
Tuning the magnetic properties of NiPS3 through organic-ion intercalation†
Daniel
Tezze
a,
José M.
Pereira
 a,
Yaiza
Asensio
a,
Mihail
Ipatov
b,
Francesco
Calavalle
a,
Felix
Casanova
ac,
Alexander M.
Bittner
ac,
Maider
Ormaza
*d,
Beatriz
Martín-García
*a,
Luis E.
Hueso
ac and
Marco
Gobbi
*ace
a,
Yaiza
Asensio
a,
Mihail
Ipatov
b,
Francesco
Calavalle
a,
Felix
Casanova
ac,
Alexander M.
Bittner
ac,
Maider
Ormaza
*d,
Beatriz
Martín-García
*a,
Luis E.
Hueso
ac and
Marco
Gobbi
*ace
aCIC nanoGUNE BRTA, 20018 San Sebastian, Spain. E-mail: m.gobbi@nanogune.eu; b.martingarcia@nanogune.eu
bSGIker Medidas Magnéticas Gipuzkoa, UPV/EHU, 20018 San Sebastian, Spain
cIKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Spain
dDepartamento de Polímeros y Materiales Avanzados: Física, Química y Tecnología, Universidad del País Vasco, Paseo Manuel de Lardizabal 3, San Sebastián 20018, Spain. E-mail: maider.ormaza@ehu.es
eMaterials Physics Center CSIC-UPV/EHU, 20018 Donostia-San Sebastian, Spain
First published on 30th December 2021
Abstract
Atomically thin van der Waals magnetic crystals are characterized by tunable magnetic properties related to their low dimensionality. While electrostatic gating has been used to tailor their magnetic response, chemical approaches like intercalation remain largely unexplored. Here, we demonstrate the manipulation of the magnetism in the van der Waals antiferromagnet NiPS3 through the intercalation of different organic cations, inserted using an engineered two-step process. First, the electrochemical intercalation of tetrabutylammonium cations (TBA+) results in a ferrimagnetic hybrid compound displaying a transition temperature of 78 K, and characterized by a hysteretic behavior with finite remanence and coercivity. Then, TBA+ cations are replaced by cobaltocenium via an ion-exchange process, yielding a ferrimagnetic phase with higher transition temperature (98 K) and higher remanent magnetization. Importantly, we demonstrate that the intercalation and cation exchange processes can be carried out in bulk crystals and few-layer flakes, opening the way to the integration of intercalated magnetic materials in devices.
 Marco Gobbi | Marco Gobbi is Research Fellow at CIC nanoGUNE, where he works as “La Caixa” Junior Leader since September 2019. Additionally, he holds an Ikerbasque Fellowship at CIC nanoGUNE and at the Materials Physics Center, in San Sebastian. Before taking up this tenure track position, Marco Gobbi was a Marie Curie Fellow at the Materials Physics Center, which he joined in 2017 after a 4-year postdoctoral contract at the Institute of Supramolecular Science and Engineering in Strasbourg (France). His research interests focus on the engineering of the physical properties of 2D Materials using molecular functionalization for electronic and spintronic applications. |
Introduction
The discovery of ferromagnetic order at the single-layer limit in CrI3 and CrGeTe3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1,2 has motivated a sudden interest towards layered magnetic materials (LMMs),3,4 in which a van der Waals (vdW) gap separates sheets of covalently bound atoms. One of the distinctive features that differentiate ultrathin LMMs from conventional bulk magnetic compounds is the tunability of their magnetic properties, which stems from their reduced dimensionality and electronic properties. While several recent studies show how the magnetic response of LMMs can be significantly altered through electrostatic gating,5–9 the impact of chemical engineering approaches10–13 remains much less explored.
1,2 has motivated a sudden interest towards layered magnetic materials (LMMs),3,4 in which a van der Waals (vdW) gap separates sheets of covalently bound atoms. One of the distinctive features that differentiate ultrathin LMMs from conventional bulk magnetic compounds is the tunability of their magnetic properties, which stems from their reduced dimensionality and electronic properties. While several recent studies show how the magnetic response of LMMs can be significantly altered through electrostatic gating,5–9 the impact of chemical engineering approaches10–13 remains much less explored.
The process of inserting guest species into the vdW gaps of a host layered material,14–17 known as intercalation, is a powerful route to manipulate the physical properties of layered materials. The occupation of the vdW gaps typically leads to an expansion of the interlayer distance and a large charge transfer.18–26 While alkali metals are often used as guest species,14–17 the chemical flexibility of organic cations offers more degrees of freedom for modulating the physical properties of the host material, as an accurate molecular selection enables a fine tuning of doping levels27 and interlayer distance.20 Intercalation has been studied for several decades28,29 to induce superconductivity in layered materials,18,20–26,28–30 and it is subject of a recent renewed interest due to the possibility of tuning the properties of atomically thin micrometric flakes27,31 and tailoring the magnetism of LMMs.26,32
Among LMMs, NiPS3 is a particularly intriguing compound, since it can be exfoliated to the single-layer limit and it exhibits an intralayer zig-zag antiferromagnetic order33 with a bulk Néel temperature of 155 K.34 The transition temperature does not dramatically vary from bulk crystals to the few-layers limit, and the magnetic order remains down to the bilayer.35 The effect of intercalation on the magnetism of NiPS3 has been so far studied by inserting different inorganic36,37 and organic ions.38,39 In particular, wet chemical approaches were employed to intercalate Li+,36,37 cobaltocene,38 and 1,10-phenanthroline39 in NiPS3, resulting in paramagnetic40 or ferrimagnetic38,39 hybrid compounds. While these works demonstrate the potential of intercalation to alter the magnetism of layered materials, they do not fully exploit the chemical versatility of organic guest species and intercalating strategies to finely tune the magnetic properties in hybrid compounds.
Here, we demonstrate a controllable manipulation of the magnetic properties of NiPS3 through the intercalation of two different organic cations, inserted using an engineered two-step procedure. A first electrochemical process leads to the intercalation of tetrabutylammonium ions (TBA+), which causes the emergence of ferrimagnetic ordering, characterized by a finite hysteresis and a transition temperature of 78 K. Then, a novel cation exchange strategy yields the insertion of cobaltocenium ions (Co(Cp)2+, where Cp is a cyclopentadienyl ring C5H5−), leading to a remarkable shift of the transition Curie temperature to 98 K and a higher remanent magnetization. Importantly, these processes were successfully carried out both in bulk crystals, as demonstrated by X-ray diffraction, and in mechanically exfoliated micrometric flakes, as monitored through micro-Raman spectroscopy. Our results open a novel route for the manipulation of the 2D magnetism of LMMs and their integration in devices.
For the insertion of TBA+ cations in NiPS3, we employ an electrochemical approach, which typically provides a fast, controllable and reproducible intercalation.16,41,42 TBA+ was chosen due to its electrochemical stability in non-aqueous solvents, which makes it one of the most frequently used compounds for electrochemical intercalation.20,32 We note that an electrochemical process was previously used to intercalate NiPS3 with alkali metals for lithium-based batteries applications43 and to exfoliate it down to the monolayer using organic ions,44,45 but not to produce bulk intercalated crystals. Fig. 1a presents a sketch of the setup used for the electrochemical process and the intercalating mechanism of TBA+ in bulk NiPS3 crystals (see also Fig. S1a in ESI section 1†). At the cathodic side of the cell, a NiPS3 crystal with a typical mass in the range of 2–3 mg is attached on a platinum plate using pressed indium dots. A silver plate serves as the anode. Both electrodes are immersed in a tetrabutylammonium bromide (TBAB) solution (2 mg mL−1) under the strict absence of oxygen and water, and are connected to an external source-measure unit, which delivers a constant current through the electrochemical cell (see ESI section 1†).
