
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast and versatile thermo-osmotic flows with a pinch of salt†
Cecilia
Herrero
 a,
Michael
De San Féliciano
a,
Samy
Merabia
*a and
Laurent
Joly
*ab
a,
Michael
De San Féliciano
a,
Samy
Merabia
*a and
Laurent
Joly
*ab
aUniv Lyon, Univ Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, F-69622, Villeurbanne, France. E-mail: laurent.joly@univ-lyon1.fr; samy.merabia@univ-lyon1.fr
bInstitut Universitaire de France (IUF), 1 rue Descartes, 75005 Paris, France
First published on 14th December 2021
Abstract
Thermo-osmotic flows – flows generated in micro and nanofluidic systems by thermal gradients – could provide an alternative approach to harvest waste heat. However, such use would require massive thermo-osmotic flows, which are up to now only predicted for special and expensive materials. Thus, there is an urgent need to design affordable nanofluidic systems displaying large thermo-osmotic coefficients. In this paper, we propose a general model for thermo-osmosis of aqueous electrolytes in charged nanofluidic channels, taking into account hydrodynamic slip, together with the different solvent and solute contributions to the thermo-osmotic response. We apply this model to a wide range of systems by studying the effects of wetting, salt type and concentration, and surface charge. We show that intense thermo-osmotic flows can be generated using slipping charged surfaces. We also predict for intermediate wettings a transition from a thermophobic to a thermophilic behavior depending on the surface charge and salt concentration. Overall, this theoretical framework opens an avenue for controlling and manipulating thermally induced flows with common charged surfaces and a pinch of salt.
Due to the increasing world energy consumption and the need for new clean energies, waste heat harvesting is a major challenge for decades to come. Some of the most common difficulties in harvesting waste heat come from the small temperature differences between the source and the environment (<50 °C)1 as well as from the need to use rare, expensive and often toxic thermoelectric materials.2 Alternatively, thermo-osmotic flows (generated at liquid–solid interfaces by temperature gradients) can be used to transform waste heat into electricity via a turbine3 or to pump water for desalination.4,5 Thermo-osmotic flows also directly generate electric current on charged surfaces6,7 by advecting the charge of the electric double layer (EDL) appearing in the liquid to screen the surface charge.8 Historically, the first experimental results on thermo-osmosis were published by Lippmann9 and Aubert10 at the beginning of the 20th century. Since then, a broad literature has been devoted to the study of aqueous solutions and various membranes, both from experiments11–15 or molecular dynamics (MD) simulations.16–20 Some disagreements have been reported for aqueous electrolytes, specifically in the flow direction (toward the hot side, so-called thermophilic flow, or toward the cold side, so-called thermophobic flow) for similar systems,21–24 or on the relation between the flow amplitude and surface charge.13 Although with exceptions, a thermophobic flow is generally expected for hydrophobic membranes and a thermophilic flow for hydrophilic membranes.25,26 Such differences cannot be understood by the classical theory27 developed by Derjaguin and Sidorenkov11,28 and by Ruckenstein for thermophoresis.29 This theory, based on the electrostatic enthalpy of the EDL, predicts that the flow is controlled by the electric surface charge, and always goes to the hot side. Other authors have recently proposed a different understanding in terms of irreversible thermodynamics by taking into account solvent contribution.15 However, their description only explains qualitatively the flow direction and does not provide a microscopic description of the effects of fluid wetting properties, salt concentration and electric surface charge.
Thermo-osmosis has seen a renewed interest due to the massive thermo-osmotic responses predicted by the use of novel materials, such as soft nanochannels,30 carbon-nanotubes5,18,31 or graphene,17 together with novel experiments by Bregulla et al.,24 who first reported a microscale manifestation of thermo-osmotic flows. Thermo-osmotic flows could, in particular, be boosted by the slip boundary condition (BC) that describes the velocity jump vs at the interface by a general expression first proposed by Navier:32,33
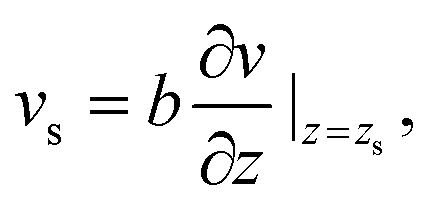 | (1) |
Following this work, we propose in this communication an analytical framework with the objective to predict thermo-osmosis of aqueous electrolytes confined by charged surfaces, extendable to thermoelectricity and thermophoresis. Solvent contribution and electrostatic ionic interactions are shown to play the leading role along with hydrodynamic slip. We apply the model to a wide range of systems by varying the wetting interaction, salt type and concentration, and surface charge. We report large thermo-osmotic responses, comparable to the highest responses predicted for special systems as inferred from previous simulations,5,16,17,31 as well as a change of sign in the flow direction. Such a change of sign cannot be predicted by only considering electrostatic interactions and can be crucial in order to interpret the different experimental results reported in the literature.
