
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic Co–Mo sulfide/carbon composites derived from polyoxometalate encapsulated polydopamine-decorated ZIF nanocubes for efficient hydrogen and oxygen evolution†
Zheng
Huang
a,
Zhuxian
Yang
a,
Quanli
Jia
b,
Nannan
Wang
c,
Yanqiu
Zhu
 a and
Yongde
Xia
*a
a and
Yongde
Xia
*a
aCollege of Engineering, Mathematics and Physical Sciences, University of Exeter, Exeter EX4 4QF, UK. E-mail: y.xia@exeter.ac.uk
bHenan Key Laboratory of High Temperature Functional Ceramics, Zhengzhou University, Zhengzhou, 450052, PR China
cGIFT (Guangxi Institute for Fullerene Technology), Key Laboratory of New Processing Technology for Nonferrous Metals and Materials, Ministry of Education, School of Resources, Environment and Materials, Guangxi University, Guangxi, 530004, China
First published on 23rd February 2022
Abstract
The increased call for carbon neutrality by 2050 makes it compelling to develop emission-free alternative energy sources. Green hydrogen produced from water electrolyzers using renewable electricity is of great importance, and the development of efficient transition-metal-based materials for hydrogen production by electrolysis is highly desirable. In this report, a new approach to produce defect-rich and ultra-fine bimetallic Co–Mo sulfides/carbon composites from polyoxometalates@ZIF-67@polydopamine nanocubes via carbonization/sulfurization, which are highly active for hydrogen and oxygen evolution reactions (HER and OER), have been successfully developed. The coating of polydopamine (PDA) on the surface of the acid-sensitive ZIF-67 cubes can prevent the over-dissociation of ZIF-67 caused by the encapsulated phosphomolybdic acid (PMA) etching through PDA chelating with the PMA molecules. Meanwhile, the partially dissociated Co2+ from ZIF-67 can be captured by the coated PDA via chelation, resulting in more evenly dispersed active sites throughout the heterogeneous composite after pyrolysis. The optimized bimetallic composite CoMoS-600 exhibits a prominent improvement in HER (with an overpotential of −0.235 V vs. RHE at a current density of 10 mA cm−2) and OER performance (with an overpotential of 0.350 V vs. RHE at a current density of 10 mA cm−2), due to the synergistic effect of ultra-fine defect-rich Co–Mo–S nanoparticle active sites and N,S-codoped porous carbons in the composites. Moreover, this synthesis approach can be readily expanded to other acidic polyoxometalates to produce HER and OER active bimetallic Co–W sulfide/carbon composites by replacing PMA with phosphotungstic acid. This new synthesis strategy to modify acid-sensitive ZIFs with selected compounds offers an alternative approach to develop novel transition metal sulfide/carbon composites for various applications.
1. Introduction
Due to its high-energy density and zero-emission, hydrogen is the most promising sustainable energy carrier for future applications. Under the increased call for carbon neutrality by 2050, green hydrogen produced from water splitting via an electrolyzer driven by renewable electricity or solar light-based photocatalysis is of great interest.1–4 The efficiency of the electrolyzer is one of the bottlenecks toward cheap and large-scale hydrogen production, which can be improved by efficient electrocatalytic materials. The current benchmark electrocatalysts for the cathodic hydrogen evolution reaction (HER) and anodic oxygen evolution reaction (OER) are Pt and IrO2/RuO2 respectively,5 but both are noble metals, which makes them undesirable for commercial applications. Therefore, developing cost-effective transition-metal-based electrocatalysts is highly desirable.Two-dimensional layered transition metal dichalcogenides (2D TMDs) have been one of the most popular material families for electrochemical energy-related applications due to their unique structures and intrinsic properties.6,7 MoS2 is one of the most well-studied materials in the 2D TMD family. Both theoretical simulations and experimental works confirm that the unsaturated chalcogens and metals on the exposed edges are the active sites for the HER.8,9 However, it is well known that MoS2 possesses poor OER activities.10 To develop bifunctional catalysts that are active towards both the HER and OER, hybridizing MoS2 with 3d transition metal (i.e. Co, Fe or Ni) sulfides has been extensively adopted.11 This multi-interface engineering strategy can also improve the conductivity and adsorption of electrochemical intermediates of the electrocatalyst.12 In particular, cobalt sulfides incorporated into MoS2 have been widely explored. For example, recently Huang et al. have synthesised mesoporous CoS/MoS2 for water splitting application,13 Ganesan et al. have created a multi-shelled CoS2–MoS2 hollow sphere for efficient water splitting,14 and Guo et al. have assembled a core–shell Co3S4@MoS2 for both the HER and OER.15
Besides introducing cobalt into MoS2, the electrochemical efficiency of the electrocatalysts can be strategically promoted by active site engineering strategies, including edge-site formation, nano-sized TMD and highly curved structures formed on the MoS2 crystals.16 Those ultra-fine open-ended nanostructures with rich defects enable the catalysts to expose a higher density of active sites for the key reactions, consequently leading to improved electrocatalytic activity.17,18 In order to maintain the fine nanostructure of electrocatalysts, to prevent heat-induced sintering19 and high surface energy induced agglomeration of the active components,20 a graphitic carbon matrix can be introduced to isolate the electrocatalytically active MoS2 nanoparticles.21,22 Meanwhile, the excellent electronic conductivity of graphitic carbon benefits fast charge transfer in the composites, resulting in enhanced activities.22,23 More interestingly, porosity can be introduced into the graphitic carbon matrix. As a result, the porous graphitic carbon matrix can not only isolate the electrocatalytically active sites to avoid agglomeration, but also promote the mass transport during the electrocatalytic reactions, thereby enabling the active sites to be readily accessible by reactants.24 Moreover, the porous carbon matrix can be doped with heteroatoms such as N and/or S, which can modulate the electronic structure of the composites and create more catalytically active sites, therefore further improving the electrocatalytic activities of the heterostructured composites.25 Built on the above considerations, the porous zeolitic imidazolate framework (i.e. ZIF-67) is an ideal precursor for the developing heteroatom functionalized carbon-rich and transition metal Co-based composites.
Previously, we reported HER and OER active WS2/Co1−xS/porous carbon nanocomposites derived from in situ synthesized polyoxometalates (POM)@ZIF-67.26 However, since ZIF-67 is unstable in an acidic environment,27 attempts to in situ synthesize its Mo-containing counterpart by a similar method were unsuccessful due to the strong acidity and the uncertainty of the molecular weight of phosphomolybdic acid hydrate (PMA, a Mo-containing POM).28 In this work, we developed an acid-resistant polydopamine (PDA) coating29 on the pre-synthesized ZIF-67 as a buffer layer to protect ZIF-67 from PMA etching, and to synthesize the PMA modified ZIF-67 composite PMA@ZIF-67@PDA. Moreover, Tris-buffer solution for dopamine polymerization is further used to avoid drastic dissociation of the ZIF when the strong acidic PMA is introduced. In this synthesis strategy, N-containing PDA can also act as another N source for N-doping besides the organic linker, 2-methylimidazole (MeIM), in ZIF-67. Since the PDA decorated on hydrophobic surfaces is less stable to acid medium than on hydrophilic surfaces,29 cetyltrimethylammonium bromide (CTAB) was added during the synthesis of ZIF-67 in order to tune the hydrophobic surface of ZIF30 into a hydrophilic surface. Meanwhile, the addition of CTAB has led to the cubical morphology of ZIF-67 crystals. In addition, PDA not only protects ZIF-67 from the strong acidity of PMA, but also provides rich functional groups that have a strong chelating capability with metal ions. Therefore PDA can chelate with free PMA molecules as well as Co2+ dissociated from the ZIF, which results in an increased amount of active sites in the final derivatives. One-step carbonization and sulfurization of the as-synthesized PMA@ZIF-67@PDA at high temperature under a hydrogen sulfide atmosphere can generate bimetallic Co–Mo sulfide supported on carbon matrixes. The optimized Co–Mo–S@N,S-doped porous carbon nanocomposites exhibit significantly enhanced HER and OER performance compared with the counterparts derived from ZIF-67 nanocubes. This synthesis approach can be further expanded to synthesize phosphotungstic acid (PTA)@ZIF-67@PDA, and the corresponding derivatives are also good electrocatalysts with enhanced performance in the HER and OER.