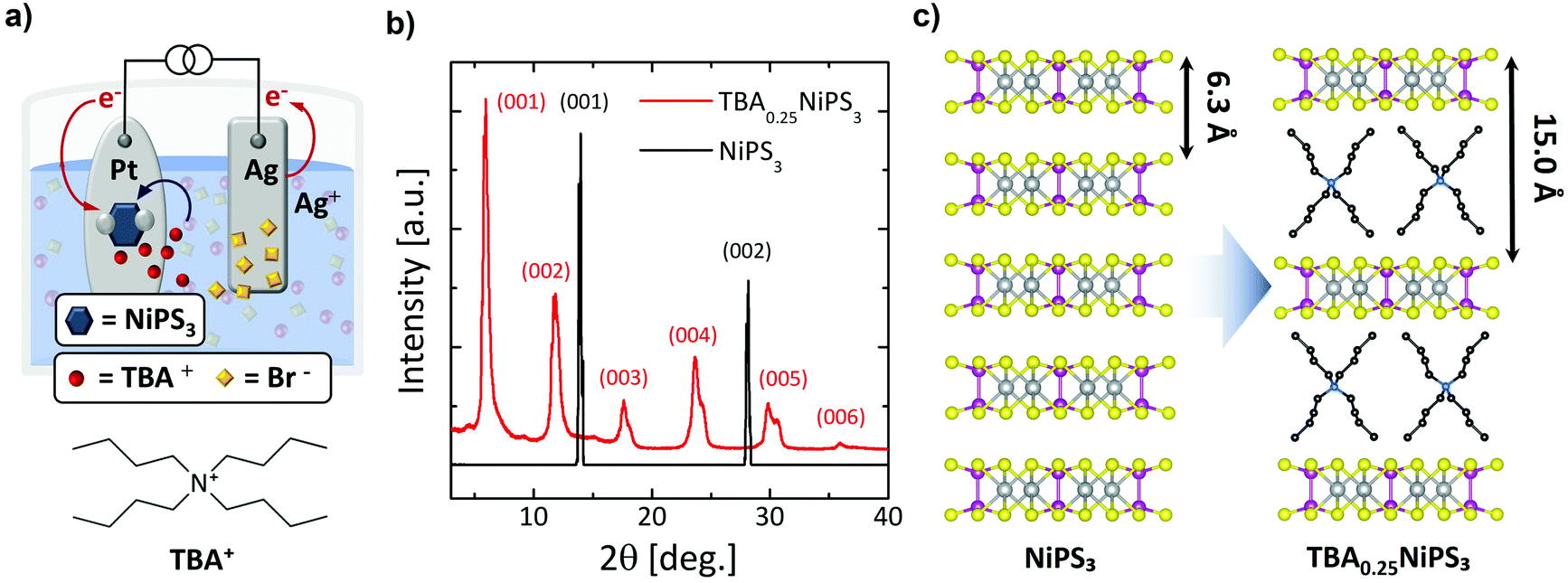 | ||
| Fig. 1 (a) Electrochemical setup used for the intercalation of TBA+ cations into NiPS3 bulk crystal. The chemical structure of tetrabutylammonium (TBA) cation is also shown. (b) X-ray diffraction patterns of pristine NiPS3 (black line) and TBA0.25NiPS3 (red line). Both patterns are normalized to the intensity of the (001) peak to facilitate their comparison. (c) Scheme of the intercalation of TBA+ cations. During the process, the interlayer distance increases from 6.3 Å to 15.0 Å due to the presence of TBA+. | ||
When the current is applied, the Br− anions in solution move to the positively biased Ag anode, and the organic TBA+ cations to the negatively biased NiPS3 crystal (Fig. 1a). At the anode, silver is oxidized, forming initially AgBr(s):20
Oxidation of Ag
| Ag → Ag+ + e− |
Precipitation of AgBr(s)
| Ag+ + Br− → AgBr(s) |
Then, AgBr(s) undergoes subsequent complexation steps.46
At the cathode, electrons are injected into low-lying partially filled 3d-block electronic bands of NiPS3.42 TBA+ guest cationic species are electrostatically attracted into the vdW gaps to balance the total electric charge of the system:
e− injection into NiPS3
| NiPS3 + e− → (NiPS3)− |
TBA+ intercalation
| TBA+ + (NiPS3)− → TBA+(NiPS3)− |
We found that the electrochemical process leads to an intercalated crystal that corresponds to a TBA0.25NiPS3 unit formula, where the 0.25 stoichiometric index is calculated through gravimetric analysis (see ESI section 2†).
Fig. 1b displays a comparison between the XRD patterns measured for a pristine NiPS3 (see also Fig. S3 and Table S1 in ESI section 3†) and an intercalated TBA0.25NiPS3 (also see Fig. S4a in ESI section 3†). The untreated NiPS3 crystal exhibits sharp (00l) peaks corresponding to a 6.34 Å interlayer distance, in good agreement with the value reported in the literature.47 After intercalation, the XRD pattern shows dramatic changes. First, the diffraction peaks (00l) corresponding to the untreated crystal cannot be detected in TBA0.25NiPS3, evidencing the absence of regions characterized by the pristine interlayer spacing in the intercalated crystal, and confirming the complete intercalation. Moreover, new (00l) diffraction peaks shifted to lower 2θ values emerge in the XRD pattern of TBA0.25NiPS3, corresponding to an increase of the interlayer distance due to the insertion of TBA+ ions (Fig. 1c). At high 2θ angles, single peaks can be resolved in multiplets, revealing the presence of more than one crystallographic phase (see Table S2 in ESI section 3†). We explain this feature by considering that the TBA+ guest molecules can adopt different orientations with respect to the ab plane of NiPS3, leading to crystallographic phases characterized by a slightly different interlayer spacing. For the crystal shown in Fig. 1b, the most abundant phase is characterized by an interlayer distance of 15.0 Å. The other crystallographic phases encountered after TBA+ intercalation are characterized by slightly different interlayer distance (in the range 14.5 Å–15.3 Å). The difference between the interlayer spacing measured for the intercalated and pristine crystals, which amounts to 8.2–8.9 Å depending on the crystallographic phase, provides an estimation of the vdW steric hindrance of the cations inserted in the host material. We note that by changing the conditions of the electrochemical intercalation, it is possible to tune the relative abundance of the different crystallographic phases, as detailed in Fig. S3 and Table S1 in ESI.†
Based on the measured interlayer distance, the estimated size of the TBA+ cations24 and the density of TBA+ molecules in the organic layer (one molecule every four formula units), we can conclude that the intercalation leads to an organic–inorganic hybrid superlattice composed of alternated single TBA+ organic layers and NiPS3 monolayers (Fig. 1c).
By comparing the XRD pattern of TBA0.25NiPS3 with those of other electrochemically intercalated compounds,48,49 we find that the TBA0.25NiPS3 peaks are relatively sharp and defined, indicating a relatively high crystalline quality.
