
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePulmonary delivery of mucosal nanovaccines
Wei
Tang
*a,
Yu
Zhang
b and
Guizhi
Zhu
 *b
*b
aDepartment of Pharmacy and Department of Diagnostic Radiology, Faculty of Science and Yong Loo Lin School of Medicine, National University of Singapore, Singapore, 119077, Singapore. E-mail: wei.tang@nus.edu.sg
bDepartment of Pharmaceutics and Center for Pharmaceutical Engineering and Sciences, Institute for Structural Biology and Drug Discovery, School of Pharmacy, The Developmental Therapeutics Program, Massey Cancer Center, Richmond, VA 23298, USA. E-mail: gzhu2@vcu.edu
First published on 6th December 2021
Abstract
Mucosal vaccination can elicit both systemic and mucosal immunity, and therefore has the potential to not only treat mucosal immune diseases, prevent the pathogen infection at the mucosal entry sites, but also treat distant or systemic immune disorders. However, only a few mucosal vaccines have been approved for human use in the clinic. Effective mucosal immunization requires the delivery of immunogenic agents to appropriate mucosal surfaces, which remains significantly challenging due to the essential biological barriers presenting at mucosal tissues. In the past decade, remarkable progress has been made in the development of pulmonary mucosal nanovaccines. The nanovaccines leverage advanced nanoparticle-based pulmonary delivery technologies on the characteristics of large surface area and rich antigen presentation cell environment of the lungs for triggering robust immune protection against various mucosal diseases. Herein, we review current methods and formulations of pulmonary delivery, discuss the design strategies of mucosal nanovaccines for potent and long-lasting immune responses, and highlight recent advances in the application of lipid-based pulmonary nanovaccines against mucosal diseases. These advances promise to accelerate the development of novel mucosal nanovaccines for the prophylaxis and therapy of infectious diseases, and cancer, as well as autoimmune disorders at mucosal tissues.
 Guizhi Zhu | Guizhi (Julian) Zhu is currently an Assistant Professor of Pharmaceutics at Virginia Commonwealth University (VCU). He was a National Institutes of Health (NIH) KL2 Scholar (2018–2021) and an NIH/NIGMS MIRA (R35) awardee (2021). He received a BS in Biotechnology from Nankai University, and PhD in Biomedical Sciences from the University of Florida, followed by a postdoc on immune drug pharmacoimaging and delivery in NIH. His multidisciplinary group (https://www.guizhizhu.org/) studies the engineering and delivery of nucleic acid immunotherapeutics and vaccines for the treatment and/or prophylaxis of cancer, infectious diseases, and autoimmune diseases. He has published >90 peer-reviewed papers with >9000 citations. |
1. Introduction