2. Experimental
All reagents, including cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 99%,), 2-methylimidazole (C4H6N2, 99%, abbreviated as MeIM), phosphomolybdic acid hydrate (H3PMo12O40·xH2O, abbreviated as PMA), phosphotungstic acid hydrate (H3PW12O40·xH2O, abbreviated as PTA), cetyltrimethylammonium bromide (C19H42BrN, 99+%, abbreviated as CTAB), Tris-buffer solution (0.5 M, pH = 8.5) and dopamine hydrochloride (C8H11NO2·HCl, 99%), were purchased from Fisher Scientific and used without further purification.2.1 Synthesis of ZIF-67 nanocubes
ZIF-67 nanocubes (Z67) were synthesized based on the previous report with slight modification to scale up the production.31 In a typical synthesis, 1.16 g of Co(NO3)2·6H2O and 0.02 g of CTAB were dissolved in 40 ml of distilled water while 18.16 g of MeIM was dissolved in 280 ml of distilled water separately. The mixture of Co(NO3)2·6H2O and CTAB was then rapidly poured into the MeIM solution under vigorous stirring at 500 rpm, followed by continuous stirring for 20 min at room temperature. The purple powder was collected by centrifugation at 7000 rpm for 5 min and washed twice with water and once with ethanol. The obtained sample was dried in a fume hood for 2 days and stored in a glass vial for further treatment.2.2 Synthesis of POM@ZIF-67@PDA nanocubes
To obtain PMA@ZIF-67@PDA nanocubes (Mo-Z67-D), three solutions were prepared in advance: solution A, 34 ml of 0.01 M Tris-buffer solution was prepared by adding 2 ml of 0.5 M Tris-buffer solution (pH = 8.5) into 32 ml of distilled water; solution B, 0.05 g of dopamine hydrochloride was dissolved in 17 mL of distilled water; solution C, 0.2 g PMA was dissolved in 50 ml of distilled water. Then 0.15 g of dried ZIF-67 powder was dispersed in 51 mL of distilled water under sonication for 5 min. An extra 1 mL of ethanol was added before adding water to achieve better dispersion of the hydrophobic ZIF nanocubes. The ZIF-67 dispersion solution was stirred at 500 rpm, followed by the addition of solutions A and B. The polydopamine started to decorate and coat on the surface of ZIF-67 nanocubes, changing the colour of the mixture from purple to dark purple. Then solution C was introduced, and the mixture was kept stirring for another 20 min. Finally, the resultant composite was collected via centrifugation at 7000 rpm and washed with ethanol three times. The obtained sample was dried in a fume hood for 2 days. PTA@ZIF-67@PDA nanocubes (W-Z67-D) were synthesized via a similar procedure for the synthesis of Mo-Z67-D, except the replacement of PMA solution by the same amount of PTA solution. In addition, ZIF-67@PDA nanocubes (Z67-D) were also prepared for comparison, following the same procedure for the synthesis of Mo/W-Z67-D, only replacing the PMA/PTA solution with the same amount of distilled water.2.3 Synthesis of bimetallic Co–Mo and Co–W sulfide/carbon composites
The as-synthesized Z67, as well as Mo-Z67-D and W-Z67-D, was then subjected to pyrolysis at three different temperatures (600, 800 and 1000 °C) to obtain a series of carbon-based nanocomposites. In a typical synthesis, 0.05 g of dry sample powder was loaded into a ceramic boat and pyrolyzed in a flow-through tube furnace in Ar with a flow rate of 50 mL min−1 and heating ramp rate of 10 °C min−1 or 2 °C min−1. After reaching the target pyrolysis temperature, H2S gas was introduced into the tube for 1 hour. Followed by the sulfurization/carbonization process, the sample was naturally cooled down to room temperature under an Ar flow. The obtained samples were named CoMoS-X or CoWS-X, where X stands for the pyrolysis temperature of the samples. For samples obtained at a heating rate of 10 °C min−1 an extra “-10” was added in the sample names. For example, CoMoS-600 refers to the sample derived from Mo-Z67-D at a pyrolysis temperature of 600 °C and heating ramp rate of 2 °C min−1, whereas CoWS-800-10 refers to the sample derived from W-Z67-D at a pyrolysis temperature of 800 °C and a heating ramp rate of 10 °C min−1. PMA and PTA were also heated at 800 °C under the same H2S/Ar atmosphere for comparison, and the obtained samples were designated as PMA-C and PTA-C respectively.2.4 Materials characterization
Powder X-ray diffraction (XRD) was performed under Cu-Kα radiation at 40 mA and 40 kV using a Bruker D8 advanced X-ray diffractometer. Fourier-transform infrared (FT-IR) spectra were recorded on a Shimadzu IRTracer-100 spectrometer in a wavelength range of 500–1300 cm−1. Samples were prepared by the KBr pellet method. Thermogravimetric analysis (TGA) was performed on a TA SDT Q600 instrument with a target temperature of 800 °C and a heating ramp rate of 10 °C min−1 in air with a flow rate of 100 mL min−1. N2 gas sorption analysis was carried out on a Quantachrome Autosorb iQ gas sorptometer with an outgas temperature of 150 °C for 3 hours. The Brunauer–Emmett–Teller (BET) method was used to calculate the surface area of the samples, based on the adsorption data in the partial pressure (P/P0) range of 0.05–0.2. Total pore volume was determined from the amount of adsorbed N2 at P/P0ca. 0.99. Raman spectra were acquired on a Renishaw inVia Raman microscope with an excitation laser beam of 532 nm using a 20× objective lens. Helios Nanolab 600i scanning electron microscope/focused ion beam DualBeam workstation was used to acquire scanning electron microscopy (SEM) images. Samples were splattered with gold to reduce the effect of charging. Transmission electron microscopy (TEM) images were obtained on a JEOL-2100 LaB6 transmission electron microscope. Samples were first dispersed by sonication in absolute ethanol for 10 min and then deposited on a holey carbon copper grid. X-ray photoelectron spectroscopy (XPS) was carried out on a Kratos Axis Ultra system with monochromated Al Kr X-ray source operated at 10 mA emission current and 15 kV anode potential.2.5 Electrocatalytic measurements
Electrochemistry measurements of the samples were carried out on a CHI660E electrochemical workstation with a three electrodes setup: 3 mm glassy carbon as the working electrode, Ag/AgCl as the reference electrode and carbon rod as the counter electrode. The HER was evaluated in 0.5 M H2SO4 solution, while the OER was tested in O2-saturated 1 M KOH solution. The O2-saturated electrolyte was prepared by purging O2 for 20 min before the test, and the working electrode was spun at 1600 rpm during the OER measurement. Sample ink was prepared by dispersing 3 mg of the sample in 1 mL of water/ethanol (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution and 5 μL of 5 wt% Nafion solution for 30 min in the ultrasonic bath. 5 μL of the ink was drop cast onto a 3 mm mirror-finished glassy carbon electrode and left to dry before the test.
:1, v/v) solution and 5 μL of 5 wt% Nafion solution for 30 min in the ultrasonic bath. 5 μL of the ink was drop cast onto a 3 mm mirror-finished glassy carbon electrode and left to dry before the test.
To activate the catalysts and obtain stable results, 30 cyclic voltammetry (CV) cycles were scanned before conducting linear sweep voltammetry (LSV) measurements. iR-correction was applied with a compensation level of 95% to eliminate the effect of uncompensated solution resistance. All the potentials are converted to and reported vs. the potentials of the reversible hydrogen electrode (RHE). The conversion is achieved using the Nernst equation (ERHE = EAg/AgCl + 0.059 × (pH) + 0.197, where pH is the pH value of the electrolyte solution). The reaction kinetic is interpreted by the Tafel slope, which is calculated from the Tafel equation (η = a + blog(j), where η is the overpotential, j is the exchange current density and a is the interception). By plotting the potential versus the current density in logarithm and fitting the linear portion of the plot, Tafel slope can be easily obtained. Electrochemical impedance spectroscopy (EIS) was conducted in the frequency range of 100 kHz–0.1 Hz, at the overpotential to achieve a current density of 10 mA cm−2 (η10) of the catalysts.
3. Results and discussion
3.1 Characterization of the as-synthesized PMA@ZIF-67@PDA nanocubes
As shown in Scheme 1, to obtain the precursor POM@ZIF-67@PDA nanocubes, cubical ZIF-67 was first synthesized at room temperature via the hydrothermal method in the presence of surfactant CTAB. ZIF-67@PDA nanocubes were then synthesized by depositing a thin layer of PDA on the surface of the ZIF-67 nanocubes via in situ polymerization. Finally, POM (i.e. PMA or PTA) was introduced into the ZIF-67@PDA solution. The acidic POM molecules can penetrate through the PDA coating layer outside the ZIF-67 cubes and reach the ZIF-67 cubes, resulting in partial dissociation of the ZIF-67 core. Meanwhile, the Co2+ dissociated from the ZIF-67 by acid etching, as well as some of the POM molecules were stuck in the PDA layer via functional group chelation. The obtained POM@ZIF-67@PDA nanocubes were then subjected to one-step carbonization/sulfurization at high temperatures under an H2S/Ar atmosphere to derive bimetallic Co–Mo–S/N,S-doped porous carbon composites as HER and OER catalysts.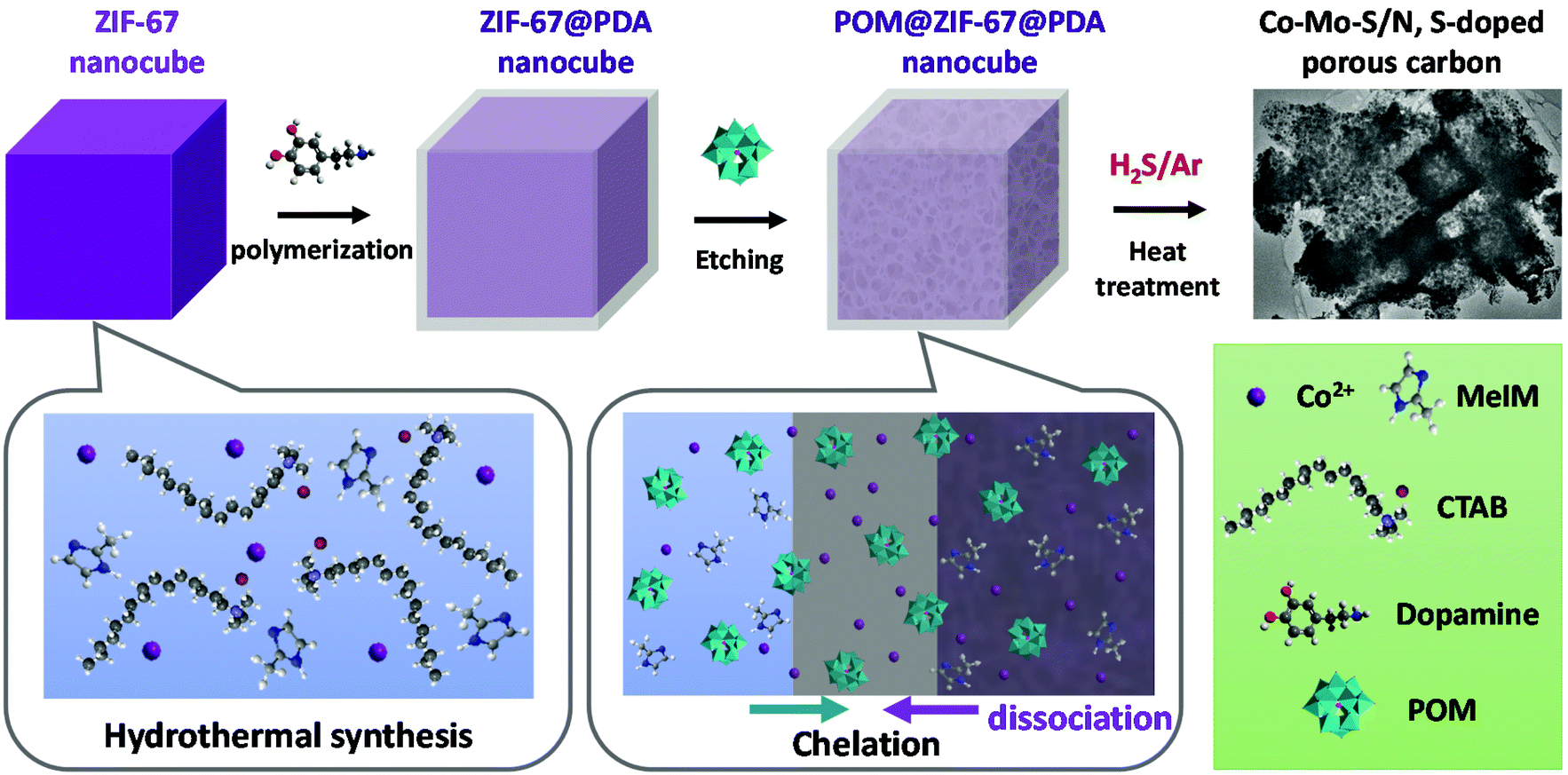 | ||
| Scheme 1 Diagram of the synthesis of the POM@ZIF-67@PDA precursor and bimetallic Co–Mo–S/C composites. | ||
The SEM images (Fig. 1) of all the as-synthesized precursors present highly uniform cubic nanoparticles with diameters of around 600 nm. The pristine ZIF-67 nanocubes (Z67) have a smooth surface compared to the other three PDA-decorated samples. With the PDA coating and POM (i.e. PMA or PTA) modification, the surfaces of the cubes have become rough and lumpy. The TEM image (inset in Fig. 1A) and elemental mapping (Fig. S1A†) show that homogeneous ZIF-67 cubes are formed with the help of surfactant CTAB. After the deposition of PDA, Z67-D is obtained with a thin layer of PDA coated on the surface of the cubes (inset in Fig. 1B). The oxygen rich outer layer is found in the corresponding elemental mapping (Fig. S1B†), which also confirms the formation of a PDA coating. With the incorporation of PMA, Mo-Z67-D shows a well-defined cubical shape with a porous interior (inset in Fig. 1C). It is believed that acidic PMA molecules can penetrate through the acid-resistant PDA coating29 and react with the acid intolerant ZIF core, leading to partial dissociation of the ZIF-67 structure, which is consistent with the previous report.31 Accumulation of Mo, O and P elements are observed on the nanocube surface from the elemental mapping image shown in Fig. S1C,† revealing that some PMA molecules have stuck in the coating layer via chelation with PDA functional groups. It is likely that the chelation mechanism of the PMA is as follows: At high pH (resulting from the Tris-buffer solution, pH = 8.5), PMA may undergo a series of hydrolysis reactions, leading to the formation of lacunary phosphomolybdates.32,33 Some of the negatively charged lacunary phosphomolybdates and the shed charged Mo species such as MoO42− and MoO22+ can then chelate with the functional groups of PDA.34,35 Moreover, small amounts of Co are found in the outer layer of cubes as well, indicating that the trace amount of free Co2+ from the dissociation of ZIF-67 have chelated with coated PDA. Elemental line scanning of the samples (Fig. S2A–C†) re-affirm the effect of PMA on the morphology of the composite and the chelation of PMA with the coated PDA.