Next, we show the substitution of TBA+ guest ions with Co(Cp)2+ ions through a non-redox heterogeneous cation exchange mechanism in solution (Fig. 2a). The mechanism can be described as follows:
| 4TBA0.25NiPS3 + Co(Cp)2+ → 4[Co(Cp)2]0.25NiPS3 + TBA+ |
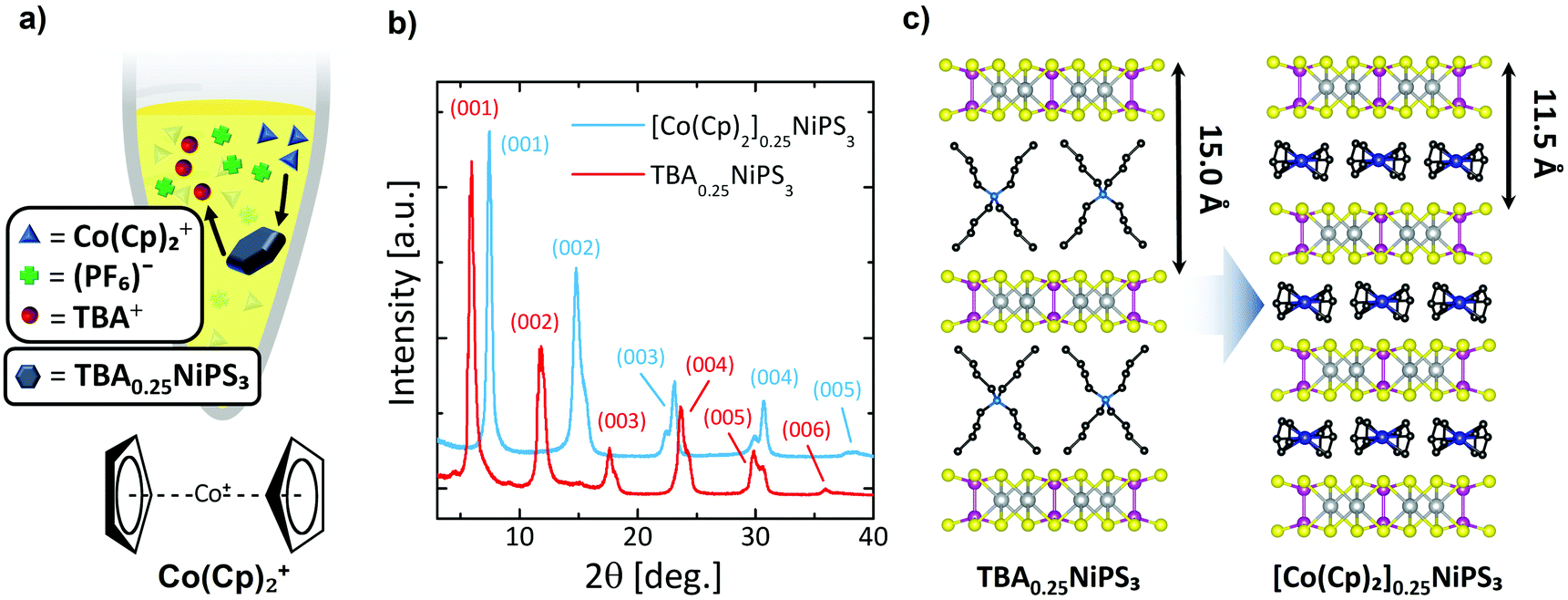 | ||
| Fig. 2 (a) Setup for the cation exchange process. A TBA0.25NiPS3 bulk crystal is immersed in a closed vial containing an acetonitrile solution of Co(Cp)2PF6. The chemical structure of bis-(cyclopentadienyl)cobalt(III), cobaltocenium (Co(Cp)2+) is also shown. (b) X-ray diffraction patterns of bulk TBA0.25NiPS3 before and after cation exchange (red and blue lines, respectively). Both patterns are normalized to the intensity of the (001) peak to facilitate their comparison. (c) Scheme of the cation exchange. During the process, the interlayer distance decreases from 15 Å to 11.5 Å due to the smaller size of Co(Cp)2+ in comparison to TBA+. | ||
To the best of our knowledge, this method has not been employed until now to tune the physical properties of an already intercalated NiPS3 crystal. Pioneering studies showed how Co(Cp)2+ is particularly prone to intercalate transition metal phosphorus trisulfides.38 In particular, a solvothermal method results in the direct insertion Co(Cp)2+ cations in NiPS3; however, this study only reported successful intercalation of powder at 130 °C.38 Our protocol follows a 2-step intercalating route that allows large (0.1 mm × 5 mm2) NiPS3 crystals (typically 2–3 mg) to be intercalated with Co(Cp)2+ cations in a similarly long process, even at room temperature. Additionally, this experiment offers the possibility to study how the use of different methods to intercalate the same organic cation affects the magnetic properties of the resulting material.
In Fig. 2b, the diffractogram of [Co(Cp)2]0.25NiPS3 (see also Fig. S5a and Table S3 in ESI section 3†) is compared with the previous one of TBA0.25NiPS3. After the ion exchange, the peaks corresponding to the TBA0.25NiPS3 phase are absent, indicating that the TBA+ cations have been successfully de-intercalated. Additionally, the (00l) diffraction peaks located at higher angles evidence a shorter interlayer distance (Fig. 2b). This observation is in agreement with the smaller size of Co(Cp)2+50 as compared to TBA+. This comparison confirms the occurrence of a complete TBA+/Co(Cp)2+ exchange, indicating that the process is thermodynamically favoured by the electrochemical potential and by the electrostatics of the system, since the positive charge in TBA+ is screened by four butyl groups, whereas in Co(Cp)2+ it is comparably more accessible for the negatively charged NiPS3 layers.
Interestingly, the XRD pattern of [Co(Cp)2]0.25NiPS3 includes two families of peaks associated with two slightly different phases with different interlayer distances, 11.9 Å, and 11.5 Å. These two different gaps correspond to an increase in the separation between layers compared to the pristine material of 5.6 Å and 5.2 Å, respectively. We understand this phenomenon as the consequence of the coexistence of two possible arrangements for the Co(Cp)2+ ions within the vdW gaps, i.e., horizontally and vertically, with the C5 symmetry axis of Cp− respectively parallel and perpendicular to the basal plane of the crystal. A 5.2 Å increase in interlayer distance was already reported for the intercalation of Co(Cp)2+ in MnPS3via cationic exchange,51 for which it was revealed that the Co(Cp)2+ is horizontally oriented (as shown in Fig. 2c). Therefore, we assign the 5.6 Å and 5.2 Å phases encountered in our sample to Co(Cp)2+ cations oriented “vertically” and “horizontally” between NiPS3 layers, i.e., with the Cp− rings parallel or perpendicular to the basal plane, respectively. Notice here that a more complex assembly of vertical and horizontal cations cannot be excluded in the 5.6 Å phase, as reported for neutral metallocenes on metallic surfaces.52 Broadening of the peaks remains almost unaltered (see Table S3 in ESI section 3†), indicating that the cation exchange process, even if highly invasive, does not affect the macroscopic lattice crystallinity.
We note that by employing different conditions for the intercalation, we obtained a Co(Cp)2+ intercalated NiPS3 displaying the single structural phase characterized by the 11.9 Å interlayer distance (see Fig. S5 and Table S3 in ESI†).