The emergency use authorization (EUA) of mRNA vaccines (mRNA-1273 from Moderna and BNT162b from Pfizer) for coronavirus disease 2019 (COVID-19) marks a milestone for nanovaccines. By taking advantage of lipid nanoparticle-based delivery technologies, the mRNA vaccines successfully protect antigen-encoding nucleic acids from degradation, facilitate their cellular uptake and release, and generate systemic neutralizing antibodies.1–3 However, the mRNA vaccines and other authorized COVID-19 vaccines are administrated via an intramuscular injection. They are able to elicit systemic immune responses to block the development of COVID-19, but not to induce mucosal immunity at the entry ports of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) to prevent the viral transmission or infection.4 To achieve robust mucosal immunization, a vaccine requires to be delivered to the appropriate mucosal surfaces,4–6 for example, the nasal or lung tissue in the case of COVID-19. Nanovaccines that leverage nanotechnologies to overcome biological barriers to antigen delivery, representing an excellent opportunity for inducing strong immune protection at mucosal surfaces (Fig. 1). | ||
| Fig. 1 Schematic illustration of pulmonary delivery of mucosal nanovaccines. (a) Representative delivery vectors of mucosal nanovaccines. (b) Illustration of biological barriers at the lung tissue. (c) Immune responses stimulated upon the delivery of mucosal vaccines. Nanoparticles can be tailored to facilitate antigens and/or adjuvants/immunostimulators to overcome the delivery barriers and elicit robust protective mucosal immune responses. | ||
Currently, mucosal nanovaccines are urgently needed for not only COVID-19, but also many other mucosal diseases caused by enteric pathogens, sexually transmitted agents, and mucosa-accessible oncogenic viruses (Fig. 2a).4,7,8 Mucosal membranes of respiratory, digestive, and urogenital tracts cover the largest exposure surface areas of a human body.5,6 These tissues form a highly specialized adaptive and innate mucosal immune system, which contains mucus as the first barrier against enteric pathogenic microorganisms and mucosa-associated lymphoid tissues (MALTs) for providing protective immune responses.5,6,9 Mucosal vaccination has potential beneficials over conventional parenteral vaccination since it stimulates both systemic and mucosal immune defenses.10 A key characteristic of mucosal adaptive immune response is the local production and secretory of immunoglobin A (IgA) antibodies. Secretory IgA (sIgA) facilitates entrapment and clearance of pathogenic antigens or microorganisms in the mucus, thus effectively excluding pathogen invasion at the mucosal sites.5,6,11 The other important characteristic is the generation of tissue-resident memory T cells (TRMs), which offer rapid protection in the tissue by recruiting circulating memory T cells and producing inflammatory cytokines/chemokines.12,13 Moreover, the crosstalk between mucosal compartments allows immunization at one mucosal surface to establish immunity at distant mucosal sites. Taken together, mucosal vaccination holds great promise in the control of mucosal diseases. Recently, a head-to-head comparison study was performed to investigate the protection activity of ChAd-SARS-CoV-2 vaccine, a chimpanzee adenoviral vaccine encoding stabilized spike protein (Fig. 2b).14 It found that a single-dose intranasal vaccination could evoke strong systemic and mucosal IgA and T cell responses in mice to almost completely prevent SARS-CoV-2 infections in both upper