 | ||
| Fig. 1 SEM and TEM images (insets) of (A) Z67, (B) Z67-D, (C) Mo-Z67-D and (D) W-Z67-D. | ||
All the modified samples show characteristic XRD peaks of ZIF-67 (Fig. S3A†), indicating that the basic ZIF-67 structure is retained in these modified samples irrespective of the PDA coating or the introduction of PMA. Both Z67-D and Mo-Z67-D show a slight decrease in the intensity of the characteristic diffraction peaks, possibly due to the partial dissociation of the ZIF core caused by PDA coating and PMA etching. The FT-IR spectra (Fig. S3B†) are in good agreement with the XRD results, revealing that the ZIF-67 structure is not significantly affected by the presence of CTAB, PDA coating and the PMA etching. Mo-Z67-D shows the characteristic peaks of both ZIF-67 and PMA, confirming the successful introduction of PMA. The slight peak shift and splitting of the PMA peaks of the modified structure are attributed to the change of the chemical environment21,26 and the dissociation of PMA itself due to the high pH value (pH = 8.5) during the synthesis process.32
Textural properties of the as-synthesized samples were analyzed by nitrogen gas sorption, and the obtained isotherms and pore size distributions are shown in Fig. S3C.† All samples exhibit the typical type I isotherm based on the IUPAC classification, which is associated with the microporous structure of ZIF-67.36 Small hysteresis loops are observed for Z67-D and Mo-Z67-D, possibly because small amounts of micropores is destroyed by the etching and hydrolysis of ZIF-67, which results in pores larger than 2 nm.37 The same conclusion can also be drawn from the pore size distributions of the modified samples (inset in Fig. S3C†), in which larger pores with diameters of 2–8 nm are observed. Moreover, macropores are found for the Mo-Z67-D sample at P/P0 ≥ 0.8,38 suggesting the formation of hierarchical pores in this sample, which is in good agreement with TEM results. The BET surface area (SBET) and total pore volume (Vtotal) of the as-synthesized samples are summarized in Table S1.† The surface area decreases from 1549 m2 g−1 for Z67 to 1123 and 1037 m−2 g−1 for Z67-D and Mo-Z67-D respectively. While Z67-D shows no significant change in Vtotal, Mo-Z67-D exhibits a prominent increase in Vtotal which further confirms that large amounts of pores are generated due to PMA etching. This observation is in good agreement with the above TEM results.
3.2 Characterization of PMA@ZIF-67 derived nanocomposites
Bimetallic Co–Mo–S/C nanocomposites can be readily produced via one-step sulfurization/carbonization of the as-synthesized Mo-Z67-D under an H2S/Ar atmosphere at three different temperatures with variable ramp rates. XRD patterns in Fig. 2A show that these Mo-containing composites mainly present characteristic diffraction peaks attributed to CoMoS3.13 (JCPDS #16-0439), accompanied by a trace amount of Mo4O11 (JCPDS #13-0142) and Mo2S3 (JCPDS #40-0972). In contrast, Z67 derived sample CoS-1000 only shows the characteristic peaks of Co1−xS (JCPDS #42-0826), which is in good agreement with our previous work.26,39 This confirms that CTAB surfactant, PDA coating and the shape of the ZIF-67 crystal do not affect the composition of the pyrolyzed samples. The XRD patterns of Mo-Z67-D derived samples at various temperatures, and heating rates are presented in Fig. 2B. It is found that the crystallinity of the metal sulfides in the composites improved with elevated pyrolysis temperature and a slower ramp rate. Moreover, there are more oxidized species in the samples that are pyrolyzed at 600 °C than those that are pyrolyzed at 1000 °C. The shift of the peak position and the broadening of the asymmetrical peak (see Fig. S4A†) of samples obtained at a lower pyrolysis temperature indicate that the metal sulfide in CoMoS-600 and CoMoS-600-10 possess smaller crystal sizes, possibly due to the non-uniform lattice distortions and crystallographic defects.40 Obviously, the introduction of PMA prominently changes the composition of the composites. It is worth mentioning that no diffraction peaks from oxides are observable in the Z67 nanocube derived samples, but diffraction peaks for both MoO2 and MoS2 appear in sample PMA-C (Fig. S4B†), implying that the oxygen element has likely originated from the coated PDA and PMA molecules.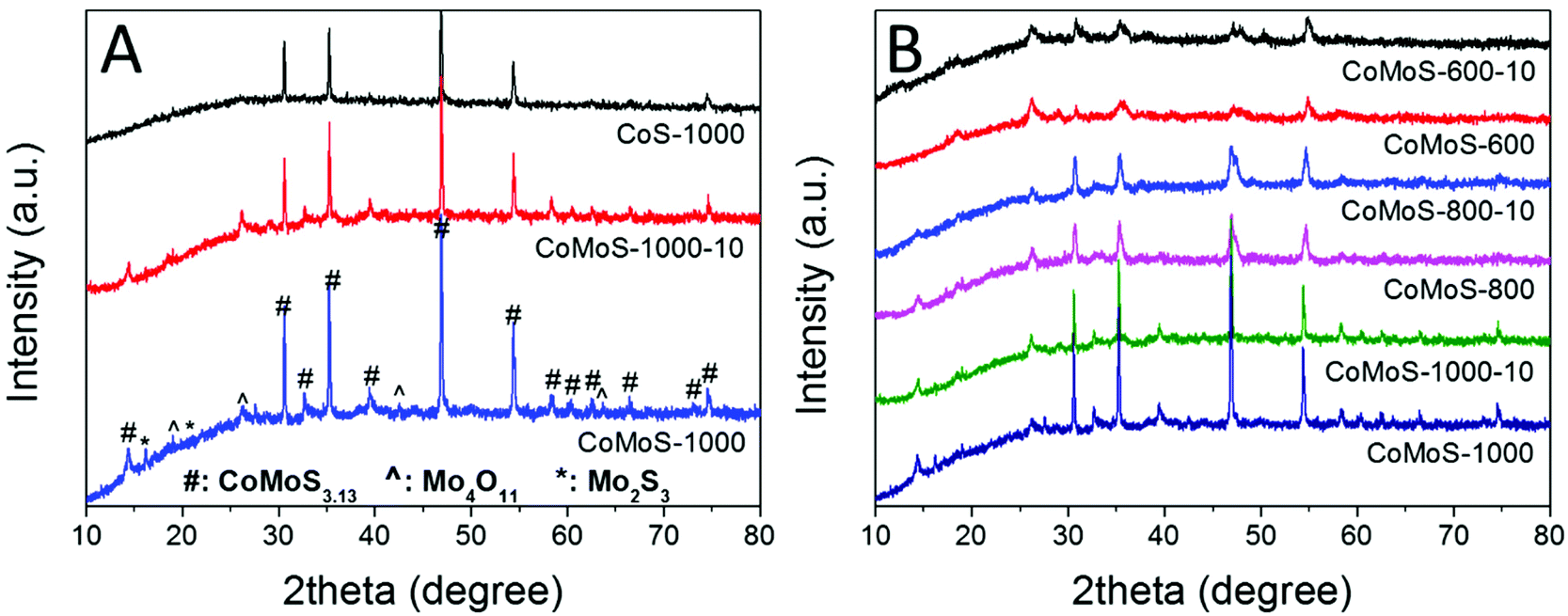 | ||
| Fig. 2 XRD patterns of (A) CoS-1000, CoMoS-1000-10 and CoMoS-1000 and (B) samples derived from Mo-Z67-D at 600, 800, and 1000 °C at a heating ramp rate of 10 and 2 °C min−1 respectively. | ||
The Raman spectra of all the bimetallic Co–Mo–S/C samples (Fig. S5A and B†) exhibit broad peaks with pronounced G band and D band. The G band at around 1593 cm−1 is associated with the in-plane stretching of the graphite lattice, whereas the D band at around 1342 cm−1 indicates the presence of a disordered band caused by the breathing mode of the graphite edges.41–43 These results indicate that the amorphous sp2-hybridized carbon with a high density of defects dominates the carbon matrix originating from the carbonization of ZIF-67. Moreover, the degree of the graphitization of carbon in the composites can be estimated by the relative intensity between the two bands (i.e. ID/IG). As shown in Table S2,† the ID/IG ratio decreases with the increase of pyrolysis temperature and slower heating ramp rate due to the formation of more graphitic carbon. It is well recognized that the degree of graphitization of carbon can affect the conductivity of the carbon matrix44 and hence alter the charge transfer kinetics of the composites, which may contribute to the better electrocatalytic performance of the carbon-based catalysts.45–47
The morphology of the composites was first investigated by SEM. As shown in Fig. 3A, CoS-600 shows a nanocube morphology similar to the parental Z67. By increasing the pyrolysis temperature to 800 °C, nanocubes with the concave surface are observed for sample CoS-800 (Fig. 3B). Further increasing the pyrolysis temperature to 1000 °C, sample CoS-1000 shows a distorted concave nanocube morphology decorated with small particles on the surface (Fig. 3C). In contrast, Mo-Z67-D derived CoMoS-600 shows mixed morphologies (Fig. 3D) of nanocubes, nanocube fragments with cracks and squared flakes. This observation further confirms that the PMA molecules not only chelate with the coated PDA, but also penetrate through the PDA coating to the inner core of the Mo-Z67-D leading to the etching of the inner core. The etching induced porous core is prone to collapse even at low pyrolysis temperature. More nanocube fragments decorated with small particles on the surface can be observed in sample CoMoS-800 (Fig. 3E). As pyrolysis temperature increased to 1000 °C, all nanocubes collapsed, and larger irregular particles appeared in sample CoMoS-1000 (Fig. 3F).