After demonstrating the TBA+ intercalation and the TBA+/Co(Cp)2+ exchange through XRD, we now discuss how micro-Raman spectroscopy can be exploited as a sensitive tool to monitor the intercalating process and to provide additional information on the phenomena accompanying the insertion of cations. The Raman spectrum of pristine NiPS3 (black curve in Fig. 3a) displays several peaks, which correspond to the eight fundamental (3A1g + 5Eg) and some higher order Raman-active modes.53
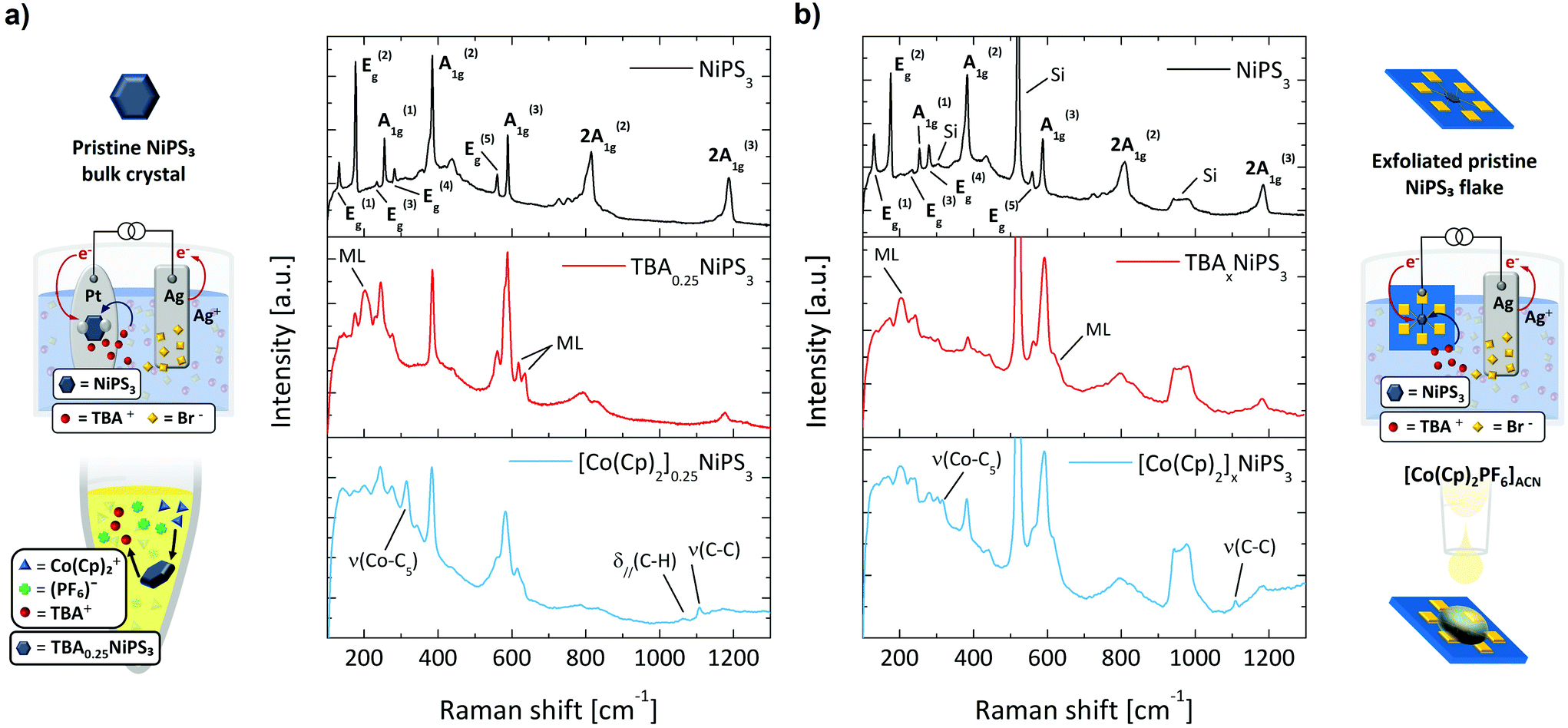 | ||
| Fig. 3 (a) Raman spectra of pristine NiPS3, TBA0.25NiPS3 and [Co(Cp)2]0.25NiPS3 bulk crystals. (b) Raman spectra of pristine NiPS3, TBAxNiPS3 and [Co(Cp)2]0.25NiPS3 flakes. On the left and right side, we display the schematics corresponding to each intercalation step. | ||
The Raman spectrum of TBA0.25NiPS3 (red curve in the middle panel of Fig. 3a) is significantly different from that of the pristine NiPS3. We highlight three main changes occurring after the TBA+ insertion which can be considered the hallmarks of successful intercalation. More detailed Raman spectra are reported in Fig. S7 and other features are present in ESI section 3.† First, the intensity of the Eg(1) peak at 132 cm−1, which is related to the translational mode of Ni(II) cations in sulfur-coordinated octahedral sites (Oh),47 is strongly suppressed for TBA0.25NiPS3. A similar effect was previously reported for Li+ intercalated NiPS3,54 and attributed to a lower density of Ni(II) in Oh sites. Indeed, Ni atoms are reduced to the metallic oxidation state (Ni0) and displaced to the tetrahedral sites (Td).36,55 Therefore, the decrease in intensity of the Eg(1) mode provides the first direct evidence of the Ni(II) → Ni0 coordination evolution induced by the intercalation. This change in the electronic configuration of Ni has a strong impact on the magnetic properties of NiPS3, as discussed below.
Second, three new peaks (indicated with ML in Fig. 3a) appear at 202 cm−1, 618 cm−1 and 635 cm−1 in the Raman spectrum of TBA0.25NiPS3. Similar spectral features were observed for ultrathin NiPS3 flakes, and their presence was explained based on the breaking of translational symmetry.35,53 The emergence of these peaks in TBA0.25NiPS3 indicates that the increased interlayer distance in the intercalated crystals produces a symmetry breaking effect similar to that generated by the isolation of monolayers. In this regard, the observation of vibrational features typical of monolayers being recovered in intercalated crystals shows the potential of intercalation to obtain monolayer-like behavior in bulk crystals.31
Lastly, we highlight the remarkable position change of the A1g(1) peak, which shifts from 254.5 cm−1 in the pristine NiPS3 to 245 cm−1 in TBA0.25NiPS3. A similar redshift but larger for A1g(1) was reported for Li+ intercalated NiPS3, attributed to the charge carrier doping introduced by intercalation.54 This can be understood as a result of the lower doping level introduced by the TBA+ ions intercalation, that is limited by their higher molecular vdW hindrance.
These three main features also appear in the Raman spectrum of the [Co(Cp)2]0.25NiPS3 (blue curve in the bottom panel of Fig. 3a). In addition to these changes, for this sample we also observe the peaks related to the ν(Co–C5), δ∥(C–H), ν(C–C) molecular modes of Co(Cp)2+, located at 314 cm−1, 1062 cm−1 and 1108 cm−1, respectively. These peaks are red-shifted to those observed for the Co(Cp)2PF6 powder, located at 317 cm−1, 1070 cm−1 and 1112.5 cm−1 (see Fig. S7 in ESI†).56 This indicates that Co(Cp)2+ ions are structurally intact and surrounded by a different chemical environment than the one provided by the coordinating PF6− in the pristine crystal lattice. Moreover, we also highlight that the main Raman feature of PF6− counterion,57ν(P − F) at 741 cm−1, is not detected in [Co(Cp)2]0.25NiPS3. This proves that the cation exchange effectively results in the swapping of TBA+ with Co(Cp)2+, and that the Co(Cp)2+ features detected in the crystals are not parasitic Co(Cp)2PF6 vibrational signatures appearing due to the presence of residuals of the salt.
Our analysis demonstrates that Raman spectroscopy is a suitable tool for characterization of the intercalation process in bulk crystals; therefore, we employ it to monitor the intercalation of micrometric mechanically exfoliated flakes. This is of particular significance, since other techniques employed to track the intercalation of bulk crystals (such as XRD) are not practicable for the characterization of few-layers-thick and few-micrometres-wide exfoliated flakes.