Theoretical framework
The thermo-osmotic response of a liquid–solid interface is quantified by the thermo-osmotic coefficient Mto, defined from the relation vto = Mto(−∇T/T), where ∇T/T is the relative temperature gradient parallel to the wall and vto is the generated thermo-osmotic velocity in the bulk region (where the liquid does not interact with the wall).27 In ref. 17, the authors propose a modification to the classical Derjaguin theory28 and show that, in order to take into account the hydrodynamic BC, the thermo-osmotic response coefficient can be expressed as (see the ESI†):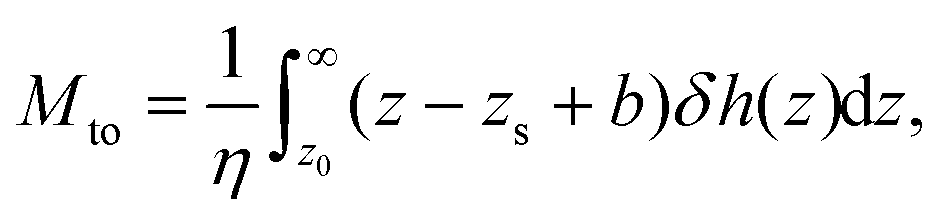 | (2) |
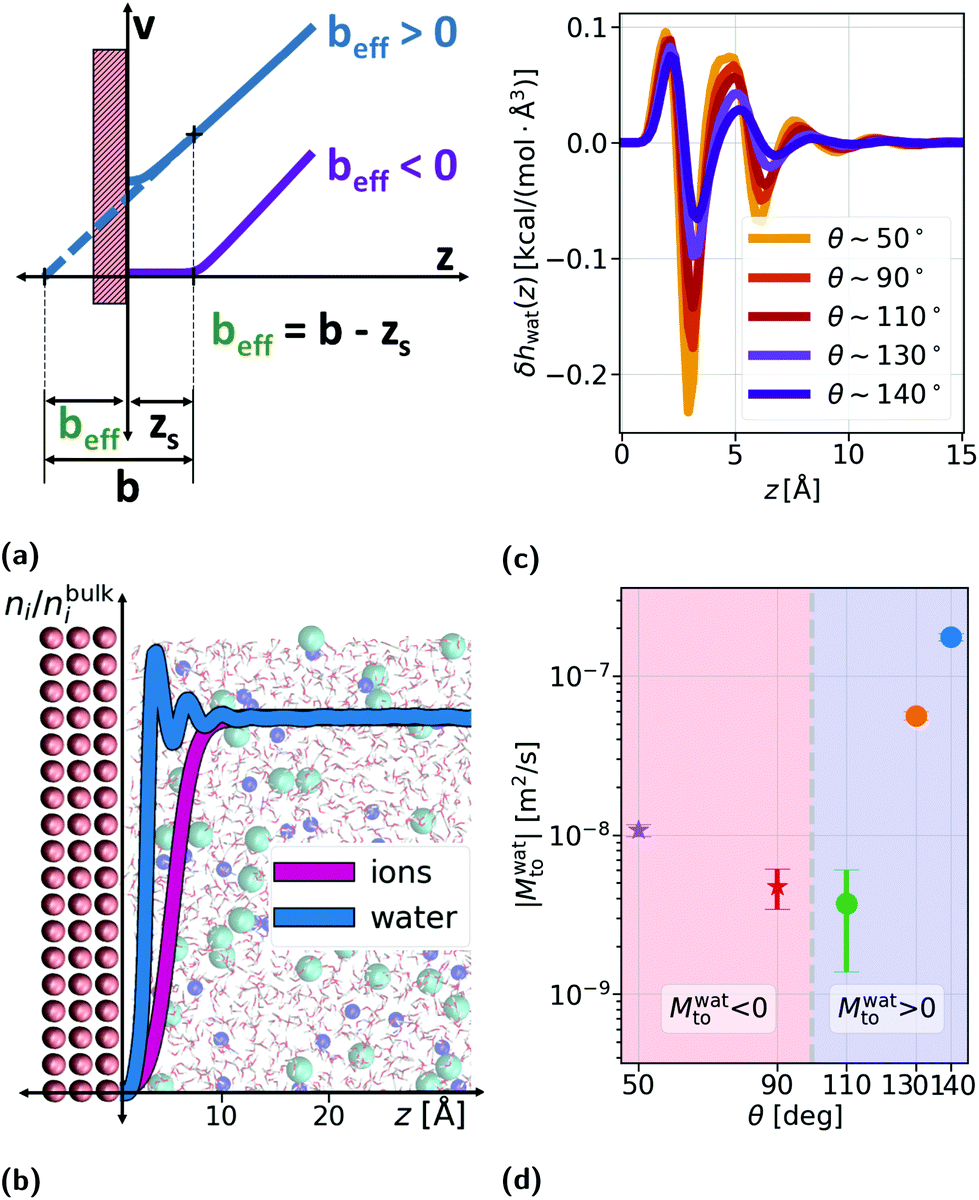 | ||
| Fig. 1 (a) Schematics of the effective slip length beff as a function of the slip length b and the shear plane position zs. We distinguish between the slip situation (beff > 0) and the stagnant layer situation (beff < 0). (b) Modelled system for the measures of water enthalpy excess density and slip, together with the normalized density profiles of water and ions (total density of cations and anions) for an uncharged surface, with z the distance to the wall. (c) Water enthalpy excess density δhwat profiles, with z the distance to the wall, for different wetting angles θ, controlled by the interaction energy between the liquid and the solid atoms εLS. (d) Water contribution to the thermo-osmotic response coefficient, Mwatto, for different wettings, determined based on water enthalpy excess and slip length computed from MD simulations; stars correspond to Mwatto < 0 and circles to Mwatto > 0. | ||
With regard to the enthalpy excess density δh, the standard approach28,29 assumes that it is mostly determined by the electrostatic enthalpy of ions in the EDL, δhel(z) = ρe(z)V(z) + p(z), where ρe is the charge density, V is the local electric potential and p is the pressure. Using the Poisson equation ρe = −εdz2V (assuming a constant solvent permittivity ε) and considering the mechanical equilibrium along the z direction, 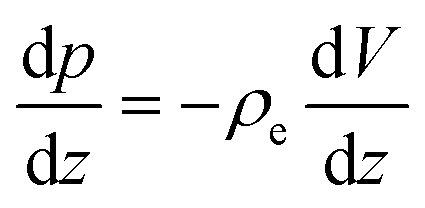 , δhel is then usually expressed in terms of the electrostatic potential as:
, δhel is then usually expressed in terms of the electrostatic potential as:
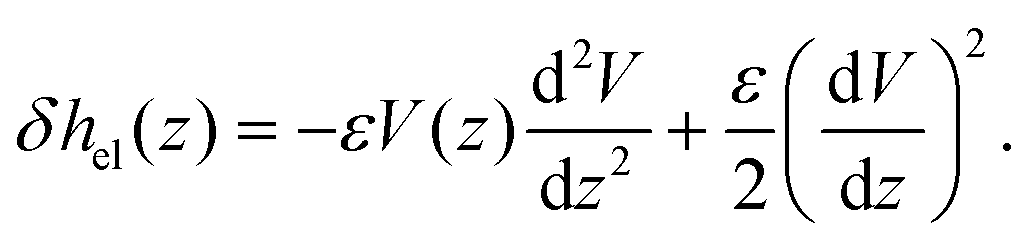 | (3) |
Accordingly, δhel vanishes outside the EDL, whose thickness is given by the Debye length λD, controlled by the salt concentration.8 The corresponding contribution to the thermo-osmotic response, Melto, can be then computed analytically within the mean-field Poisson–Boltzmann framework,37 considering a semi-infinite channel (see the ESI†).