and lower respiratory tracts, whereas intramuscular immunization was only able to activate systemic immune responses that partially reduced lung infections.14 This study clearly suggests that the route of immunization exerts a substantial impact on vaccine efficacy. However, a majority of vaccines in use today are administrated by an intramuscular or subcutaneous route. There are only nine mucosal vaccines are approved for human use, including eight oral vaccines against cholera, rotavirus, poliovirus, and salmonella, and one intranasally administrated live attenuated influenza vaccine.4 However, oral vaccines need to survive harsh acidic environment in the stomach as well as to overcome variable pH conditions and the tight monolayer of endothelial cells lining throughout the gastrointestinal tract.15 Intranasal delivery have risks of exposing therapeutics to the central nervous system (CNS),16,17 which may cause adverse side effects. Therefore, continuous efforts are highly desired to develop safe and effective mucosal vaccines. Recently, pulmonary vaccine delivery has attracted increasing attention due to the large surface area and the rich antigen presentation cells (APCs) as desired vaccine target cells in the microenvironment of lung tissue.18 Pulmonary vaccination has demonstrated successful control of respiratory pathogen infections, lung cancer metastasis, and even sexually transmitted viral infections.13,19
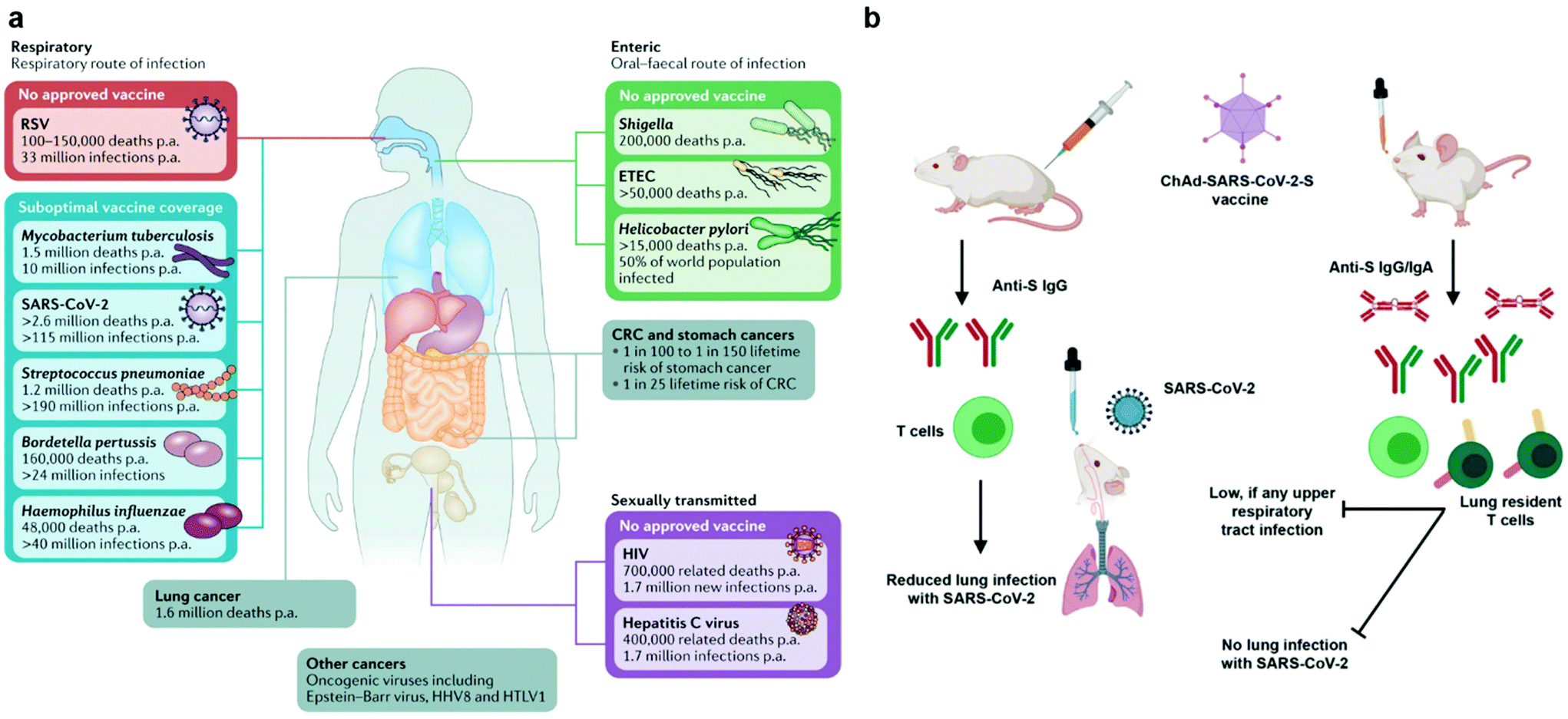 | ||
| Fig. 2 Mucosal vaccines are urgently needed for mucosal diseases and they have potential benefits over conventional parenteral vaccination. (a) Burden of mucosal diseases with unmet vaccine needs. In addition to their centrality in the pathogenesis of infectious disease, mucosal tissues are frequent sites of infectious disease and tumor development. CRC, colorectal cancer; ETEC, enterotoxigenic Escherichia coli; p.a., per annum; RSV, respiratory syncytial disease; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2. Reproduced with permission.4 Copyright 2021, Nature Publishing Group. (b) Comparison of immunogenicity of single-dose administration of ChAd-SARS-CoV-2-S, a chimpanzee adenoviral vaccine encoding stabilized spike protein, by intramuscular and intranasal delivery routes. The mucosal immunity generated by intranasal inoculation of ChAd-SARS-CoV-2 likely controlled infections at both upper and lower respiratory tract, potentially protecting against SARS-CoV-2 infection and transmission. Reproduced with permission.14 Copyright 2020, Elsevier Inc. | ||
In this review, we briefly overview the methods and formulations of pulmonary drug delivery. Then, we discuss the key factors that need to be considered in the design of delivery vectors, including the overcoming of pulmonary mucosal barriers, specific targeting of pulmonary epithelial and immune cells, as well as co-delivery of adjuvants and/or cytokines with antigens. We also highlight some significant achievements of lipid-based pulmonary mucosal nanovaccines in literature. Finally, we provide our perspectives on the future possibilities and challenges in pulmonary delivery technologies as well as in the development of mucosal nanovaccines.