To intercalate micrometric NiPS3 crystals, we stamped a mechanically exfoliated flake onto prepatterned Au contacts (see ESI section 1†). After that, we performed the electrochemical intercalation of TBA+ cations by contacting one of the electrodes to the external circuitry, as shown in Fig. 3b (real apparatus shown in Fig. S1c in ESI section 1†). Subsequently, the TBA+ → Co(Cp)2+ cation exchange was achieved by simply drop-casting a Co(Cp)2PF6 solution onto the TBA+ intercalated flake under strict exclusion of oxygen and water. In Fig. 3b, we compare the Raman spectra measured (i) on a pristine flake transferred onto prepatterned contacts, (ii) on the same flake after the electrochemical intercalation of TBA+ cations, and (iii) for the same flake after the TBA+/Co(Cp)2+ cation exchange. The Raman spectrum of the pristine flake is analogous to that of the untreated bulk crystal, displaying all the Raman-active modes. The peaks of the exfoliated flake are generally broader than those of the bulk crystal, which can be ascribed to lattice strain due to the stamping,58 or interaction with the SiO2 substrate.59 Since the flake is thin, the typical Raman features of the silicon substrate appear at 303 cm−1, 520 cm−1 and around 960 cm−1. Remarkably, after intercalating the flake with TBA+, we observe the same modifications found in the Raman spectrum previously described for bulk crystals – including the lower intensity in the Ni(II) vibrational mode Eg(1), the emergence of new peaks characteristic of the reduced symmetry, and the doping-related redshift in A1g(1). Moreover, even after the cation exchange, we observe the spectral features previously discussed for its corresponding bulk crystal, which include the appearance of the characteristic vibrational modes of Co(Cp)2+.
These results demonstrate that (i) micro-Raman spectroscopy is an ideal technique to monitor the intercalation process for ultrathin micrometric exfoliated flakes, and that (ii) the electrochemical and ion exchange processes developed here lead to efficient intercalation not only in bulk crystals, but also in mechanically exfoliated flakes. While several works on intercalation focus on bulk crystals which are not suitable for devices, the successful intercalation of exfoliated flakes offers the possibility of taking advantage from the materials changes associated to the intercalation in novel nanoscale devices.
Finally, we show how the intercalation and ion exchange processes modify the magnetic properties of NiPS3. Fig. 4a shows the temperature dependence of the field-cooled magnetization M(T) measured within an in-plane magnetic field H = 500 Oe for pristine, TBA+ and Co(Cp)2+ intercalated NiPS3. For the pristine NiPS3 crystal, the M(T) displays the reported trend,33,35 characterized by a decrease in the magnetization associated with the antiferromagnetic transition at TNéel = 155 K. A very different behavior is observed for TBA+ and Co(Cp)2+ intercalated NiPS3. In both cases, we find a sudden increase of the magnetization at 78 K for TBA0.25NiPS3 and at 98 K for [Co(Cp)2]0.25NiPS3, characteristic of a magnetic phase transition. The magnetization values achieved at low temperatures, which are for both compounds on the order of 10−3μB per Ni atom, are low compared to the expected magnetic moment of Ni(II) (μ[Ni(II)] = 2.83μB). Therefore, we conclude that at low-temperature TBA0.25NiPS3 and [Co(Cp)2]0.25NiPS3 are not ferromagnetic, but rather ferrimagnetic. Moreover, in the M(T) we observe an upturn of the magnetization below 20 K for both intercalated compounds, suggesting the co-existence of paramagnetic and ferrimagnetic phases. We note that the magnetization reached at low temperature for the two compounds is quite different, being 7.5 emu mol(Ni)−1 for TBA0.25NiPS3 and 20 emu mol(Ni)−1 for [Co(Cp)2]0.25NiPS3, indicating that the latter is characterized by a larger spontaneous magnetization (see also Fig. S8 in ESI†).
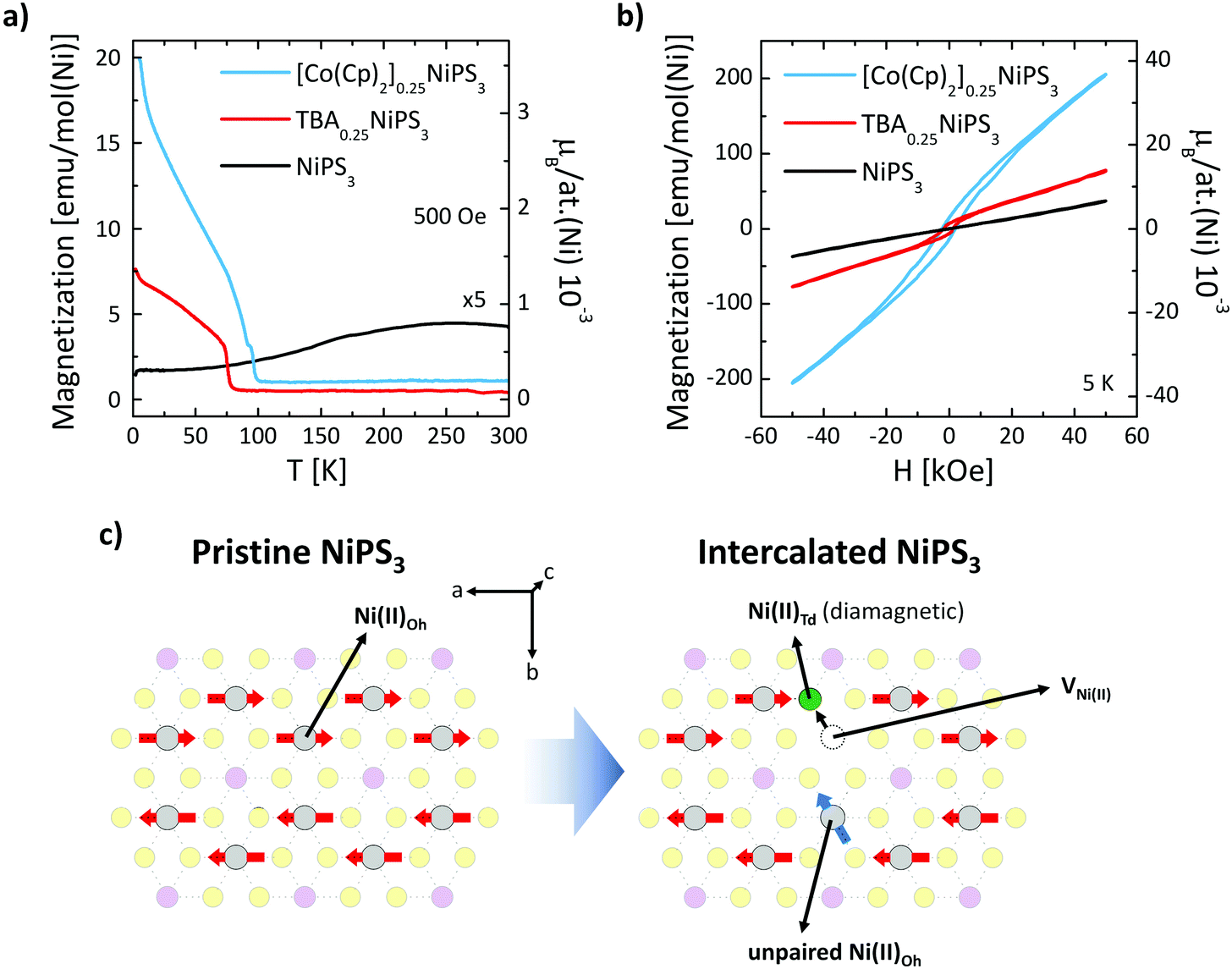 | ||
| Fig. 4 (a) Field-cooled molar magnetization vs. temperature for bulk NiPS3, TBA0.25NiPS3 and [Co(Cp)2]0.25NiPS3 crystals. The applied field (500 Oe) is oriented parallel to the ab plane of the crystal. (b) Hysteresis loops at 5 K of bulk pristine NiPS3, TBA0.25NiPS3 and [Co(Cp)2] 0.25NiPS3 crystals. (c) Scheme of the structural change in the NiPS3 layer accompanying the reduction of the Ni atoms. | ||
This scenario is confirmed by the hysteresis loops M(H) measured at 5 K for TBA+ and Co(Cp)+ intercalated NiPS3 displayed in Fig. 4b, compared with the loop recorded for pristine NiPS3. Magnetization of pristine NiPS3 increases linearly with the applied field H. This trend can be understood considering that NiPS3 is an antiferromagnet with an in-plane anisotropy, and that the application of an in-plane field causes a slight tilting of the spins along its direction. Conversely, the M(H) curves of TBA0.25NiPS3 and [Co(Cp)2]0.25NiPS3 show a clear hysteresis with finite coercive fields of H = 1.5 kOe and 2.2 kOe, as well as remanent magnetization Mr = 6.6 emu mol−1 and 14.2 emu mol−1, respectively. In both cases, the hysteretic behavior demonstrates the emergence of (ferri)magnetic ordering. For [Co(Cp)2]0.25NiPS3, hysteresis loops measured with an in-plane and out-of-plane magnetic field (Fig. S9 in ESI†) reveal that the crystal possesses a magnetic anisotropy and displays an in-plane magnetic easy axis. We highlight that also in pristine NiPS3 crystals the spins are oriented in plane.35
At large magnetic fields, the magnetization does not saturate, confirming the presence of a paramagnetic signal in addition to the ferrimagnetic one. The coexistence of different magnetic responses can be ascribed to the different structural phases found in our intercalated samples. This conclusion is supported by a combined structural and magnetic characterization of other crystals intercalated using different conditions (see ESI section 7†). In particular, the magnetic response in TBA+ intercalated NiPS3 was found to depend on the details of the structural phases generated in the intercalation process. Besides, the single-phase [Co(Cp)2]0.25NiPS3 crystal characterized by the 11.9 Å interlayer distance did not display the ferrimagnetic behavior (Fig. S10 in ESI†). Therefore, we conclude that for this compound, the paramagnetic and the ferromagnetic phases are related to the phases with 11.9 Å and 11.5 Å interlayer distance, respectively.