Although the model proposed by Derjaguin et al. can predict Mto experimental orders of magnitude under certain conditions,24 it fails to describe the amplitude of the responses reported in the literature,5,16,17,31 the thermo-osmotic response reported for weakly charged membranes,13 and the experimental discrepancies observed in Mto sign.21–24 Aside from the electrostatic ionic interactions, other contributions to δh can be important. Such contributions are related to the solvent (water in the present work), ion solvation, and water dipole orientation in the electric double layer. After comparing all the different contributions to Mto (see the ESI†), the two main ones are (in the case of symmetric salts such as NaCl or KCl):
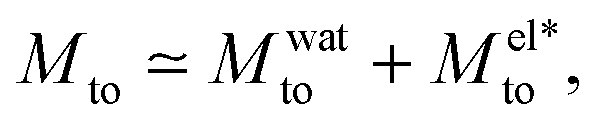 | (4) |
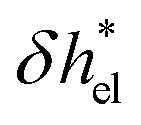 , accounting for the depletion of the ions in the vicinity of the wall (see the density profiles in Fig. 1b).
, accounting for the depletion of the ions in the vicinity of the wall (see the density profiles in Fig. 1b).
Defining the characteristic depletion length as d![[small script l]](https://www.rsc.org/images/entities/i_char_e146.gif) , one can effectively account for this effect by imposing a vanishing enthalpy excess in the interfacial region where there are no ions:
, one can effectively account for this effect by imposing a vanishing enthalpy excess in the interfacial region where there are no ions: 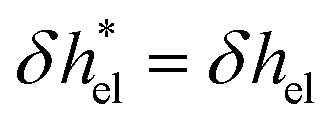 for z > d and
for z > d and 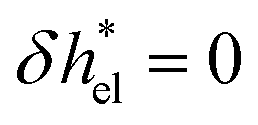 otherwise. With regard to the water contribution, δhwat can be computed as the sum of the different atomic contributions, δhwat = δhO + δhH, where the atomic enthalpy density for an element i is defined as:
otherwise. With regard to the water contribution, δhwat can be computed as the sum of the different atomic contributions, δhwat = δhO + δhH, where the atomic enthalpy density for an element i is defined as:
| δhi(z) = [δui(z) + δpi(z)]ni(z), | (5) |
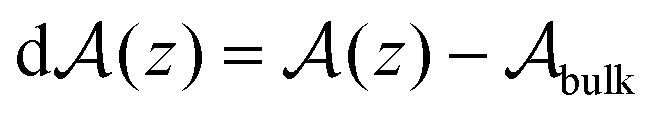 , with
, with 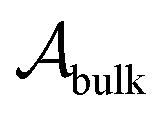 denoting the bulk value of the physical property
denoting the bulk value of the physical property 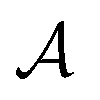 , ui denoting the energy per atom, pi denoting the stress per atom (a practical difficulty with measuring this term is discussed in the ESI†), and ni denoting the atomic number density profile.
, ui denoting the energy per atom, pi denoting the stress per atom (a practical difficulty with measuring this term is discussed in the ESI†), and ni denoting the atomic number density profile.
To compute the solvent term δhwat and the hydrodynamic BC as a function of wetting, we ran MD simulations using the LAMMPS package.38 The system consisted of an aqueous electrolyte (constituted by SPC/E water39 and NaCl, such that the bulk salt concentration was ns ∼ 1 M, following ref. 40), confined between generic uncharged Lennard–Jones (LJ) walls or graphene (see Fig. 1b and details in the ESI†). From these simulations with uncharged walls, we confirmed that in the case of symmetric salts the solute enthalpy, even at large concentrations, did not affect the total enthalpy profile and thus the enthalpy excess density is controlled by the solvent for neutral surfaces (see the ESI†).