2. Formulations and methods for pulmonary delivery
Unlike parenteral vaccination, pulmonary immunization does not need the use of a needle and syringe, thus offering improved safety, lowered cost per dose, reduced pain for patients, and increased feasibility of mass vaccinations. Formulations for pulmonary delivery can be (1) solutions/suspensions that can be administrated by nebulizers or pressurized metered dose inhalers, or (2) dry powder formulations that administrated by active or passive pressurized dry powder inhalers (DPIs). The nebulizers and inhalers have their unique advantages. In general, nebulizers can consistently deliver high doses of drugs with deeper deposition, while inhalers are portable and less expensive. The most important requirement for the formulations is that antigens are present in a particulate form and the particles can be dispersed in fine aerosol with an aerodynamic particle size below 5 μm.20 Details of the pulmonary delivery technologies and devices have been reviewed elsewhere.20–22For liquid preparations, stability is a major challenge due to the high molecular mobility, which requires the use of cold chains for storage and transportation. One solution to enhance the stability is to carefully adjust the pH and osmolarity of the vehicles, as well as the ionic strength of the buffering salts.20,23 In most cases, the vaccines remain their maximum activity in the pH range of 5–8, and have a better stability at low osmolarity of the vehicles as well as relatively low ionic strength (I < 0.15) of the buffering salts.24,25 Another solution is to first freeze dry the vaccines and then reconstitute them before use. In this approach, both antigen and delivery vectors need to avoid intermolecular reactions and remain stable during the freezing and dehydration. Recently, the Anderson group synthesized hyperbranched poly(beta amino esters) (hPBAEs), hC32-118 and hDD90-118 (Fig. 3a), and loaded them with firefly luciferase-encoding mRNA.26 The resulting mRNA-hPBAE polyplexes (0.5 mg mL−1 mRNA) were nebulized to mice by using a vibrating mesh nebulizer connected to a whole-body chamber (Fig. 3b). This nebulization process didn't change the average size and polydispersity index (PDI) of mRNA-hPBAE polyplexes (Fig. 3c and d). In control groups, naked firefly luciferase-encoding mRNA or branched 25 kDa polyethylenimine (bPEI)-packed mRNA were subjected to nebulized delivery to mice. After 24 hours, all