We note that (i) the ferrimagnetism in TBA+ intercalated NiPS3, which has not been previously reported, proves that an electrochemical approach can be used to modulate the magnetism of NiPS3; and (ii) the phase transition at 98 K measured in our Co(Cp)2+ intercalated NiPS3 is significantly higher than the one previously reported for the same compound intercalated using a wet chemistry approach.38 This indicates that not only the guest molecule, but also the intercalation method can strongly impact the resulting magnetic properties of NiPS3.
The dramatic change in the magnetic properties of NiPS3 can be explained based on the nanoscale phenomena taking place in the NiPS3 layers. In particular, the intercalation induces the Ni(II) → Ni0 reduction and displacement from the Oh to the Td lattice site36,55 (see Fig. 4c). This reductive displacement is accompanied by a dramatic change in the magnetic properties of the Ni atoms. Ni(II)-Oh atoms are characterized by a [Ar]3d8s0 electronic configuration (high spin) bearing a magnetic moment μ[Ni(II)] = 2.83μB, whereas Ni0-Td atoms possess a [Ar]3d10s0 zero-spin configuration (low spin). As discussed previously, Raman spectroscopy evidences that the Ni(II)-Oh → Ni0-Td reduction occurs for a significant fraction of Ni(II) atoms, which can be quantified based on charge balance considerations. The reduction of each Ni(II) atom requires two transferred electrons, which are provided by two guest molecules. Therefore, from the 0.25 stoichiometric index in TBA0.25NiPS3, we estimate that 12.5% of Ni atoms are reduced and displaced. As a result, the antiferromagnetism in the intercalated compounds is not fully compensated, as part of the spins that give rise to the antiferromagnetic order in the pristine crystal are suppressed during the intercalation, or analogously, unpaired spins are introduced in the system (see Fig. 4c).
While this argument accounts for the presence of unpaired spins in the intercalated NiPS3 compounds, understanding their magnetic coupling is more complex. Our M(T) and M(H) data indicate that different structural phases of crystals intercalated with the same molecule are characterized by diverse magnetic responses, demonstrating that the overall magnetic properties are determined by the details of the nanoscale interactions between the guest species and the inorganic layers. For instance, the molecules intercalated between the layers may form ordered self-assembled structures, which might induce a periodicity in the displacement of Ni atoms, resulting in the measured magnetic interactions. Therefore, our results indicate that the intercalation of organic cations provides a readily available “tuning wheel” to engineer the magnetic properties of layered materials.
Finally, we highlight that while layered materials intercalated with organic compounds are typically reactive at ambient conditions,29 only minor changes in the magnetic properties of a [Co(Cp)2]0.25NiPS3 crystal were observed after storing it in air for more than one month (Fig. S9 in ESI†).
Materials and methods
Materials
NiPS3 crystals were purchased from HQ graphene. Acetonitrile (anhydrous <0.001% H2O), tetrabutyl-ammonium bromide (TBAB – purity ≥98%) and bis-(cyclopentadienyl)cobalt(III) hexafluorophosphate (Co(Cp)2PF6 – purity 98%) were purchased at Aldrich. Platinum and silver electrode plates were obtained by pressing metallic pellets (purity 99.99%, Kurt J. Lesker). Before usage, salts were dehydrated at 100° C in vacuum (1 mbar) overnight. Electrodes were polished with sandpaper and sonicated for 2 minutes in acetone and then in isopropanol for surface cleaning.Device fabrication
To intercalate NiPS3 flakes, devices have been fabricated by stamping mechanically exfoliated flakes onto gold contacts prepatterned in a Hall configuration on Si/SiO2 substrates. The stamping is performed by exfoliating NiPS3 on a polydimethylsiloxane (PDMS) elastomer. Target flakes with a thickness of a few layers (typically 5–15) are identified through optical microscopy and transferred onto the prepatterned contacts using a micromanipulator coupled to an optical microscope.TBA+ electrochemical intercalation
Self-limiting tetrabutylammonium (TBA+) electrochemical intercalation is achieved using a custom-made cell. This cell is composed of two electrodes, a platinum holder for NiPS3 as the cathode and a silver plate as the anode, both immersed in a TBAB solution in acetonitrile (2 mg mL−1–3.1 mM) (Fig. 1a).Bulk NiPS3 crystals (typical mass m = 2–3 mg) were electrically anchored to the platinum plate by utilizing indium strips and subsequently fully dipped into the electrolyte. In this case, the intercalation was achieved by applying a constant current (30 μA or 50 μA) using a Keithley 2635 source meter. The voltage drop caused by the current was monitored during the intercalation, and the full intercalation was identified as an increase in the voltage (see also ESI 1†). Micrometric flakes were intercalated by connecting one of the Au electrodes to the external circuitry using a Cu wire attached through an indium connection. In this case, the intercalation was achieved by applying a constant current of 2 μA for 5 minutes.