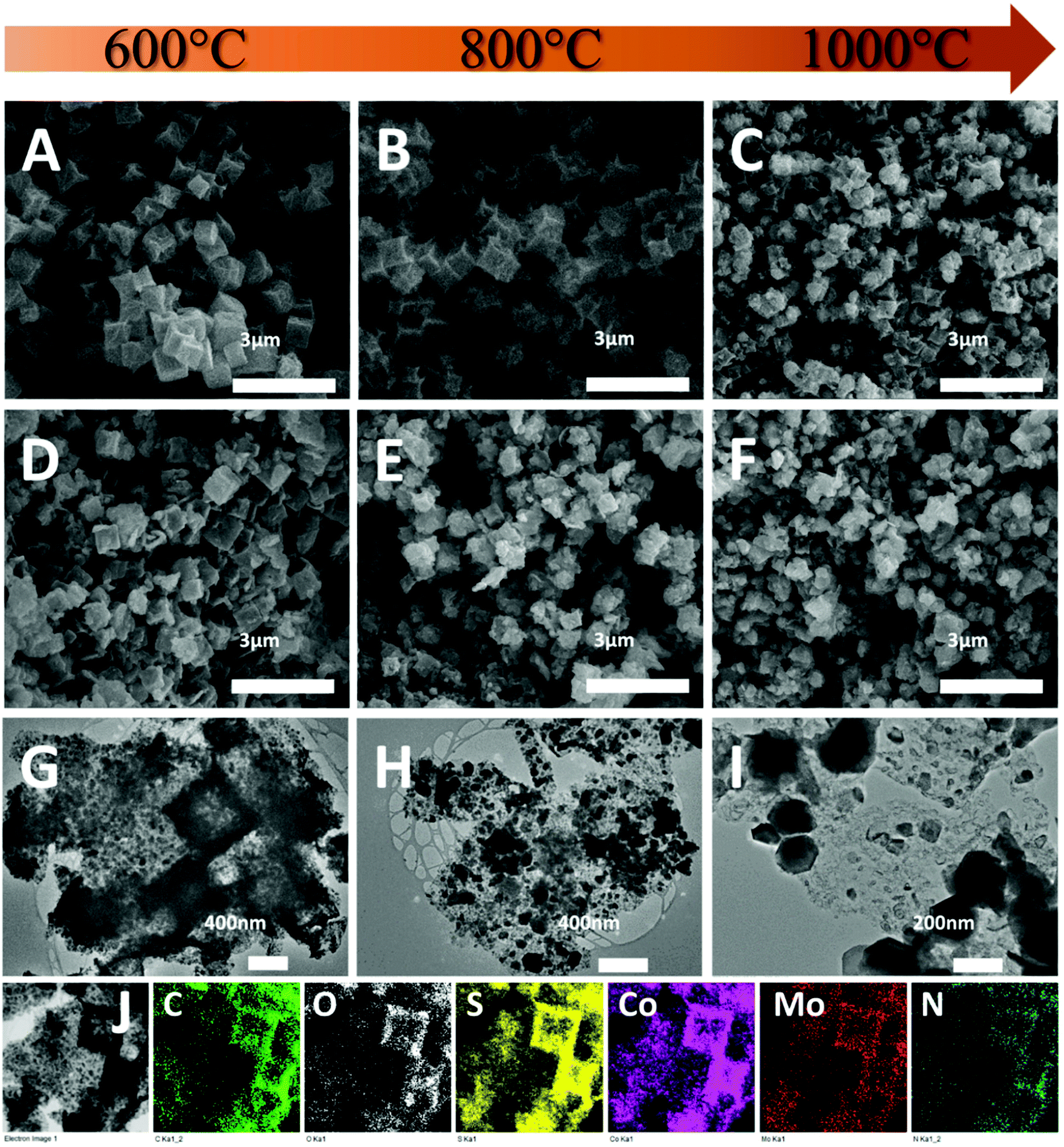 | ||
| Fig. 3 SEM images of (A) CoS-600, (B) CoS-800, (C) CoS-1000, (D) CoMoS-600, (E) CoMoS-800, and (F) CoMoS-1000; TEM images of (G) CoMoS-600, (H) CoMoS-800, and (I) CoMoS-1000 and (J) elemental mappings of CoMoS-600 obtained by TEM. | ||
The morphologies of Mo-Z67-D derived composites were further investigated by TEM. As shown in Fig. 3G, large numbers of fine nanoparticles are formed at 600 °C, which are evenly distributed in the carbon matrix. The elemental mapping images of sample CoMoS-600 in Fig. 3J show that all elements including C, O, S, Co, Mo and N are evenly distributed throughout the carbon matrix, but no P element is detected in the sample, possibly due to the high volatility of elemental phosphorus at high temperature. When the pyrolysis temperature increases to 800 °C (Fig. 3H), the small particles agglomerate to form larger particles with sizes up to 200 nm. Smaller particles are still embedded in the carbon matrix, while larger ones accumulate at the edge of the matrix. For the sample obtained at 1000 °C (Fig. 3I), severe agglomeration is observed, and most of the metal sulfide particles migrate away from the carbon matrix to form aggregated large particles with sizes up to 500 nm. The elemental mappings of both CoMoS-800 and CoMoS-1000 are shown in Fig. S6.† Similar to the sample obtained at 600 °C, elements C, O, S, Co, Mo and N are detected for both CoMoS-800 and CoMoS-1000 composites. The agglomerated metal sulfide particles are dominated by Co, Mo and S elements, which is in good agreement with the aforementioned XRD results. Small amounts of thin flakes that mainly contain Mo and S are observed for the sample CoMoS-1000, which may be the formed Mo2S3 particles.48 Based on the TEM-EDX results (see Table S3†), the Co/Mo ratio for CoMoS-600 is approximately 4.6:1, very close to 4.5:1 for the parental Mo-Z67-D.
The fine microstructures of the Mo-Z67-D derived bimetallic Co–Mo–S/C composites were studied via TEM and the images are shown in Fig. 4. Comparing the samples obtained at different temperatures (Fig. 4A–C), it is obvious that large numbers of nanoparticles with maximum diameters of 20 nm are embedded in or supported on the amorphous carbon matrix for sample CoMoS-600. A higher pyrolysis temperature results in remarkably increased particle sizes. As shown in Fig. 4A and A1–A3, sample CoMoS-600 contains large amounts of curved lattices and grain boundaries. The lattice spacing of three kinds of nanoparticles are found as labelled in the images, where a d-spacing of 0.56 nm corresponds to Mo4O11;49 0.54 nm belongs to Mo2S3,50 while 0.62 and 0.29 nm are related to CoMoS3.13.51 The interlayer distances of CoMoS3.13 vary in the range of 0.62–0.75 nm, which could be due to the formation of large amounts of lattice distortions and defects in the 1–3 layers of CoMoS3.13 crystals, which may benefit the electrochemical performance.52,53 The SAED pattern shown in the inset of Fig. 4A indicates the polycrystalline nature of sample CoMoS-600 and that CoMoS3.13 is the dominant crystalline phase of the composite. In samples CoMoS-800 and CoMoS-1000 (Fig. 4B and C), larger and thicker metal sulfide particles are clearly observed. The high-resolution TEM image in Fig. S7A† for sample CoMoS-800 shows that a more intact CoMoS3.13 crystal structure is formed with increased numbers of layers and reduced interlayer distances (ranging from 0.62–0.66 nm). Compared to the composite obtained at 600 °C, sample CoMoS-800 exhibits fewer defects in the crystals. For sample CoMoS-1000, most metal sulfides agglomerate to form large particles (Fig. S7B and C†). Moreover, compared with samples CoMoS-600/800, sample CoMoS-1000 exhibits CoMoS3.13 crystals with further increased thickness inside the carbon matrix (Fig. S7D†). In addition, the interlayer distance of CoMoS3.13 in sample CoMoS-1000 is exactly 0.62 nm, indicating the reduction of defects at high pyrolysis temperatures. Graphitic carbon is observable in CoMoS-1000, as shown in Fig. S7E.† These TEM observations are well in line with the above XRD results.
 | ||
| Fig. 4 TEM images of (A) CoMoS-600, (B) CoMoS-800 and (C) CoMoS-1000. The inset in (A) is the SAED pattern of CoMoS-600. (A1–A3) Are high resolution TEM images of CoMoS-600. | ||
Textural properties of the three Co–Mo–S/C composites obtained at different pyrolysis temperatures are summarized in Table S4.† Both the BET surface area and total pore volume are in the order of CoMoS-800 > CoMoS-1000 > CoMoS-600, with CoMoS-800 possessing the highest surface area and pore volume, which can benefit mass and ion transportation during the electrochemical reactions.