mice were sacrificed and major organs were collected for bioluminescence imaging. As shown in Fig. 3e, positive signals were only localized in the lungs but not in the spleen and liver, which was significantly different from the biodistribution after intravenous injection. The luminescent radiance in lungs was quantified to be 4.8 × 105 p s−1 cm−2 sr−1 and 4.3 × 104 p s−1 cm−2 sr−1 in the hDD90-118 and hC32-118 groups, respectively, which were much higher than that in the bPEI control group (2.9 × 104 p s−1 cm−2 sr−1) (Fig. 3e). In good agreement with the radiance values, mRNA-hPBAE polyplexes induced a more efficient luciferase protein production than mRNA-bPEI (Fig. 3e). This study demonstrated that hPBAE is an effective pulmonary delivery vector for nucleic acid therapeutics, which could find broad applications in the control of many diseases.27,28
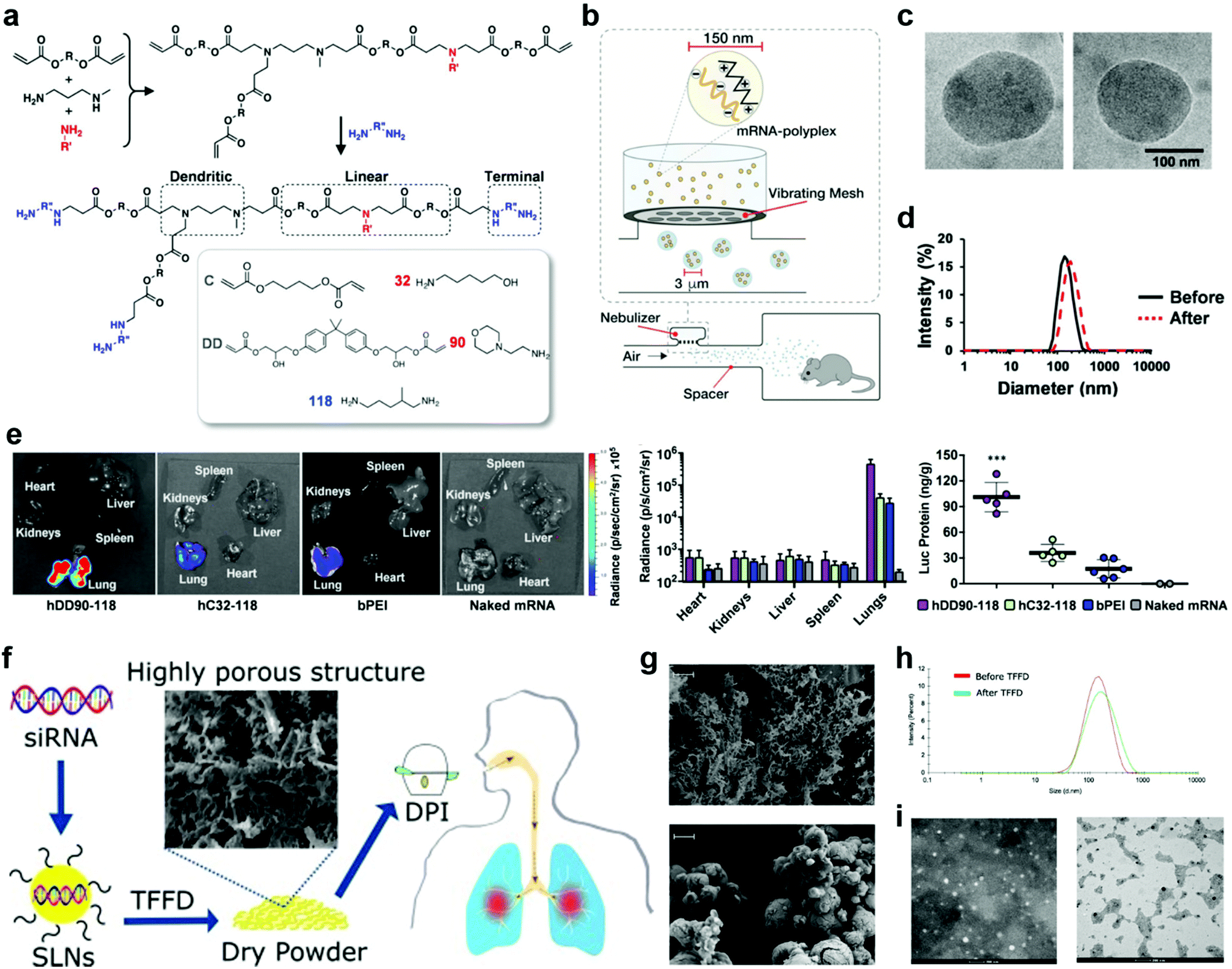 | ||
| Fig. 3 Pulmonary delivery with different devices and formulations. (a–e) Delivery of liquid formulations with a nebulizer. (a) Synthesis scheme of hDD90-118 and hC32-118 hyperbranched poly(beta amino esters) (PBAEs). (b) A vibrating mesh nebulizer connected to a whole-body chamber was used to deliver in vitro transcribed (IVT)-mRNA encoding for firefly luciferase to mice. The nebulizer generates micrometer sized droplets optimal for lung deposition, containing nanoparticles for intracellular delivery. (c) Electron microscopy of hDD90-118 particles before (left) and after (right) nebulization, particles had an average size of 137 nm (±21) and 146 nm (±40), respectively (±SD, n = 13–15). (d) Particles have narrow size distribution with polydispersity indices of 0.10 before (black) and 0.11 after (red, dashed) nebulization. (e) Bioluminescence 24 h after inhalation of polyplexes, hDD90-118 vectors produced significantly higher level of radiance (±SD, n = 4) and luciferase production (***P < 0.001, ±SD, n = 5–6) in the lungs, compared to hC32-118 and bPEI. Statistical analysis using one-way ANOVA with post-hoc Tukey test. Reproduced with permission.19 Copyright 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (f–i) Delivery of dry powder formulations with a dry powder inhaler (DPI). (f) Schematic illustration of the preparation, thin-film freeze-drying (TFFD), and pulmonary delivery of aerosolizable siRNA-encapsulated solid lipid nanoparticles (LNPs). (g) Representative scanning electron microscope (SEM) images of dry