TBA+/Co(Cp)2+ cation exchange
Bulk TBA0.25NiPS3 crystal was immersed in 1 mL of Co(Cp)2PF6 solution in acetonitrile at a concentration of 50 mg mL−1. The process was carried out in a sealed vial kept in an oven at 50 °C located in a glovebox for 2 days to ensure the exchange for the whole crystal (see also ESI 1†).Characterization
X-ray diffractometry is carried out with an XPERT-PRO diffractometer on bulk crystals in a ω/ω configuration. A copper cathode λ(Kα1) = 1.540598 Å is used as X-ray source. Micro-Raman characterization is performed with a Renishaw inVia Qontor system at room temperature using a 633 nm HeNe laser. Magnetization measurements vs. temperature or field H are carried out using a physical properties measurement system (PPMS) in vibrating sample magnetometer mode.Conclusions
We have employed an electrochemical approach to intercalate TBA+ cations in the LMM NiPS3, leading to a hybrid organic/inorganic superlattice compound composed of alternating TBA+ and NiPS3 single layers. Moreover, immersing the TBA+ intercalated crystals in a Co(Cp)2PF6 solution results in the complete exchange of the TBA+ cations with Co(Cp)2+.These processes, which are confirmed through XRD for large crystals, can be also monitored through Raman spectroscopy, which provides important insight into the nanoscale phenomena accompanying the intercalation. Importantly, we used micro-Raman spectroscopy to demonstrate the successful intercalation of micrometric exfoliated flakes, which could not be accessed via conventional XRD. Finally, we show that the magnetic properties of NiPS3 are dramatically modified by the intercalation in a way that depends on the molecular guests and the intercalation process. In particular, pristine NiPS3 is antiferromagnetic, whereas a ferrimagnetic transition at 78 K and 98 K is recorded in TBA+ and Co(Cp)2+ intercalated compounds, respectively. Our results show how molecular intercalation can be tailored to generate hybrid superlattice compounds with novel magnetic properties, offering the possibility to introduce additional compounds with tunable magnetic properties in the LMM family. Moreover, the successful intercalation of exfoliated flakes opens the way to the integration of intercalated NiPS3 in devices.
Author contributions
D. T., M. O., M. G. conceived the study. D. T. and A. B. performed the electrochemical intercalation, the ion-exchange and XRD measurements. J. M. P., Y. A., F. C. and F. C. fabricated the devices for the intercalation of flakes. J. M. P. and B. M.-G. performed the Raman characterization. M. I. carried out the magnetic measurements. M. G., B. M.-G. and L. E. H. supervised the work. D. T. and M. G. wrote the manuscript, with inputs from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge R. Llopis and A. Eleta for technical support. This work is supported by “la Caixa” Foundation (ID 100010434), under the agreement LCF/BQ/PI19/11690017, by the Spanish MICINN under Project PID2019-108153GA-I00, RTI2018-094861-B-100 and under the María de Maeztu Units of Excellence Program (MDM-2016-0618). B. M.-G. thanks Gipuzkoa Council (Spain) in the frame of Gipuzkoa Fellows Program.Notes and references
- B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire, D. H. Cobden, W. Yao, D. Xiao, P. Jarillo-Herrero and X. Xu, Nature, 2017, 546, 270–273 CrossRef CAS PubMed.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z. Q. Qiu, R. J. Cava, S. G. Louie, J. Xia and X. Zhang, Nature, 2017, 546, 265–269 CrossRef CAS PubMed.
- M. Gibertini, M. Koperski, A. F. Morpurgo and K. S. Novoselov, Nat. Nanotechnol., 2019, 14, 408–419 CrossRef CAS PubMed.
- M. Och, M.-B. Martin, B. Dlubak, P. Seneor and C. Mattevi, Nanoscale, 2021, 13, 2157–2180 RSC.
- Y. Deng, Y. Yu, Y. Song, J. Zhang, N. Z. Wang, Z. Sun, Y. Yi, Y. Z. Wu, S. Wu, J. Zhu, J. Wang, X. H. Chen and Y. Zhang, Nature, 2018, 563, 94–99 CrossRef CAS PubMed.
- S. Jiang, J. Shan and K. F. Mak, Nat. Mater., 2018, 17, 406–410 CrossRef CAS PubMed.
- S. Jiang, L. Li, Z. Wang, K. F. Mak and J. Shan, Nat. Nanotechnol., 2018, 13, 549–553 CrossRef CAS PubMed.
- Z. Wang, T. Zhang, M. Ding, B. Dong, Y. Li, M. Chen, X. Li, J. Huang, H. Wang, X. Zhao, Y. Li, D. Li, C. Jia, L. Sun, H. Guo, Y. Ye, D. Sun, Y. Chen, T. Yang, J. Zhang, S. Ono, Z. Han and Z. Zhang, Nat. Nanotechnol., 2018, 13, 554–559 CrossRef CAS PubMed.
- I. A. Verzhbitskiy, H. Kurebayashi, H. Cheng, J. Zhou, S. Khan, Y. P. Feng and G. Eda, Nat. Electron., 2020, 3, 460–465 CrossRef CAS.
- D. Voiry, A. Goswami, R. Kappera, C. d. C. Castro e Silva, D. Kaplan, T. Fujita, M. Chen, T. Asefa and M. Chhowalla, Nat. Chem., 2015, 7, 45–49 CrossRef CAS PubMed.
- L. Daukiya, J. Teyssandier, S. Eyley, S. El Kazzi, M. C. Rodríguez González, B. Pradhan, W. Thielemans, J. Hofkens and S. De Feyter, Nanoscale, 2021, 13, 2972–2981 RSC.
- I. Gómez-Muñoz, S. Laghouati, R. Torres-Cavanillas, M. Morant-Giner, N. V. Vassilyeva, A. Forment-Aliaga and M. Giménez-Marqués, ACS Appl. Mater. Interfaces, 2021, 13, 36475–36481 CrossRef PubMed.
- M. Gobbi, E. Orgiu and P. Samori, Adv. Mater., 2018, 30 Search PubMed.
- J. Wan, S. D. Lacey, J. Dai, W. Bao, M. S. Fuhrer and L. Hu, Chem. Soc. Rev., 2016, 45, 6742–6765 RSC.
- M. S. Stark, K. L. Kuntz, S. J. Martens and S. C. Warren, Adv. Mater., 2019, 31, 1808213 CrossRef PubMed.
- J. Zhou, Z. Lin, H. Ren, X. Duan, I. Shakir, Y. Huang and X. Duan, Adv. Mater., 2021, 33, 2004557 CrossRef CAS PubMed.
- S. J. Sung, J. W. Yang, P. R. Lee, J. G. Kim, M. T. Ryu, H. M. Park, G. Lee, C. C. Hwang, K. S. Kim, J. S. Kim and J. W. Chung, Nanoscale, 2014, 6, 3824–3829 RSC.
- C. A. Formstone, E. T. FitzGerald, D. O'Hare, P. A. Cox, M. Kurmoo, J. W. Hodby, D. Lillicrap and M. Goss-Custard, J. Chem. Soc., Chem. Commun., 1990, 501–503 RSC.
- Z. Li, Y. Zhao, K. Mu, H. Shan, Y. Guo, J. Wu, Y. Su, Q. Wu, Z. Sun, A. Zhao, X. Cui, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 16398–16404 CrossRef CAS PubMed.
- L. K. Ma, M. Z. Shi, B. L. Kang, K. L. Peng, F. B. Meng, C. S. Zhu, J. H. Cui, Z. L. Sun, D. H. Ma, H. H. Wang, B. Lei, T. Wu and X. H. Chen, Phys. Rev. Mater., 2020, 4, 124803 CrossRef CAS.
- H. Wu, S. Li, M. Susner, S. Kwon, M. Kim, T. Haugan and B. Lv, 2D Mater., 2019, 6, 045048 CrossRef CAS.
- M. Z. Shi, N. Z. Wang, B. Lei, C. Shang, F. B. Meng, L. K. Ma, F. X. Zhang, D. Z. Kuang and X. H. Chen, Phys. Rev. Mater., 2018, 2, 074801 CrossRef.
- B. Rendenbach, T. Hohl, S. Harm, C. Hoch and D. Johrendt, J. Am. Chem. Soc., 2021, 143, 3043–3048 CrossRef CAS PubMed.
- M. Z. Shi, N. Z. Wang, B. Lei, J. J. Ying, C. S. Zhu, Z. L. Sun, J. H. Cui, F. B. Meng, C. Shang, L. K. Ma and X. H. Chen, New J. Phys., 2018, 20, 123007 CrossRef CAS.