The surface chemistry of the samples was analyzed by XPS. The XPS element survey is presented in Fig. S8A,† which confirms the presence of O, C, N, S and Co in the samples derived from both the Mo-modified and PDA decorated ZIF67 and the pristine Z67, whereas the Mo element appears only in the Mo-modified samples. This is consistent with the XRD and elemental mapping results. In addition, only a trace amount of P is detected in sample CoMoS-600, which is negligible in composites CoMoS-800 and CoMoS-1000 (Fig. S8B†) due to the high volatility of elemental P during the pyrolysis. This small amount of P may lead to extra active sites in the Co–Mo–S/C composites, which contributes to the extraordinary electrochemistry performance of the composites.54,55
High-resolution XPS spectra of Mo 3d for samples CoMoS-600, CoMoS-800 and CoMoS-1000 are presented in Fig. 5A. The peak at ca. 227 eV is assigned to the S 2s peak due to the overlapping of Mo 3d and S 2s. Three doublets at around 229.4/232.5 eV, 231.8/234.9 eV and 233.4/236.5 eV are assigned to Mo 3d5/2 and 3d3/2 peaks of Mo4+, Mo5+ and Mo6+ respectively, which are close to previously reported binding energies56 with a spin–orbital splitting of 3.1 eV.57 As the pyrolysis temperature increases, the amount of Mo6+ and Mo5+ decreases while that of Mo4+ increases. The high intensity of Mo4+ peaks in all samples evidences the presence of CoMoS3.13.51,58 Interestingly, the binding energy of the Mo4+ 3d5/2 peak for sample CoMoS-600 red shifts 0.3 eV compared to that of samples CoMoS-800 and CoMoS-1000, revealing the increase in the electron density around Mo due to the electrons transferred from the less electronegative Co to Mo,58 resulting in a stronger interaction between Co and Mo in CoMoS-600, which is beneficial for its electrochemistry performance.59
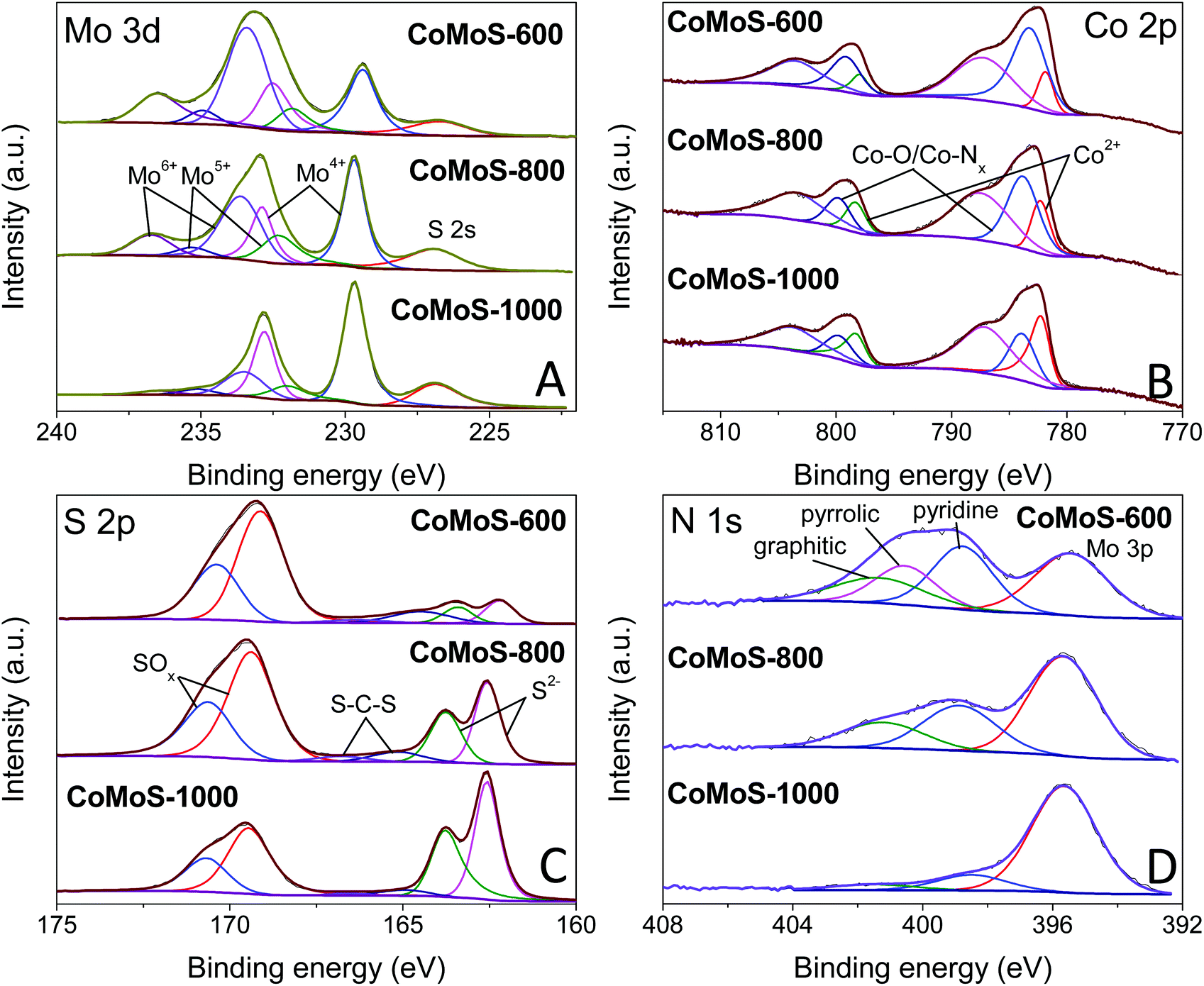 | ||
| Fig. 5 High-resolution XPS spectra of (A) Mo 3d, (B) Co 2p, (C) S 2p and (D) N 1s for CoMoS-600, CoMoS-800 and CoMoS-1000. | ||
Moreover, the XPS spectra of Co 2p for all the CoMoS samples are presented in Fig. 5B. The Co 2p peaks can be deconvoluted into three Co 2p3/2 and 2p1/2 doublets with the spin–orbit splitting of 16 eV. The peaks that appear at around 781.9/798.0 eV are ascribed to 2p3/2 and 2p1/2 of Co2+ respectively,26 whereas the doublets at around 783.2/799.1 eV represent Co with a higher oxidation state, which is reduced as pyrolysis temperature increases. The latter can be ascribed to the formed intermediate cobalt oxide species60 at low pyrolysis temperature, and Co–Nx species61 that originates from the combination of Co ions with the N element from PDA and MeIM. It is believed that OER performance can be enhanced by the presence of Co–Nx.46,62,63 The last pair of doublets at around 787.2/803.5 eV are the shake-up satellite peaks of Co 2p.
Furthermore, high-resolution XPS spectra of S 2p are presented in Fig. 5C. Peaks at around 162.2/163.4 eV correspond to the 2p3/2 and 2p1/2 of S2−,26 confirming the formation of the Co–Mo–S phase. The other set of doublets at 163.4/164.5 eV is attributed to the C–S–C bond,64,65 indicating the doping of the S element in the carbon matrix. It is obvious that an increased amount of S doping in the carbon matrix at a low pyrolysis temperature results in enhanced modification of the electronic structure and improved catalytic active sites, which are expected to improve the HER and OER performance. The doublets at 169.1/170.4 eV are attributed to oxidized S species, which is partly due to the intermediate oxide formed during the pyrolysis and partly due to the surface oxidation of the samples during the XPS inspection.21,26,66
Finally, the N 1s spectra of the composites are shown in Fig. 5D. Except for the Mo 3p peak at around 395.4 eV, the N 1s spectra can be deconvoluted into three peaks at around 398.8, 400.5 and 401.3 eV, which can be assigned to pyridinic N, pyrrolic N and graphitic N, respectively.51 These N species indicate that the carbon matrix in the Co–Mo–S/C composites has been doped with the N element, which can change the electronic structure of neighbouring carbon atoms, promoting the formation of increased catalytic active sites that can improve the HER and OER activities.25,26 Similar to S-doping, the level of N-doping in the composites decreases with increasing pyrolysis temperature.