powders of LNPs prepared by TFFD (upper panel) or spray drying (lower panel). The scale bar indicates 2 μm. (h) A comparison of siRNA-encapsulated LNPs particle size distribution by intensity. (i) Transmission electron microscopy (TEM) images of siRNA-encapsulated LNPs before (left panel) and after (right panel) they were subjected to TFFD and reconstitution. Reproduced with permission.32 Copyright 2021, Elsevier B.V. | ||
Further, dry powder formulations are also associated with the complication of stability. This can be mitigated by adding stabilizers such as carbohydrate matrices.20 Moreover, their particle size is an essential concern. It requires careful adjustment to achieve good dispersion, low aggregation, and low-adhesive properties, in order to allow efficient release from an inhaler as well as to achieve desired deposition and distribution in the lungs.20,22,29–31 Common drying processes used in vaccine preparations include spray drying, freeze drying, air drying, or a hybrid of them. Very recently, Cui et al. judiciously studied the impact of drying processes on the aerosol performance properties of solid lipid nanoparticles (LNPs) (Fig. 3f).32 They prepared LNP powders by thin-film freeze drying (TFFD), spray-freeze drying, or conventional shelf freeze drying. The TFFD-dried SLNs had a fluffy porous microstructure, showing approximately 20 times larger surface areas than the beads-like spray-freeze dried LNPs (Fig. 3g). After reconstitution, the TFFD-dried SLNs retained their particle size and PDI (0.24 ± 0.09); while the spray-freeze drying process induced slightly increase in the particle size and 50% enhancement in PDI (0.41 ± 0.06); in contrast, the shelf freeze drying process almost tripled particle size and PDI (0.82 ± 0.19), which required further processing such as milling for size reduction in order to be used in inhalable delivery. In addition, small interfering RNA (siRNA)-encapsulated LNPs can be successfully converted into aerosolizable dry powder by TFFD and remained their particle size, size distribution (Fig. 3h and i) and the function of siRNA unchanged after reconstitution. Therefore, TFFD represented the optimal drying process for aerosolizable SLNs. Despite excellent dispersion and low aggregation, the LNP composition and TFFD parameters need to be further optimized for animal studies. And the TFFD may not be superior to other drying methods for delivery vectors that different from LNPs.