- N. Z. Wang, M. Z. Shi, C. Shang, F. B. Meng, L. K. Ma, X. G. Luo and X. H. Chen, New J. Phys., 2018, 20, 023014 CrossRef.
- Z. Li, X. Zhang, X. Zhao, J. Li, T. S. Herng, H. Xu, F. Lin, P. Lyu, X. Peng, W. Yu, X. Hai, C. Chen, H. Yang, J. Martin, J. Lu, X. Luo, A. H. Castro Neto, S. J. Pennycook, J. Ding, Y. Feng and J. Lu, Adv. Mater., 2020, 32, 1907645 CrossRef CAS PubMed.
- Q. He, Z. Lin, M. Ding, A. Yin, U. Halim, C. Wang, Y. Liu, H.-C. Cheng, Y. Huang and X. Duan, Nano Lett., 2019, 19, 6819–6826 CrossRef CAS PubMed.
- Y. Koike, H. Suematsu, K. Higuchi and S. Tanuma, Solid State Commun., 1978, 27, 623–627 CrossRef CAS.
- T. E. Weller, M. Ellerby, S. S. Saxena, R. P. Smith and N. T. Skipper, Nat. Phys., 2005, 1, 39–41 Search PubMed.
- I. A. Verzhbitskiy, D. Voiry, M. Chhowalla and G. Eda, 2D Mater., 2020, 7, 035013 Search PubMed.
- C. Wang, Q. He, U. Halim, Y. Liu, E. Zhu, Z. Lin, H. Xiao, X. Duan, Z. Feng, R. Cheng, N. O. Weiss, G. Ye, Y.-C. Huang, H. Wu, H.-C. Cheng, I. Shakir, L. Liao, X. Chen, W. A. Goddard III, Y. Huang and X. Duan, Nature, 2018, 555, 231–236 CrossRef CAS PubMed.
- N. Wang, H. Tang, M. Shi, H. Zhang, W. Zhuo, D. Liu, F. Meng, L. Ma, J. Ying, L. Zou, Z. Sun and X. Chen, J. Am. Chem. Soc., 2019, 141, 17166–17173 CrossRef CAS PubMed.
- A. R. Wildes, V. Simonet, E. Ressouche, G. J. McIntyre, M. Avdeev, E. Suard, S. A. J. Kimber, D. Lançon, G. Pepe, B. Moubaraki and T. J. Hicks, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 224408 CrossRef.
- P. A. Joy and S. Vasudevan, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 5425–5433 CrossRef CAS PubMed.
- K. Kim, S. Y. Lim, J.-U. Lee, S. Lee, T. Y. Kim, K. Park, G. S. Jeon, C.-H. Park, J.-G. Park and H. Cheong, Nat. Commun., 2019, 10, 345 CrossRef PubMed.
- C. Berthier, Y. Chabre and M. Minier, Solid State Commun., 1978, 28, 327–332 CrossRef CAS.
- G. Giunta, V. Grasso, F. Neri and L. Silipigni, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 8189–8194 CrossRef CAS PubMed.
- E. Manova, A. Leaustic, I. Mitov, D. Gonbeau and R. Clement, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1998, 311, 155–160 CrossRef.
- X. Ma, L. Zhang, C. Xu, Q. Dong, R. I. Walton, Z. Li, H. Shi, G. Chen, J. Hu, J. Li and H. Yang, Chem. Commun., 2020, 56, 4603–4606 RSC.
- R. Brec, D. M. Schleich, G. Ouvrard, A. Louisy and J. Rouxel, Inorg. Chem., 1979, 18, 1814–1818 CrossRef CAS.
- M. Rajapakse, B. Karki, U. O. Abu, S. Pishgar, M. R. K. Musa, S. M. S. Riyadh, M. Yu, G. Sumanasekera and J. B. Jasinski, npj 2D Mater. Appl., 2021, 5, 30 CrossRef CAS.
- R. Brec, Solid State Ionics, 1986, 22, 3–30 CrossRef CAS.
- I. Saikh, C. Julien and M. Balkanski, in Solid State Microbatteries, ed. J. R. Akridge and M. Balkanski, Springer US, Boston, MA, 1990, pp. 297–300 Search PubMed.
- X. Li, Y. Fang, J. Wang, B. Wei, K. Qi, H. Y. Hoh, Q. Hao, T. Sun, Z. Wang, Z. Yin, Y. Zhang, J. Lu, Q. Bao and C. Su, Small, 2019, 15, 1902427 CrossRef PubMed.
- J. Luxa, Š. Cintl, L. Spejchalová, J.-Y. Lin and Z. Sofer, ACS Appl. Energy Mater., 2020, 3, 11992–11999 CrossRef CAS.
- D. C. Luehrs, R. T. Iwamoto and J. Kleinberg, Inorg. Chem., 1966, 5, 201–204 CrossRef CAS.
- B. E. Taylor, J. Steger and A. Wold, J. Solid State Chem., 1973, 7, 461–467 CrossRef CAS.
- C. Wan, X. Gu, F. Dang, T. Itoh, Y. Wang, H. Sasaki, M. Kondo, K. Koga, K. Yabuki, G. J. Snyder, R. Yang and K. Koumoto, Nat. Mater., 2015, 14, 622–627 CrossRef CAS PubMed.
- C. Wan, Y. Kodama, M. Kondo, R. Sasai, X. Qian, X. Gu, K. Koga, K. Yabuki, R. Yang and K. Koumoto, Nano Lett., 2015, 15, 6302–6308 CrossRef CAS PubMed.
- D. O′hare and J. S. O. Evans, Comments Inorg. Chem., 1993, 14, 155–206 CrossRef.
- K. Kim and D. A. Cleary, J. Phys. Chem., 1990, 94, 3816–3819 CrossRef CAS.
- M. Ormaza, P. Abufager, N. Bachellier, R. Robles, M. Verot, T. Le Bahers, M.-L. Bocquet, N. Lorente and L. Limot, J. Phys. Chem. Lett., 2015, 6, 395–400 CrossRef CAS PubMed.
- C.-T. Kuo, M. Neumann, K. Balamurugan, H. J. Park, S. Kang, H. W. Shiu, J. H. Kang, B. H. Hong, M. Han, T. W. Noh and J.-G. Park, Sci. Rep., 2016, 6, 20904 CrossRef CAS PubMed.
- I. Kerrache, C. Julien and C. Sourisseau, Solid State Ionics, 1996, 92, 37–43 CrossRef CAS.
- G. Ouvrard, E. Prouzet, R. Brec, S. Benazeth and H. Dexpert, J. Solid State Chem., 1990, 86, 238–248 CrossRef CAS.
- D. Hartley and M. J. Ware, J. Chem. Soc. A, 1969, 138–142 RSC.
- R. Aroca, M. Nazri, G. A. Nazri, A. J. Camargo and M. Trsic, J. Solution Chem., 2000, 29, 1047–1060 CrossRef CAS.
- C. Neumann, S. Reichardt, P. Venezuela, M. Drögeler, L. Banszerus, M. Schmitz, K. Watanabe, T. Taniguchi, F. Mauri, B. Beschoten, S. V. Rotkin and C. Stampfer, Nat. Commun., 2015, 6, 8429 CrossRef CAS PubMed.
- B. J. Robinson, C. E. Giusca, Y. T. Gonzalez, N. D. Kay, O. Kazakova and O. V. Kolosov, 2D Mater., 2015, 2, 015005 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary methods, Fig. S1–S9. See DOI: 10.1039/d1nr07281a |
| This journal is © The Royal Society of Chemistry 2022 |