The solid wall atoms were frozen and the oxygen–solid (LS) interactions were varied between the hydrophobic and hydrophilic values given in ref. 40 for LJ walls, corresponding to contact angles on uncharged surfaces of θ ∼ 140° and θ ∼ 50°, respectively (the values for θ and beff can be found in the ESI†). In Fig. 1c, one can observe the typical shape of the δhwat profile for different wettings. We note that the most hydrophilic situation (θ ∼ 50°) is considered a no-slip situation with b = 0.0 Å, corresponding to a stagnant layer (beff < 0). For simplicity, we also did not take into account in our model the coupling between the surface charge and slip.41 Using the proposed analytical framework, we explored a range of experimentally accessible values for the surface charge density Σ and the salt concentration ns: Σ was varied between −1 and −300 mC m−2 and ns ∈ {10−4,1} M corresponding to a Debye length λD ∈ {0.3,30} nm.
The objective of this communication is to present a general simple model, and in that regard, some approximations are applied in order to explore a broad range of parameters. Nevertheless, the validity of the approximations we use is consistent with the range of parameters we explored, such as the choice of a lower boundary for λD comparable to the size of water's first absorption layer (where water solvent properties should be accounted for in the calculations, and solvation and water properties should not be considered separately) and the upper boundary for Σ, under which the mean-field Poisson–Boltzmann description should remain valid37 (see the ESI†).
Results and discussion
From eqn (4), we expect that Mto is controlled by the competition between water and electrostatic contributions, depending on wetting, Σ and ns (or analogously λD). In Fig. 2, the total thermo-osmotic response is represented for all the wettings considered, together with the water contribution Mwatto (which, by construction, is independent of λD and Σ) and the modified electrostatic contribution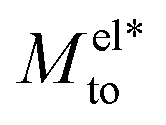 (which is only weakly affected by wetting, mostly through the change in beff). We observed from the figure the rich behavior resulting from that competition, where the water term mostly dominates for the most hydrophobic surfaces (θ ≳ 110°), while for the less hydrophobic surfaces (θ ≲ 110°), the electrostatic contribution can dominate for the larger λD. We can also see a large variation in Mto values for different wettings, ranging from 10–9 to 10−7 m2 s−1 for the most hydrophobic case.
(which is only weakly affected by wetting, mostly through the change in beff). We observed from the figure the rich behavior resulting from that competition, where the water term mostly dominates for the most hydrophobic surfaces (θ ≳ 110°), while for the less hydrophobic surfaces (θ ≲ 110°), the electrostatic contribution can dominate for the larger λD. We can also see a large variation in Mto values for different wettings, ranging from 10–9 to 10−7 m2 s−1 for the most hydrophobic case.
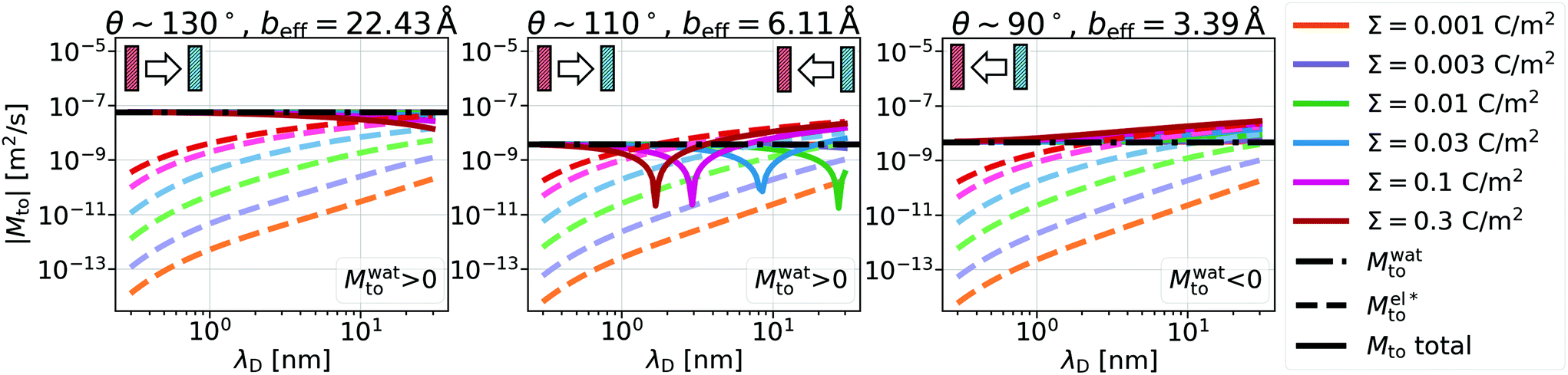 | ||
Fig. 2 Thermo-osmotic response coefficient Mto (solid lines) as a function of the Debye length for different wettings and surface charges. In all the graphs, the two main contributions, water Mwatto (dash-dotted lines) and modified electrostatic 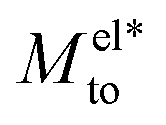 (dashed lines), are also represented. While (dashed lines), are also represented. While 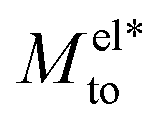 is always negative, the sign of Mwatto depends on wetting such that the total response can be thermophilic or thermophobic depending on wetting, surface charge and Debye length. is always negative, the sign of Mwatto depends on wetting such that the total response can be thermophilic or thermophobic depending on wetting, surface charge and Debye length. | ||
A striking result from Fig. 2 is the transition for intermediate wettings from a thermophobic behavior (Mto > 0) at high salt concentrations (small λD) to a thermophilic behavior (Mto < 0) at low salt concentrations; see for instance, θ ∼ 110°. In agreement with previous predictions,24 the electrostatic contribution 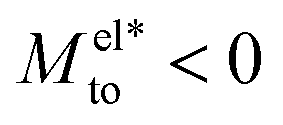 yields a thermophilic behavior independent of the sign of the surface charge. In contrast, the water term exhibits a change of sign when varying the wetting (see Fig. 1d and the ESI†). Such change of sign of Mwatto happens at θ ∼ 110° and thus, for θ ≳ 110°,
