- B. N. Balzer, M. Gallei, M. V. Hauf, M. Stallhofer, L. Wiegleb, A. Holleitner, M. Rehahn and T. Hugel, Angew. Chem., Int. Ed., 2013, 52, 6541–6544 CrossRef CAS PubMed.
- S. Lv, D. M. Dudek, Y. Cao, M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69–73 CrossRef CAS PubMed.
- W. Cai, D. Xu, L. Qian, J. Wei, C. Xiao, L. Qian, Z.-Y. Lu and S. Cui, J. Am. Chem. Soc., 2019, 141, 9500–9503 CrossRef CAS PubMed.
- M. Grandbois, M. Beyer, M. Rief, H. Clausen-Schaumann and H. E. Gaub, Science, 1999, 283, 1727–1730 CrossRef CAS PubMed.
- P. Schwaderer, E. Funk, F. Achenbach, J. Weis, C. Bräuchle and J. Michaelis, Langmuir, 2008, 24, 1343–1349 CrossRef CAS PubMed.
- S. W. Schmidt, M. K. Beyer and H. Clausen-Schaumann, J. Am. Chem. Soc., 2008, 130, 3664–3668 CrossRef CAS PubMed.
- S. Krysiak, Q. Wei, K. Rischka, A. Hartwig, R. Haag and T. Hugel, Beilstein J. Org. Chem., 2015, 11, 828–836 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- G. D. Degen, P. R. Stow, R. B. Lewis, R. C. Andresen Eguiluz, E. Valois, K. Kristiansen, A. Butler and J. N. Israelachvili, J. Am. Chem. Soc., 2019, 141, 18673–18681 CrossRef CAS PubMed.
- Y. Mu, P. Mu, X. Wu and X. Wan, Appl. Surf. Sci., 2020, 530, 146973 CrossRef CAS.
- P. Das and M. Reches, Nanoscale, 2016, 8, 15309–15316 RSC.
- L. Han, L. Gong, J. Chen, J. Zhang, L. Xiang, L. Zhang, Q. Wang, B. Yan and H. Zeng, ACS Appl. Mater. Interfaces, 2018, 10, 2166–2173 CrossRef CAS PubMed.
- P. Steinbauer, A. Rohatschek, O. Andriotis, N. Bouropoulos, R. Liska, P. J. Thurner and S. Baudis, Langmuir, 2020, 36, 13292–13300 CrossRef CAS PubMed.
- Y. Li, H. Liu, T. Wang, M. Qin, Y. Cao and W. Wang, ChemPhysChem, 2017, 18, 1466–1469 CrossRef CAS PubMed.
- Y. Li, J. Cheng, P. Delparastan, H. Wang, S. J. Sigg, K. G. DeFrates, Y. Cao and P. B. Messersmith, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- T. Utzig, P. Stock and M. Valtiner, Angew. Chem., 2016, 128, 9676–9680 CrossRef.
- A. Wislez, D. Sluysmans, N. Giamblanco, N. Willet, F. Bano, C. Van De Weerdt, C. Detrembleur and A.-S. Duwez, Biomacromolecules, 2020, 22, 183–189 CrossRef PubMed.
- J. L. Cuellar-Camacho, S. Bhatia, V. Reiter-Scherer, D. Lauster, S. Liese Jr., P. Rabe, A. Herrmann and R. Haag, J. Am. Chem. Soc., 2020, 142, 12181–12192 CrossRef CAS PubMed.
- T. A. Sulchek, R. W. Friddle, K. Langry, E. Y. Lau, H. Albrecht, T. V. Ratto, S. J. DeNardo, M. E. Colvin and A. Noy, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16638–16643 CrossRef CAS PubMed.
- V. Reiter-Scherer, J. L. Cuellar-Camacho, S. Bhatia, R. Haag, A. Herrmann, D. Lauster and J. P. Rabe, Biophys. J., 2019, 116, 1037–1048 CrossRef CAS PubMed.
- M. Delguste, C. Zeippen, B. Machiels, J. Mast, L. Gillet and D. Alsteens, Sci. Adv., 2018, 4, eaat1273 CrossRef CAS PubMed.