3.3 Electrochemistry performance of Co–Mo–S/C composites
The hydrogen evolution reaction (HER) performance of all the Mo-Z67-D derived samples was evaluated in the 0.5 M H2SO4 electrolyte with a three-electrode setup. The benchmark HER catalyst 20% Pt/C, as well as Z67 derived CoS-600 is also assessed for comparison. As shown by the linear sweep voltammogram (LSV) curves in Fig. 6A, the onset potential (ηonset) and overpotential to achieve a current density of 10 mA cm−2 (η10) for sample CoMoS-600 is −0.167 and −0.235 V, respectively, which are apparently lower than those of samples CoMoS-800 (ηonset = −0.206 V, η10 = −0.274 V) and CoMoS-1000 (ηonset = −0.251 V, η10 = −0.327 V). Compared with CoS-600 (ηonset = −0.205 V, η10 = −0.359 V), CoMoS-600 shows 38 mV and 124 mV more positive onset potential and η10 respectively, which indicates that the introduction of the PDA coating and PMA molecules have introduced large numbers of active sites into the derived composites.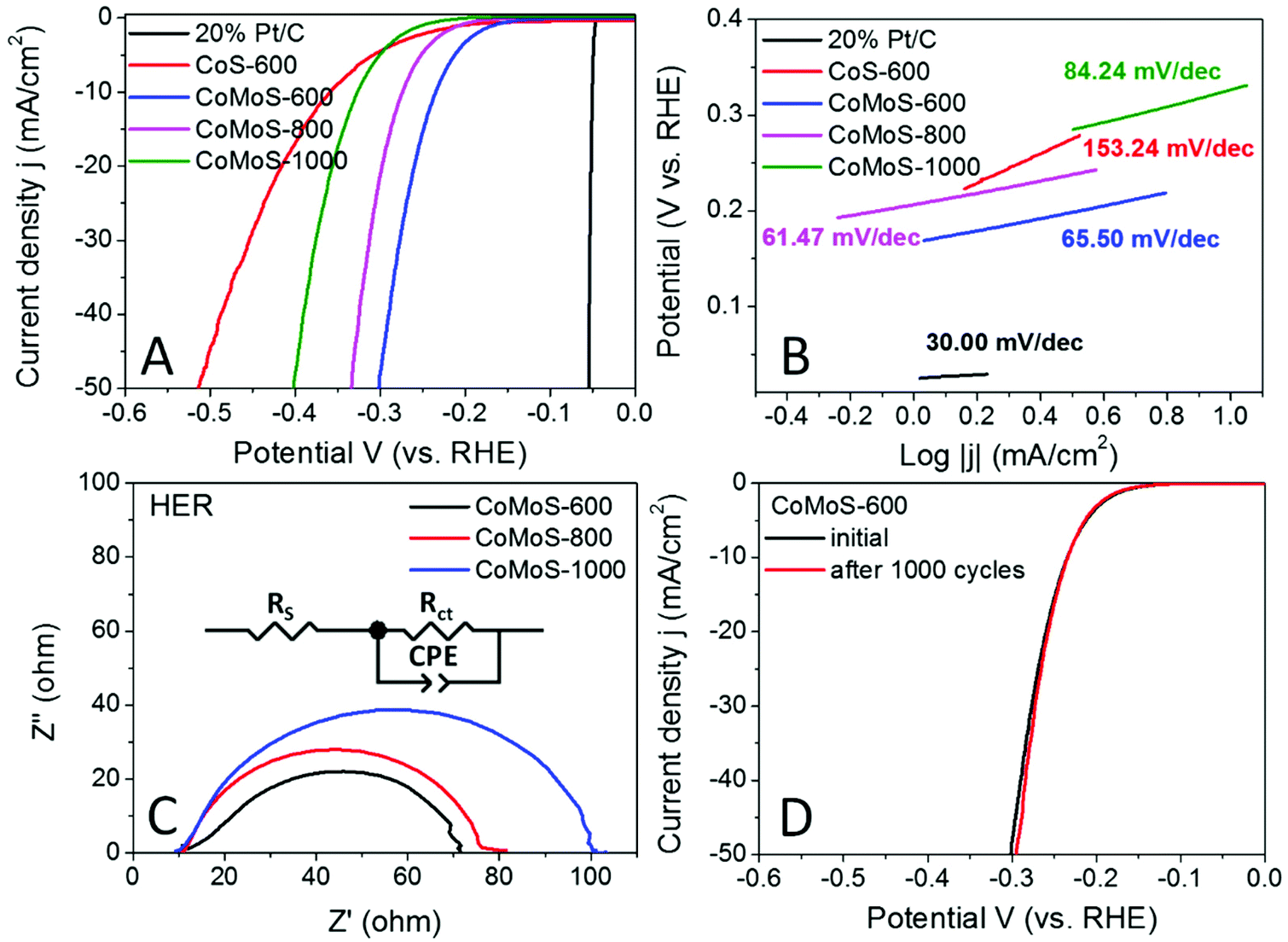 | ||
| Fig. 6 (A) HER polarization curves and (B) corresponding Tafel plots of benchmark 20% Pt/C, CoS-600, CoMoS-600, CoMoS-800 and CoMoS-1000; (C) EIS of CoMoS-600, CoMoS-800 and CoMoS-1000 and (D) polarization curves of CoMoS-600 before and after 1000 CV cycles. All polarization curves are iR corrected. | ||
Moreover, the reaction kinetics of the catalysts are estimated by linear fitting of Tafel plots (Fig. 6B) derived from the polarization curves. Generally, the smaller the Tafel slope, the faster the reaction kinetics. A dramatic improvement in the Tafel slope is observed for CoMoS-600 (65.50 mV dec−1) compared to its Mo-free counterpart (153.24 mV dec−1). Interestingly, sample CoMoS-800 shows a slightly smaller Tafel slope (61.47 mV dec−1) than sample CoMoS-600, possibly due to the much higher surface area and the better conductivity of the graphitized carbon matrix of CoMoS-800. However, overall CoMoS-600 still shows the best performance among all Mo-containing composites since the electrocatalytic activities of the bimetallic Co–Mo–S/C composites are the synergistic coordination effect of the graphitization degree of the carbon matrix, the exposed active sites originated from the ultra-small nanoparticles and the number of heteroatoms N, S and even P doping in the carbon matrix.
Electrochemical impedance spectroscopy (EIS) of the Co–Mo–S/C composites were examined to evaluate the kinetic difference of the catalysts. As shown in the Nyquist plot (Fig. 6C), all samples show depressed semi-circles, which can be modelled by an equivalent circuit consisting of series resistance (RS), a constant phase element (CPE) and a charge-transfer resistance (Rct). Sample CoMoS-600 exhibits the smallest Rct of 54.75 Ω than that of CoMoS-800 (62.96 Ω) and CoMoS-1000 (86.92 Ω), which possesses a higher degree of graphitization and a better conductive substrate. This indicates that, firstly, sample CoMoS-600 possesses better electron transferability at the interface between the electrocatalyst and the electrolyte; secondly, the number of exposed active sites from the ultra-small defect-rich nanoparticles are the dominant factor towards a faster HER process for sample CoMoS-600.
In addition, the long-term stability of sample CoMoS-600 was also evaluated by the polarization curves before and after 1000 cyclic voltammetry (CV) cycles. Fig. 6D shows that the difference between the two polarization curves is negligible, which suggests good stability for sample CoMoS-600.
The oxygen evolution reaction (OER) catalytic activities of these bimetallic Co–Mo–S/C composites were examined in an O2 saturated 1 M KOH electrolyte at 1600 rpm, and the results are presented in Fig. 7A. All the Co–Mo–S/C composites exclusively exhibit dramatically improved OER performance compared to that of CoS-600 and the benchmark IrO2 catalysts. Sample CoMoS-600 displays OER performance with ηonset = 0.248 V and η10 = 0.350 V, while sample CoMoS-800 exhibits OER performance with ηonset = 0.293 V and η10 = 0.359 V, which are significantly lower than that of sample CoMoS-1000 (ηonset = 0.340 V, η10 = 0.396 V). Compared with Mo-free sample CoS-600 (ηonset = 0.409 V, η10 = 0.481 V), sample CoMoS-600 shows a prominent improvement of 161 mV downshift in the onset potential and 131 mV decrease in η10, which indicates that the introduced Mo-containing active sites are favourable for OER performance.
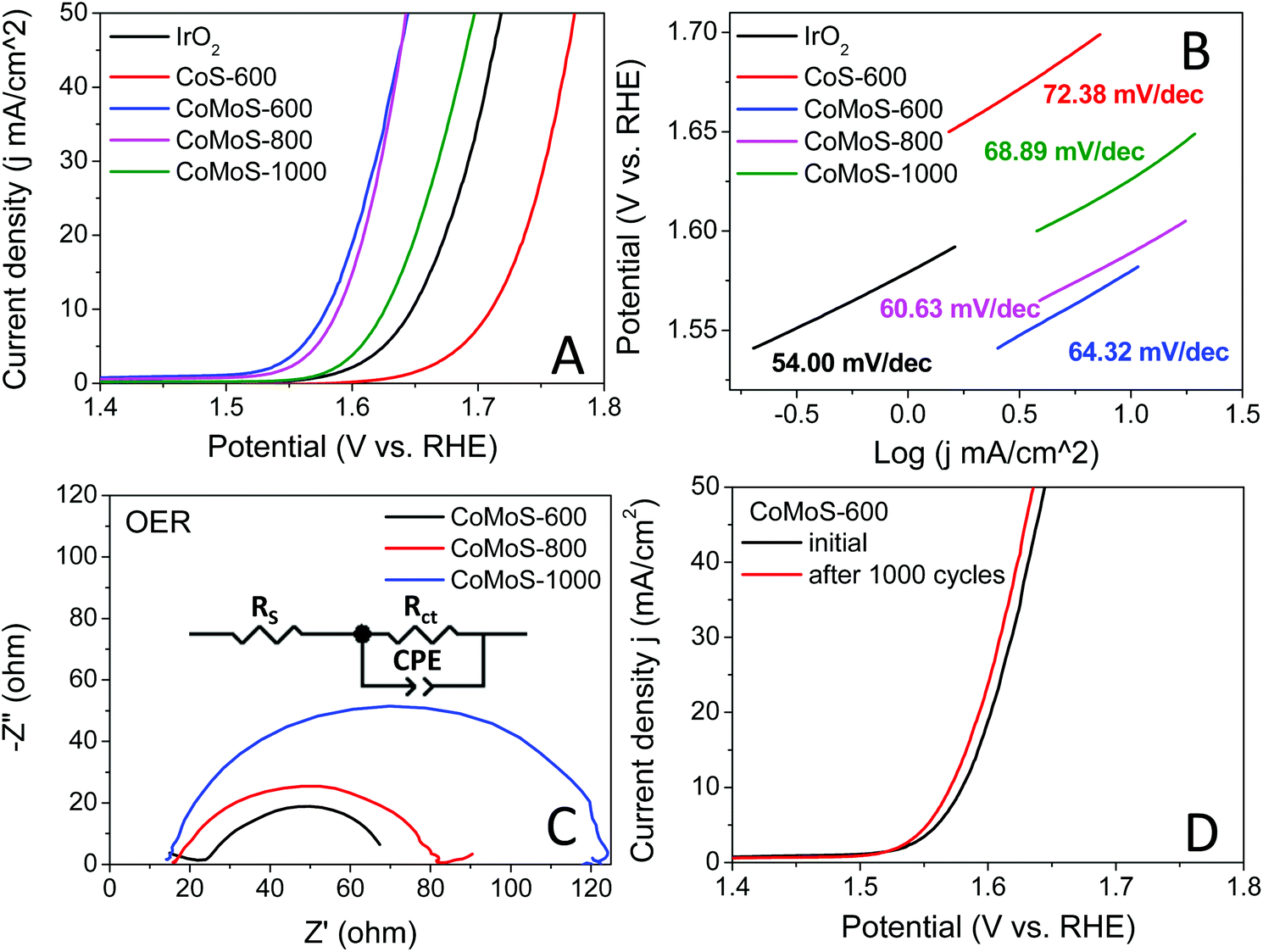 | ||
| Fig. 7 (A) OER polarization curves and (B) corresponding Tafel plots of the benchmark IrO2, CoS-600, CoMoS-600, CoMoS-800 and CoMoS-1000. (C) EIS of CoMoS-600, CoMoS-800 and CoMoS-1000. (D) Polarization curves of CoMoS-600 before and after 1000 CV cycles. All polarization curves are iR corrected. | ||
Moreover, the reaction kinetics of the catalysts towards the OER are demonstrated in Fig. 7B. Sample CoMoS-800 exhibits a slightly faster reaction kinetics than sample CoMoS-600, as indicated by a smaller Tafel slope. However, overall sample CoMoS-600 is the best performing OER catalyst among the three Mo-containing composites. EIS of these Co–Mo–S/C composites were examined and fitted with a similar equivalent circuit used for the above HER (Fig. 7C). Sample CoMoS-600 exhibits the smallest Rct of 45.1 Ω amongst the three Co–Mo–S/C samples, indicating that CoMoS-600 possesses the highest electron transferability in the OER process.