yields a thermophilic behavior independent of the sign of the surface charge. In contrast, the water term exhibits a change of sign when varying the wetting (see Fig. 1d and the ESI†). Such change of sign of Mwatto happens at θ ∼ 110° and thus, for θ ≳ 110°, 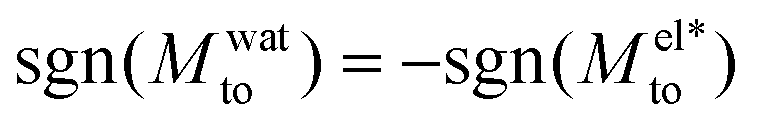 resulting in a change of sign of Mto when λD is such that
resulting in a change of sign of Mto when λD is such that 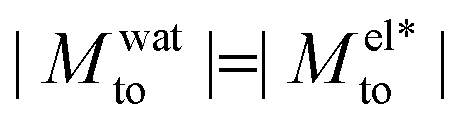 . Although this change of behavior happens for all θ ≳ 110°, for the most hydrophobic cases it takes place for λD values higher than the ones considered in this study and corresponding to extremely low salt concentrations. Even so, within our parameter range, we can still observe for θ ∼ 130° a decrease of the total response for high λD, which goes against the standard expectation and can only happen if water and electrostatic contributions have opposite signs. In contrast, for the less hydrophobic cases (e.g. θ ∼ 90°),
. Although this change of behavior happens for all θ ≳ 110°, for the most hydrophobic cases it takes place for λD values higher than the ones considered in this study and corresponding to extremely low salt concentrations. Even so, within our parameter range, we can still observe for θ ∼ 130° a decrease of the total response for high λD, which goes against the standard expectation and can only happen if water and electrostatic contributions have opposite signs. In contrast, for the less hydrophobic cases (e.g. θ ∼ 90°), 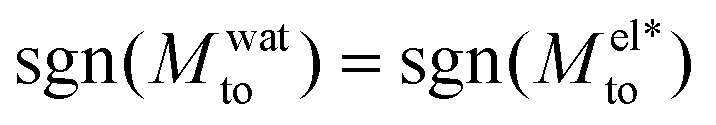 and Mto does not change sign for any λD value.
and Mto does not change sign for any λD value.
It is interesting to note that a similar change of sign has been found in the context of thermophoresis experiments.42–44 This change of sign is commonly attributed to the so-called thermopotential ψ0.45 Such a thermopotential appears when cations and anions have different mobilities and when the channel boundary conditions impose a vanishing flux of each ion type in the bulk.45 The thermopotential generates an electro-osmotic flow that can go against the thermo-osmotic flow and reverse the total flow direction. Nevertheless, ψ0 should disappear by allowing ionic fluxes through the channel, and as a consequence, the change of sign would disappear. By introducing the water contribution to the thermo-osmotic response, we propose a more fundamental understanding of such change of sign, which should persist independently of the boundary conditions on the fluxes through the channel.
The proposed 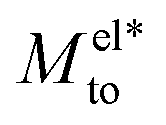 and Mwatto decomposition allows us to obtain an order of magnitude in agreement with the experimental results of Mto,24 in the order of 10−10–10−9 m2 s−1. Such an agreement is especially significant for hydrophilic surfaces and in the stagnant layer situation (see the ESI†), typical of experiments due to the presence of imperfections in the solid surface, when Mwatto decreases and
and Mwatto decomposition allows us to obtain an order of magnitude in agreement with the experimental results of Mto,24 in the order of 10−10–10−9 m2 s−1. Such an agreement is especially significant for hydrophilic surfaces and in the stagnant layer situation (see the ESI†), typical of experiments due to the presence of imperfections in the solid surface, when Mwatto decreases and 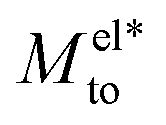 may dominate for a broader range of Debye lengths. Because Mwatto increases when increasing the slip, one interesting surface is the one constituted by graphene, with an effective slip length of beff = 538.77Å, which we obtained in MD simulations for NaCl aqueous solution at room temperature (see the ESI†). From the left part of Fig. 3, one can observe a significant increase in both electrostatic and water Mto contributions, resulting in a large value of the total response (Mto ∼ 10−6 m2 s−1) for this interface. Because Melto does not vary significantly with wetting and the order of magnitude of the total response is given by the water contribution, one can expect Mto ∼ Mwatto for graphene. In the right part of Fig. 3, one can see how Mwatto is affected by the effective slip. From this figure, one can observe that a large Mto value may be obtained for very slipping systems (as CNT, where slip values of b ∼ 300 nm have been reported at room temperature for a tube radius of R ∼ 15 nm (ref. 46)), although it is important to note that the presence of a stagnant layer or defects in the surface (resulting in smaller beff) may decrease the large predicted thermo-osmotic response, down to 10−9 m2 s−1.