- Q. Hu, H. Yang, Y. Wang and S. Xu, Chem. Commun., 2016, 52, 3705–3708 RSC.
- E. Peterson, C. Joseph, H. Peterson, R. Bouwman, S. Tang, J. Cannon, K. Sinniah and S. K. Choi, Langmuir, 2018, 34, 7135–7146 CrossRef CAS PubMed.
- A. Gomez-Casado, H. H. Dam, M. D. Yilmaz, D. Florea, P. Jonkheijm and J. Huskens, J. Am. Chem. Soc., 2011, 133, 10849–10857 CrossRef CAS PubMed.
- F. Oesterhelt, M. Rief and H. Gaub, New J. Phys., 1999, 1, 6 CrossRef.
- S. Liese, M. Gensler, S. Krysiak, R. Schwarzl, A. Achazi, B. Paulus, T. Hugel Jr., P. Rabe and R. R. Netz, ACS Nano, 2017, 11, 702–712 CrossRef CAS PubMed.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 CrossRef CAS.
- S. Zou, H. Schönherr and G. J. Vancso, J. Am. Chem. Soc., 2005, 127, 11230–11231 CrossRef CAS PubMed.
- S. Kawai, T. Nishiuchi, T. Kodama, P. Spijker, R. Pawlak, T. Meier, J. Tracey, T. Kubo, E. Meyer and A. S. Foster, Sci. Adv., 2017, 3, e1603258 CrossRef PubMed.
- W. Cai, C. Xiao, L. Qian and S. Cui, Nano Res., 2019, 12, 57–61 CrossRef CAS.
- H.-J. Butt, Biophys. J., 1991, 60, 1438–1444 CrossRef CAS PubMed.
- T. Pirzer and T. Hugel, ChemPhysChem, 2009, 10, 2795–2799 CrossRef CAS PubMed.
- M. Rief, H. Clausen-Schaumann and H. E. Gaub, Nat. Struct. Biol., 1999, 6, 346–349 CrossRef CAS PubMed.
- M. Geisler, S. Xiao, E. M. Puchner, F. Gräter and T. Hugel, J. Am. Chem. Soc., 2010, 132, 17277–17281 CrossRef CAS PubMed.
- F. Kilchherr, C. Wachauf, B. Pelz, M. Rief, M. Zacharias and H. Dietz, Science, 2016, 353, 1116 CrossRef CAS PubMed.
- D. Sluysmans, L. Zhang, X. Li, A. Garci, J. F. Stoddart and A.-S. Duwez, J. Am. Chem. Soc., 2020, 142, 21153–21159 CrossRef CAS PubMed.
- A. Erbas, D. Horinek and R. R. Netz, J. Am. Chem. Soc., 2012, 134, 623–630 CrossRef CAS PubMed.
- J. Blass, M. Albrecht, B. L. Bozna, G. Wenz and R. Bennewitz, Nanoscale, 2015, 7, 7674–7681 RSC.
- X. Zhang and V. T. Moy, Biophys. Chem., 2003, 104, 271–278 CrossRef CAS PubMed.
- M. Salomäki, L. Marttila, H. Kivelä, T. Ouvinen and J. Lukkari, J. Phys. Chem. B, 2018, 122, 6314–6327 CrossRef PubMed.
- N. Umek, B. Geršak, N. Vintar, M. Šoštarič and J. Mavri, Front. Mol. Neurosci., 2018, 11, 467 CrossRef CAS PubMed.
- R. Pinnataip and B. P. Lee, ACS Omega, 2021, 6, 5113–5118 CrossRef CAS PubMed.
- T. D. Jacobs and R. W. Carpick, Nat. Nanotechnol., 2013, 8, 108–112 CrossRef CAS PubMed.
- C. Schoeler, R. C. Bernardi, K. H. Malinowska, E. Durner, W. Ott, E. A. Bayer, K. Schulten, M. A. Nash and H. E. Gaub, Nano Lett., 2015, 15, 7370–7376 CrossRef CAS PubMed.
- S. W. Benson, J. Chem. Educ., 1965, 42, 502 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr08338d |
| This journal is © The Royal Society of Chemistry 2022 |