Furthermore, the stability of the best-performing sample, CoMoS-600, was also evaluated, and the polarization curves before and after 1000 CV cycles are presented in Fig. 7D. A 0.01 V decrease in η10 is observed, implying a mild improvement in OER activities after 1000 CV cycles. Moreover, XRD and SEM of the sample CoMoS-600 after 1000 CV cycles of OER measurements (see Fig. S9A and B†) still show the same characteristic peaks and morphologies without obvious difference compared with the original sample, which further confirms the stability of the catalyst.
It is worth noting that the Co–Mo–S/C composites derived from Mo-Z67-D at a heating ramp rate of 2 °C min−1 generally exhibit slightly better electrocatalytic HER and OER performance than those obtained at a heating ramp rate of 10 °C min−1 (Fig. S10A and B†). This may be due to the relatively higher level of graphitization of the carbon matrix obtained at a slower heating ramp rate (Table S2†), which provides a relatively better charge transferability during the electrocatalytic process compared to the sample obtained at a fast heating ramp rate.
Based on the electrocatalytic performance and the material characterization results, the remarkable improvement in electrocatalytic performance of the bimetallic Co–Mo–S/C composite obtained at 600 °C can be ascribed to the synergistic effect of following factors: (1) the evenly distributed ultra-small defect-rich Co–Mo–S phase nanoparticles that maximize the active-surface-area-to-volume-ratio and increase the number of exposed active sites; the abundant lattice distortions, defects in the ultra-small nanoparticles are active sites for the HER and OER;16,17 (2) heteroatoms including the S element from H2S gas, as well as the N element from the PDA coating and the organic linker in ZIF-67, even a trace amount of P derived from PMA, leads to heteroatom N and S-doped composites, which modulates the electronic structure of neighbouring C atoms and provides enhanced active sites for the HER and OER;25,26 (3) the strong interactions between Co and Mo in the Co–Mo–S phase improve the electron conductivity of the composites and is favourable for charge transfer during the electrocatalytic reaction;58,59 and (4) the porous carbon support offers high surface area and provides access routes to the reaction active sites, facilitate the easy mass transportation during the electrocatalytic reaction.22,23
3.4 Characterization and electrocatalytic performance of bimetallic Co–W–S/C composites derived from PTA@ZIF-67@PDA nanocubes
The same synthesis approach is further expanded to synthesize phosphotungstic acid (PTA)@ZIF-67@PDA to demonstrate the generalization of this material synthesis method. As shown in Fig. 1D, the TEM image of the as-obtained PTA@ZIF-67@PDA, i.e. W-Z67-D, shows a slightly different morphology from the parental Z67-D sample that the lamellar petal-like W–PDA complex with greater thickness is coated on the cubes with protruding random free-end, no hollow cavity is observed in the core. Obviously, no observable dissociation of ZIF-67 occurred. The presence of a homogeneous and intact core in W-Z67-D indicates that the addition of the same weight amount of PTA does not etch the core of the composite nor alter the structure and morphology of ZIF-67. The elemental mapping of W-Z67-D (Fig. S1D†) shows that O, P and W elements mostly accumulate on the surface of the nanocubes, whereas N and Co elements distribute evenly across the cubes, indicating that the majority of PTA molecules are chelated with the coated PDA. The corresponding elemental line scanning (Fig. S2D†) also supports this conclusion. Both XRD patterns (Fig. S3A†) and FT-IR spectra (Fig. S3B†) support that PTA has been successfully introduced into the PDA decorated ZIF-67 composites without changing the structure of ZIF-67. W-Z67-D possesses a SBET close to that of Z67-D, suggesting that PTA etching does not occur and extra hierarchical pores are not generated by the introduction of PTA.The as-obtained W-Z67-D was also subjected to pyrolysis at different temperatures under an H2S/Ar atmosphere with different heating ramp rates. The XRD patterns (Fig. S11†) suggest that the derivatives are composed of defect-rich Co1−xS and WS2 nanocrystals, and no metal oxide is observed. The Raman spectra (Fig. S5C and D†) of the W-Z67-D derivatives and their ID/IG ratios (Table S2†) are similar to the Mo-Z67-D derivatives, and slower heating ramp rate results in a higher degree of graphitization of the carbon matrix. Compared with Mo-Z67-D derived composites, W-Z67-D derived Co–W–S/C composites show a similar agglomeration and crystallization trend as the pyrolysis temperature increases. The TEM images and elemental mappings of the Co–W–S/C composites are presented in Fig. S12 and S13† respectively. TEM images suggest the presence of Co1−xS crystals and WS2 flakes in the Co–W–S/C composites, and elemental mappings confirm the homogeneous distribution of all elements.
Unsurprisingly, Co–W–S/C composites also show enhanced electrocatalytic activities towards both HER and OER but with completely different performance orders for the samples (Fig. S14 and Table S5†). Composite CoWS-800 exhibits the best HER activity, while composite CoWS-1000 displays the highest OER performance amongst the samples obtained at different pyrolysis temperatures, which is consistent with the performance of one-pot synthesized POM@ZIF-67 derived WS2/Co1−xS/N,S co-doped porous carbon reported by our group previously.26 It is believed that the different orders of performance between PMA and PTA@ZIF-67@PDA derivatives are due to (i) different types of active sites in the composites and (ii) different morphologies of the composites derived at the same temperature. However, the effect of the heating ramp rate on the electrocatalytic performance of the Mo-Z67-D derived samples and W-Z67-D derivatives are the same. The sample obtained at a slow heating ramp rate shows a slightly better electrocatalytic performance (Fig. S10C and D†). It is again likely due to the relatively higher degree of graphitization of the carbon matrix obtained at a slower heating ramp rate (Table S2†) that provides a relatively better charge transferability during the electrochemical reaction process.
4. Conclusions
In summary, we have successfully developed a approach to synthesize both HER and OER active, defect-rich ultra-fine bimetallic Co–Mo sulfide nanoparticles supported on N,S-doped porous carbon composites from PMA@ZIF-67@PDA nanocubes via carbonization/sulfurization. PDA decorated on ZIF-67 cubes can prevent the over-dissociation of ZIF-67 from PMA etching by chelating with the PMA molecules, and acts as an additional N source for N-doping. Moreover, the partially dissociated Co2+ from ZIF-67 can be captured by the coated PDA via chelation, resulting in more evenly dispersed active sites throughout the heterogeneous composite after pyrolysis. The resulting bimetallic CoMoS-600 composite exhibits remarkable improvement in HER (with an overpotential of −0.235 V vs. RHE at a current density of 10 mA cm−2 and a Tafel slope of 65.50 mV dec−1) and OER performance (with an overpotential of 0.350 V vs. RHE at a current density of 10 mA cm−2 and a Tafel slope of 64.32 mV dec−1). The outstanding performance can be attributed to the synergistic effect of ultra-fine and defect-rich Co–Mo–S nanoparticle exposed active sites and N,S-doped porous carbons in the composites. Moreover, HER and OER active W-containing bimetallic Co–W–S/C composites can be readily obtained by replacing PMA with PTA, proving the universality of this generalized synthesis approach. This work offers a new synthesis strategy to modify acid-sensitive ZIFs with acidic compounds, providing an alternative approach for developing other advanced metal sulfides/porous carbon functional materials for energy and environmental applications.Author contributions
Zheng Huang: investigation, methodology, data curation, and writing – original draft. Zhuxian Yang: investigation, data curation, validation, and writing – review & editing. Quanli Jia: investigation, validation, and data curation. Nannan Wang: investigation, validation, and data curation. Yanqiu Zhu: supervision, and writing – review & editing. Yongde Xia: supervision, conceptualization, methodology, validation, and writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the EPSRC Doctoral Training Partnership at the University of Exeter, Leverhulme Trust (RPG-2018-320) and the Royal Society (IEC\NSFC\201121).References
- G. Kakoulaki, I. Kougias, N. Taylor, F. Dolci, J. Moya and A. Jäger-Waldau, Energy Convers. Manage., 2021, 228, 113649 CrossRef CAS.
- M. Z. Hussain, B. van der Linden, Z. Yang, Q. Jia, H. Chang, R. A. Fischer, F. Kapteijn, Y. Zhu and Y. Xia, J. Mater. Chem. A, 2021, 9, 4103–4116 RSC.
- M. Z. Hussain, Z. Yang, Z. Huang, Q. Jia, Y. Zhu and Y. Xia, Adv. Sci., 2021, 8, 2100625 CrossRef CAS PubMed.
- M. Z. Hussain, Z. Yang, B. van der Linden, Z. Huang, Q. Jia, E. Cerrato, R. A. Fischer, F. Kapteijn, Y. Zhu and Y. Xia, J. Energy Chem., 2021, 57, 485–495 CrossRef.
- N. K. Oh, J. Seo, S. Lee, H.-J. Kim, U. Kim, J. Lee, Y.-K. Han and H. Park, Nat. Commun., 2021, 12, 4606 CrossRef CAS PubMed.
- X. Chia, A. Y. S. Eng, A. Ambrosi, S. M. Tan and M. Pumera, Chem. Rev., 2015, 115, 11941–11966 CrossRef CAS PubMed.
- X. Wu, H. Zhang, J. Zhang and X. W. Lou, Adv. Mater., 2021, 33, 2008376 CrossRef CAS PubMed.
- P. Zhuang, Y. Sun, P. Dong, W. Smith, Z. Sun, Y. Ge, Y. Pei, Z. Cao, P. M. Ajayan, J. Shen and M. Ye, Adv. Funct. Mater., 2019, 29, 1901290 CrossRef.