may dominate for a broader range of Debye lengths. Because Mwatto increases when increasing the slip, one interesting surface is the one constituted by graphene, with an effective slip length of beff = 538.77Å, which we obtained in MD simulations for NaCl aqueous solution at room temperature (see the ESI†). From the left part of Fig. 3, one can observe a significant increase in both electrostatic and water Mto contributions, resulting in a large value of the total response (Mto ∼ 10−6 m2 s−1) for this interface. Because Melto does not vary significantly with wetting and the order of magnitude of the total response is given by the water contribution, one can expect Mto ∼ Mwatto for graphene. In the right part of Fig. 3, one can see how Mwatto is affected by the effective slip. From this figure, one can observe that a large Mto value may be obtained for very slipping systems (as CNT, where slip values of b ∼ 300 nm have been reported at room temperature for a tube radius of R ∼ 15 nm (ref. 46)), although it is important to note that the presence of a stagnant layer or defects in the surface (resulting in smaller beff) may decrease the large predicted thermo-osmotic response, down to 10−9 m2 s−1.
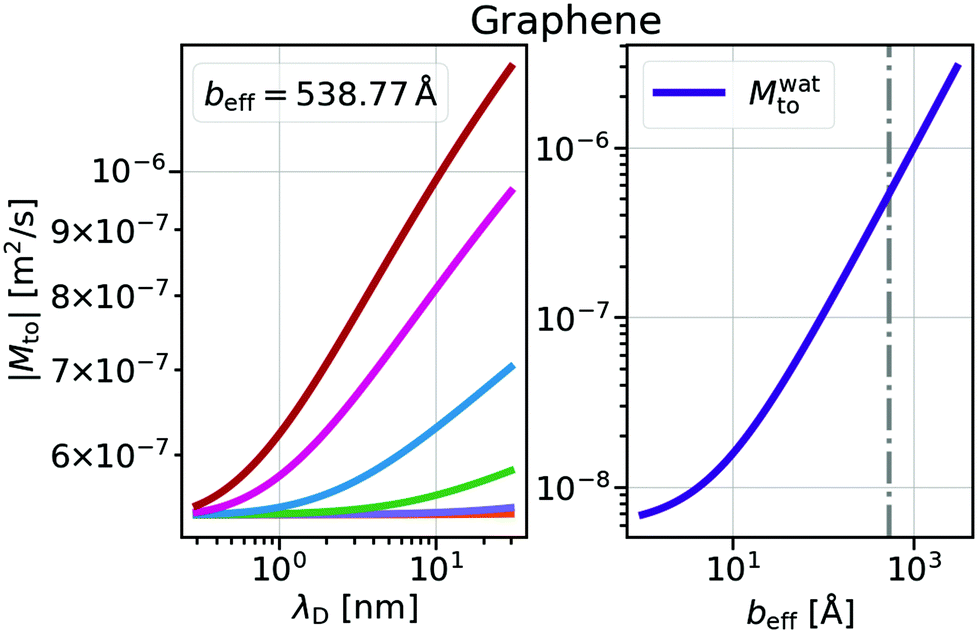 | ||
| Fig. 3 Thermo-osmotic response coefficient for graphene walls. Left: total response as a function of the Debye length for different surface charges as in Fig. 2. Right: water contribution Mwatto as a function of the effective slip length. The dash-dotted line indicates the beff value obtained from MD simulations. For graphene walls, Mwatto and Mto are always negative, corresponding to a thermophilic flow. | ||
Conclusions
We proposed here an analytical framework aimed at predicting the thermo-osmotic response of aqueous electrolytes for a wide range of nanofluidic systems and experimental conditions. While the standard picture relates the response to the ion electrostatic enthalpy in the electrical double layer close to charged walls, we show firstly that this contribution to the interfacial enthalpy may be negligible when compared to the water contribution for a broad range of parameters, and secondly that it should be slightly lowered due to the depletion of ions from the solid surface.The competition between the modified electrostatic and water contributions and the impact of the hydrodynamic boundary condition leads to a rich phenomenology that we have illustrated here. First, our theory predicts a higher thermo-osmotic response at low λD than the one expected from only considering the electrostatic contribution. Second, we predict a thermophobic flow for hydrophobic systems and a thermophilic flow for hydrophilic systems, in agreement with the typical tendencies observed in the experiments.15,25,26 Third, for intermediate wettings, the proposed model also predicts a transition between a thermophobic behavior at low salt concentrations to a thermophilic behavior at high salt concentrations. Such a transition has also been observed in thermophoresis experiments42–44 and is commonly attributed to the existence of a thermopotential which is, however, limited to particular boundary conditions imposing no ionic fluxes in the bulk liquid. In contrast, our interpretation of the change of sign is more general and independent of the nanofluidic channel boundary conditions, and opens the way to manipulate thermally induced nanoscale flows with a pinch of salt. Finally, we predict intense thermally induced flows for slipping systems, with orders of magnitude comparable to the ones reported from MD simulations of water thermo-osmosis in CNTs5,31 or on uncharged planar walls.16,17 Such large flows require a significant interfacial enthalpy excess, which we have shown can be obtained for a wide range of wettings, and a large slip length, favored by hydrophobicity. While very hydrophobic materials can pose practical issues in waste heat harvesting with nanoporous membranes, large slip lengths have also been reported for mildly hydrophobic carbon-based materials.46,47 Promising hydroelectric energy conversion performance is predicted for such materials,41 and our model suggests that thermoelectric energy conversion should also be excellent.