- X. Zhang, F. Zhou, S. Zhang, Y. Liang and R. Wang, Adv. Sci., 2019, 6, 1900090 CrossRef PubMed.
- E. German and R. Gebauer, Appl. Surf. Sci., 2020, 528, 146591 CrossRef CAS.
- Y. Li, Z. Yin, M. Cui, X. Liu, J. Xiong, S. Chen and T. Ma, J. Mater. Chem. A, 2021, 9, 2070–2092 RSC.
- H. Wang, W. Zhang, X. Zhang, S. Hu, Z. Zhang, W. Zhou and H. Liu, Nano Res., 2021, 14, 4857–4864 CrossRef CAS.
- W.-H. Huang, X.-M. Li, X.-F. Yang, H.-B. Zhang, F. Wang and J. Zhang, Chem. Commun., 2021, 57, 4847–4850 RSC.
- V. Ganesan and J. Kim, Int. J. Hydrogen Energy, 2020, 45, 13290–13299 CrossRef CAS.
- Y. Guo, J. Tang, Z. Wang, Y.-M. Kang, Y. Bando and Y. Yamauchi, Nano Energy, 2018, 47, 494–502 CrossRef CAS.
- C.-A. Chen, C.-L. Lee, P.-K. Yang, D.-S. Tsai and C.-P. Lee, Catalysts, 2021, 11, 151 CrossRef CAS.
- R. Abinaya, J. Archana, S. Harish, M. Navaneethan, S. Ponnusamy, C. Muthamizhchelvan, M. Shimomura and Y. Hayakawa, RSC Adv., 2018, 8, 26664–26675 RSC.
- H. Wang, Z. Xu, Z. Zhang, S. Hu, M. Ma, Z. Zhang, W. Zhou and H. Liu, Nanoscale, 2020, 12, 22541–22550 RSC.
- L. Wang, L. Wang, X. Meng and F.-S. Xiao, Adv. Mater., 2019, 31, 1901905 CrossRef CAS PubMed.
- Y. Jazaa, T. Lan, S. Padalkar and S. Sundararajan, Lubricants, 2018, 6, 106 CrossRef.
- Z. Huang, Z. Yang, M. Z. Hussain, Q. Jia, Y. Zhu and Y. Xia, J. Mater. Sci. Technol., 2021, 84, 76–85 CrossRef.
- X. Zheng, J. Xu, K. Yan, H. Wang, Z. Wang and S. Yang, Chem. Mater., 2014, 26, 2344–2353 CrossRef CAS.
- H. Li, K. Yu, C. Li, Z. Tang, B. Guo, X. Lei, H. Fu and Z. Zhu, Sci. Rep., 2015, 5, 18730 CrossRef CAS PubMed.
- T. Rodenas, S. Beeg, I. Spanos, S. Neugebauer, F. Girgsdies, G. Algara-Siller, P. P. M. Schleker, P. Jakes, N. Pfänder, M. Willinger, M. Greiner, G. Prieto, R. Schlögl and S. Heumann, Adv. Energy Mater., 2018, 8, 1802404 CrossRef.
- X. Yu, M. Zhang, J. Chen, Y. Li and G. Shi, Adv. Energy Mater., 2016, 6, 1501492 CrossRef.
- Z. Huang, Z. Yang, M. Z. Hussain, B. Chen, Q. Jia, Y. Zhu and Y. Xia, Electrochim. Acta, 2020, 330, 135335 CrossRef CAS.
- S. Gao, J. Hou, Z. Deng, T. Wang, S. Beyer, A. G. Buzanich, J. J. Richardson, A. Rawal, R. Seidel, M. Y. Zulkifli, W. Li, T. D. Bennett, A. K. Cheetham, K. Liang and V. Chen, Chem, 2019, 5, 1597–1608 CAS.
- J. Javidi, M. Esmaeilpour, Z. Rahiminezhad and F. N. Dodeji, J. Cluster Sci., 2014, 25, 1511–1524 CrossRef CAS PubMed.
- H. Wei, J. Ren, B. Han, L. Xu, L. Han and L. Jia, Colloids Surf., B, 2013, 110, 22–28 CrossRef CAS PubMed.
- P. Zhou, J. Cheng, Y. Yan, S. Xu and C. Zhou, Sep. Purif. Technol., 2021, 272, 118871 CrossRef CAS.
- H. B. Wu, B. Y. Guan, P. He and X.-Y. Yu, J. Mater. Chem. A, 2018, 6, 19338–19341 RSC.
- Z. Zhu, R. Tain and C. Rhodes, Can. J. Chem., 2003, 81, 1044–1050 CrossRef CAS.
- A. Patel, N. Narkhede and A. Patel, Catal. Surv. Asia, 2019, 23, 257–264 CrossRef CAS.
- K. M. Im, T.-W. Kim and J.-R. Jeon, ACS Biomater. Sci. Eng., 2017, 3, 628–636 CrossRef CAS PubMed.
- L. Sun, C. Wang, X. Wang and L. Wang, Small, 2018, 14, 1800090 CrossRef PubMed.
- J. Qin, S. Wang and X. Wang, Appl. Catal., B, 2017, 209, 476–482 CrossRef CAS.
- W. Kong, J. Li, Y. Chen, Y. Ren, Y. Guo, S. Niu and Y. Yang, Appl. Surf. Sci., 2018, 437, 161–168 CrossRef CAS.
- M. Zhang, C. Wang, R. Luo, W. Zhang, S. Chen, X. Yan, J. Qi, X. Sun, L. Wang and J. Li, J. Mater. Chem. A, 2019, 7, 5173–5178 RSC.
- B. Chen, R. Li, G. Ma, X. Gou, Y. Zhu and Y. Xia, Nanoscale, 2015, 7, 20674–20684 RSC.
- P. Muhammed Shafi and A. Chandra Bose, AIP Adv., 2015, 5, 057137 CrossRef.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- D. B. Schuepfer, F. Badaczewski, J. M. Guerra-Castro, D. M. Hofmann, C. Heiliger, B. Smarsly and P. J. Klar, Carbon, 2020, 161, 359–372 CrossRef CAS.
- L. Deng, Z. Yang, R. Li, B. Chen, Q. Jia, Y. Zhu and Y. Xia, Front. Chem. Sci. Eng., 2021, 15, 1487–1499 CrossRef CAS.
- D. H. Kweon, M. S. Okyay, S.-J. Kim, J.-P. Jeon, H.-J. Noh, N. Park, J. Mahmood and J.-B. Baek, Nat. Commun., 2020, 11, 1278 CrossRef CAS PubMed.
- H. Han, Z. Bai, T. Zhang, X. Wang, X. Yang, X. Ma, Y. Zhang, L. Yang and J. Lu, Nano Energy, 2019, 56, 724–732 CrossRef CAS.
- F. Yang, P. Zhao, X. Hua, W. Luo, G. Cheng, W. Xing and S. Chen, J. Mater. Chem. A, 2016, 4, 16057–16063 RSC.
- Y. Zhong, Y. Zhang, G. Zhang, R. Li and X. Sun, Appl. Surf. Sci., 2012, 263, 410–415 CrossRef CAS.
- P. Mallet, H. Guyot, J. Y. Veuillen and N. Motta, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 165428 CrossRef.
- Z. Chen, A. Liao, Z. Guo, F. Yu, T. Mei, Z. Zhang, M. S. Irshad, C. Liu, L. Yu and X. Wang, Electrochim. Acta, 2020, 353, 136561 CrossRef CAS.
- S. H. Yang, S.-K. Park, J. K. Kim and Y. C. Kang, J. Mater. Chem. A, 2019, 7, 13751–13761 RSC.
- R. Wei, K. Zhang, P. Zhao, Y. An, C. Tang, C. Chen, X. Li, X. Ma, Y. Ma and X. Hao, Appl. Surf. Sci., 2021, 549, 149327 CrossRef CAS.
- J. Xie, X. Yang and Y. Xie, Nanoscale, 2020, 12, 4283–4294 RSC.
- W. Hong, M. Kitta and Q. Xu, Small Methods, 2018, 2, 1800214 CrossRef.
- F. Bu, W. Chen, M. F. Aly Aboud, I. Shakir, J. Gu and Y. Xu, J. Mater. Chem. A, 2019, 7, 14526–14535 RSC.
- Y. Sun, J. Wang, B. Zhao, R. Cai, R. Ran and Z. Shao, J. Mater. Chem. A, 2013, 1, 4736–4746 RSC.
- J.-W. Shi, Y. Zou, D. Ma, Z. Fan, L. Cheng, D. Sun, Z. Wang, C. Niu and L. Wang, Nanoscale, 2018, 10, 9292–9303 RSC.
- C. Ray, S. C. Lee, K. V. Sankar, B. Jin, J. Lee, J. H. Park and S. C. Jun, ACS Appl. Mater. Interfaces, 2017, 9, 37739–37749 CrossRef CAS PubMed.
- X. Ma, W. Chen, Q. Li, L. Xue and C. Peng, Energy Environ. Mater., 2021, 4, 658–663 CrossRef CAS.
- J. Zhang, D. Meng, H. Huang, H. Cai, Q. Huang, J. Wang, Y. Yang, X. Zhai, Z. Fu and Y. Lu, AIP Adv., 2018, 8, 025322 CrossRef.
- X. Cui, W. Li, K. Junge, Z. Fei, M. Beller and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 7501–7507 CrossRef CAS PubMed.
- A. Macedo Andrade, Z. Liu, S. Grewal, A. J. Nelson, Z. Nasef, G. Diaz and M. H. Lee, Dalton Trans., 2021, 50, 5473–5482 RSC.
- B. Chen, X. He, F. Yin, H. Wang, D.-J. Liu, R. Shi, J. Chen and H. Yin, Adv. Funct. Mater., 2017, 27, 1700795 CrossRef.
- J. Cui, Y. Qiu, H. Zhang, Z. Yao, W. Zhao, Y. Liu and J. Sun, Solid State Ionics, 2021, 361, 115548 CrossRef CAS.
- A. Abdul Razzaq, Y. Yao, R. Shah, P. Qi, L. Miao, M. Chen, X. Zhao, Y. Peng and Z. Deng, Energy Storage Mater., 2019, 16, 194–202 CrossRef.
- G. Li, J. Sun, W. Hou, S. Jiang, Y. Huang and J. Geng, Nat. Commun., 2016, 7, 10601 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr07913a |
| This journal is © The Royal Society of Chemistry 2022 |