The importance of solvent contribution in thermo-osmosis of aqueous electrolytes, together with a modification of the classical electrostatic term, opens the way to several perspectives. With respect to the electrostatic term, a more accurate description of thermo-osmosis should take into account spatial heterogeneities of the dielectric and viscosity profiles at the interface.48–50 In addition, when salt ions adsorb specifically to the surface, e.g. iodide for NaI, the ion-size-dependent hydrophobic solvation term should be considered, e.g. through the modified Poisson–Boltzmann framework described in ref. 40 and 51. Regarding the water term, it is left to determine the impact of the surface charge and its distribution on water contribution to the response. In addition, one could consider more realistic surfaces than the apolar walls described in this study52 and, by following the same methodology, we propose to establish the effect of different charge distributions on the total thermo-osmotic response through their effect on the enthalpy excess and on the slip.41 Besides, one should take into account the limits of considering pure water simulations as an approximation of the water enthalpy contribution. For high concentrations, steric effects should be accounted for and ions can affect water viscosity.53 Nevertheless, such effects correspond to extreme ns values37 and they should not understate one of the main results of this communication: the great Mto value found for slipping surfaces. Finally, it is straightforward to extend the current model to the Debye-overlap regime (when the system height is smaller than λD), as long as the Poisson–Boltzmann framework still holds. In the same line, one can also follow the same procedure here proposed to predict the thermoelectric6,54,55 and thermodiffusive56 response, with promising applications for electricity production from waste heat or to refine large-scale continuum descriptions.57 Overall, our predictions call for future experimental verification and could be exploited for the design of innovative solutions for heat harvesting applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Aymeric Allemand, Anne-Laure Biance, Li Fu and Christophe Ybert for fruitful discussions. The authors are also grateful for HPC resources from GENCI/TGCC (grants A0070810637 and A0090810637) and the PSMN mesocenter in Lyon. This work is supported by the ANR (Project ANR-16-CE06-0004-01 NECtAR). LJ is supported by the Institut Universitaire de France.References
- A. P. Straub, N. Y. Yip, S. Lin, J. Lee and M. Elimelech, Nat. Energy, 2016, 1, 1–6 CrossRef.
- K. R. Kristiansen, V. M. Barragán and S. Kjelstrup, Phys. Rev. Appl., 2019, 11, 044037 CrossRef CAS.
- A. P. Straub and M. Elimelech, Environ. Sci. Technol., 2017, 51, 12925–12937 CrossRef CAS PubMed.
- K. Zhao and H. Wu, Nano Lett., 2015, 15, 3664–3668 CrossRef CAS PubMed.
- E. Oyarzua, J. H. Walther, C. M. Megaridis, P. Koumoutsakos and H. A. Zambrano, ACS Nano, 2017, 11, 9997–10002 CrossRef CAS.
- M. Dietzel and S. Hardt, Phys. Rev. Lett., 2016, 116, 225901 CrossRef PubMed.
- L. Fu, L. Joly and S. Merabia, Phys. Rev. Lett., 2019, 123, 1–15 Search PubMed.
- T. Markovich, D. Andelman and R. Podgornik, arXiv preprint arXiv:1603.09451, 2016.
- G. Lippmann, Compt. Rend., 1907, 145, 104–105 CAS.
- M. Aubert, PhD thesis, Verlag nicht ermittelbar, 1912.
- B. Derjaguin and G. Sidorenkov, C. R. Acad. Sci., 1941, 32, 622–626 Search PubMed.
- M. S. Dariel and O. Kedem, J. Phys. Chem., 1975, 79, 336–342 CrossRef CAS.
- J. I. Mengual, J. Aguilar and C. Fernandez-Pineda, J. Membr. Sci., 1978, 4, 209–219 CrossRef CAS.
- R. Piazza, J. Phys.: Condens. Matter, 2004, 16, S4195 CrossRef CAS.
- V. M. Barragán and S. Kjelstrup, J. Non-Equilib. Thermodyn., 2017, 42, 217–236 Search PubMed.
- R. Ganti, Y. Liu and D. Frenkel, Phys. Rev. Lett., 2017, 119, 1–5 CrossRef PubMed.
- L. Fu, S. Merabia and L. Joly, Phys. Rev. Lett., 2017, 119, 214501 CrossRef PubMed.
- R. Rajegowda and S. P. Sathian, Phys. Chem. Chem. Phys., 2018, 20, 30321–30330 RSC.
- R. Ganti, Y. Liu and D. Frenkel, Phys. Rev. Lett., 2018, 121, 68002 CrossRef CAS PubMed.
- K. Prakash, K. Dheeraj, S. K. Kannam and S. P. Sathian, Nanotechnology, 2020, 31, 425403 CrossRef CAS PubMed.
- B. Derjaguin, Pure Appl. Chem., 1980, 52, 1163–1178 CAS.
- R. Rusconi, L. Isa and R. Piazza, J. Opt. Soc. Am. B, 2004, 21, 605–616 CrossRef CAS.
- S. Nedev, S. Carretero-Palacios, P. Kühler, T. Lohmüller, A. S. Urban, L. J. Anderson and J. Feldmann, ACS Photonics, 2015, 2, 491–496 CrossRef CAS PubMed.
- A. P. Bregulla, A. Würger, K. Günther, M. Mertig and F. Cichos, Phys. Rev. Lett., 2016, 116, 1–5 CrossRef PubMed.
- J. Villaluenga, B. Seoane, V. Barragán and C. Ruiz-Bauzá, J. Membr. Sci., 2006, 274, 116–122 CrossRef CAS.
- S. Kim and M. Mench, J. Membr. Sci., 2009, 328, 113–120 CrossRef CAS.
- J. Anderson, Annu. Rev. Fluid Mech., 1989, 21, 61–99 CrossRef.
- B. V. Derjaguin, N. V. Churaev and V. M. Muller, Surface Forces, Springer, 1987 Search PubMed.
- E. Ruckenstein, J. Colloid Interface Sci., 1981, 83, 77–81 CrossRef CAS.
- R. S. Maheedhara, H. Jing, H. S. Sachar and S. Das, Phys. Chem. Chem. Phys., 2018, 20, 24300–24316 RSC.
- L. Fu, S. Merabia and L. Joly, J. Phys. Chem. Lett., 2018, 9, 2086–2092 CrossRef CAS PubMed.
- C. Navier, Mémoires de l'Académie Royale des Sciences de l'Institut de France, 1823, vol. 6, pp. 389–440 Search PubMed.
- B. Cross, C. Barraud, C. Picard, L. Léger, F. Restagno and É. Charlaix, Phys. Rev. Fluids, 2018, 3, 1–9 Search PubMed.
- C. Herrero, T. Omori, Y. Yamaguchi and L. Joly, J. Chem. Phys., 2019, 151, 041103 CrossRef PubMed.
- L. Bocquet and J.-L. Barrat, Soft Matter, 2007, 3, 685 RSC.
- X. Wang, M. Liu, D. Jing, A. Mohamad and O. Prezhdo, Nano Lett., 2020, 20, 8965–8971 CrossRef CAS PubMed.
- C. Herrero and L. Joly, arXiv preprint arXiv:2105.00720, 2021.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- H. J. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- D. M. Huang, C. Cottin-Bizonne, C. Ybert and L. Bocquet, Phys. Rev. Lett., 2007, 98, 1–4 Search PubMed.
- Y. Xie, L. Fu, T. Niehaus and L. Joly, Phys. Rev. Lett., 2020, 125, 014501 CrossRef CAS PubMed.
- F. S. Gaeta, G. Perna, G. Scala and F. Bellucci, J. Phys. Chem., 1982, 86, 2967–2974 CrossRef CAS.
- S. A. Putnam and D. G. Cahill, Langmuir, 2005, 21, 5317–5323 CrossRef CAS PubMed.
- A. Würger, Phys. Rev. Lett., 2008, 101, 108302 CrossRef.
- A. Würger, Rep. Prog. Phys., 2010, 73, 126601 CrossRef.
- E. Secchi, S. Marbach, A. Niguès, D. Stein, A. Siria and L. Bocquet, Nature, 2016, 537, 210–213 CrossRef CAS PubMed.
- B. Radha, A. Esfandiar, F. Wang, A. Rooney, K. Gopinadhan, A. Keerthi, A. Mishchenko, A. Janardanan, P. Blake and L. Fumagalli, et al. , Nature, 2016, 538, 222–225 CrossRef CAS PubMed.
- H. Hoang and G. Galliero, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 021202 CrossRef PubMed.
- D. J. Bonthuis and R. R. Netz, J. Phys. Chem. B, 2013, 117, 11397–11413 CrossRef CAS PubMed.
- M. Rezaei, B. G. Mitterwallner, P. Loche, Y. Uematsu, R. R. Netz and D. J. Bonthuis, J. Phys. Chem. B, 2021, 125, 4767–4778 CrossRef CAS PubMed.
- D. M. Huang, C. Cottin-Bizonne, C. Ybert and L. Bocquet, Langmuir, 2008, 24, 1442–1450 CrossRef CAS PubMed.
- C. Wang, H. Lu, Z. Wang, P. Xiu, B. Zhou, G. Zuo, R. Wan, J. Hu and H. Fang, Phys. Rev. Lett., 2009, 103, 137801 CrossRef PubMed.
- J. S. Kim, Z. Wu, A. R. Morrow, A. Yethiraj and A. Yethiraj, J. Phys. Chem. B, 2012, 116, 12007–12013 CrossRef CAS PubMed.
- A. Härtel, M. Janssen, D. Weingarth, V. Presser and R. V. Roij, Energy Environ. Sci., 2015, 8, 2396–2401 RSC.
- Y. Jin, R. Tao, S. Luo and Z. Li, J. Phys. Chem. Lett., 2021, 12, 1144–1149 CrossRef CAS PubMed.
- S. Di Lecce, T. Albrecht and F. Bresme, Nanoscale, 2020, 12, 23626–23635 RSC.
- M. Dietzel and S. Hardt, J. Fluid Mech., 2017, 813, 1060–1111 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical framework derivation; molecular dynamics simulation details; discussion on the generality of the proposed model; thermo-osmotic response results for all wettings. See DOI: 10.1039/d1nr06998e |
| This journal is © The Royal Society of Chemistry 2022 |