3. Nanovaccine design strategies for improved pulmonary immunization
Nanovaccines, either alone or in combination with other cancer immunotherapies, such as chimeric antigen receptor (CAR) T-cell therapy33 and immune checkpoint blockade (ICB) therapy,34,35 have achieved great success in cancer control.36–39 By employing nanoparticles to deliver antigens and/or adjuvants, nanovaccines have multiple advantages over subunit vaccines, including enhancing the in vivo half-lives of vaccines, protecting antigens from degradation for increased stability, providing opportunities to co-deliver multiple antigens and adjuvants for enhanced immunogenicity, promoting APC uptake and cytoplasmic delivery of antigens for improved antigen cross-presentation, and offering specific targeting to lymphoid tissues or APCs for precision immunomodulation.40 Mucosal nanovaccines inherit all the above merits. Meanwhile, mucosal vaccination can elicit both systemic and mucosal immune responses by eliciting not only circulating IgG, but also sIgA and TRMs at mucosal tissues.5,6,11,12 However, a number of challenges are associated with effective pulmonary vaccine delivery for potent and long-term immune protection. Nanotechnologies can be leveraged to overcome biological barriers to drug delivery.41 In this part, we will provide our insights on nanovaccine design strategies to potentiate mucosal immunity from three perspectives: (1) to overcome mucosal barriers to nanoparticulate delivery vectors by modulating their surface chemistry such as charge and hydrophilicity; (2) to increase specific recognition and uptake by lung epithelial cells and immune cells through surface functionalizing nanocarriers with active targeting ligands; (3) to elicit sufficiently strong mucosal immune responses for long-term protection by co-delivering adjuvants and/or immunostimulators with antigens.Mucus presents a highly heterogeneous and viscous microenvironment. It functions as the body's first line of defense to entrap and rapidly remove foreign invaders such as pathogens and nanovaccines. To effectively overcome the mucosal barriers, nanoparticles must find a balance between mucoadhesion and mucosal penetration, which strongly depends on the residence time of mucosal layers as well as their thickness.42 Nanoparticles need to across the mucosal layers before being subjected to mucus clearance. In general, nanoparticles with neutral surface charge and great hydrophilicity would have enhanced mucosal penetration, while positively charged or highly hydrophobicity promotes mucoadhesion due to their interactions with negatively charged mucus layers.43,44 For example, surface modification of nanoparticles with surfactant poloxmer 188 increased mucosal diffusion and cellular uptake.23 Polyethylene glycol (PEG) coating influenced both mucoadhesion and mucosal penetration performance according to the length of chains and surface density.45 It was demonstrated that nanoparticles with long PEG chains (≥10 kDa) had more interactions with mucus glycoproteins for improved adhesion, and those with shorter PEG chains (≤2 kDa) were associated with better mucosal diffusion. Moreover, increasing the surface PEG density of nanoparticles could efficiently enhance mucosal penetration.46 On this basis, Suk et al. shielded tetra(piperazino)fullerene epoxide (TPFE)-based nanoparticles with 5 kDa PEG polymers by chemical conjugation.47 The cationic carbon nanocluster TPFE is an attractive delivery vector for gene transfer agents,48,49 for example, plasmid DNA encoding fluorescent Zsgreen protein (pZG). The resulting pZG/TPFE or pZG/TPFE-PEG showed cluster-shaped morphology under TEM (Fig. 4a). Upon incubation in mucin solution, the pZG/TPFE rapidly aggregated, while the hydrodynamic diameters of pZG/TPFE-PEG remained unchanged (Fig. 4b). This was attributed to the PEGylation, which helped to resist mucoadhesive interactions. To further investigate the PEGylation effect in vivo, TPFE nanoparticles were engineered with either pZG or plasmid DNA encoding luciferase (pGL3) for pulmonary administration. The nanoparticle distribution in lungs was examined by confocal imaging, which showed widespread and uniform transgene expression in lung tissue sections of pZG/TPFE-PEG-treated wild-type C57BL/6 mice (>65% coverage). In contrast, negligible fluorescent signals were observed in both pZG and pZG/TPFE control groups (<2% coverage) (Fig. 4c and d). These results well agreed with the overall level of transgene expression that was measured by lung homogenate-based luciferase assays. Pulmonary delivery of pGL3/TPFE-PEG induced two orders of magnitude higher luciferase activity compared to naked pGL3 or pGL3/TPFE in lungs of wild-type C57BL/6 mice (Fig. 4d). Similar results were observed in a chronic lung disease transgenic Scnn1b-Tg mouse model, which featured mucus accumulation and stasis. Although encountering more challenging delivery barriers, the TPFE-PEG delivery system remained their mucosal penetration capacity and induced a widespread (∼35% coverage) as well as high-level overall transgene expression in the lungs of Scnn1b-Tg mice (Fig. 4c and d). Collectively, PEG surface coating could endow nanoparticles with the capacity to overcome essential mucosal barriers in the lungs, even in pathological conditions characterized by the strengthened barrier properties.
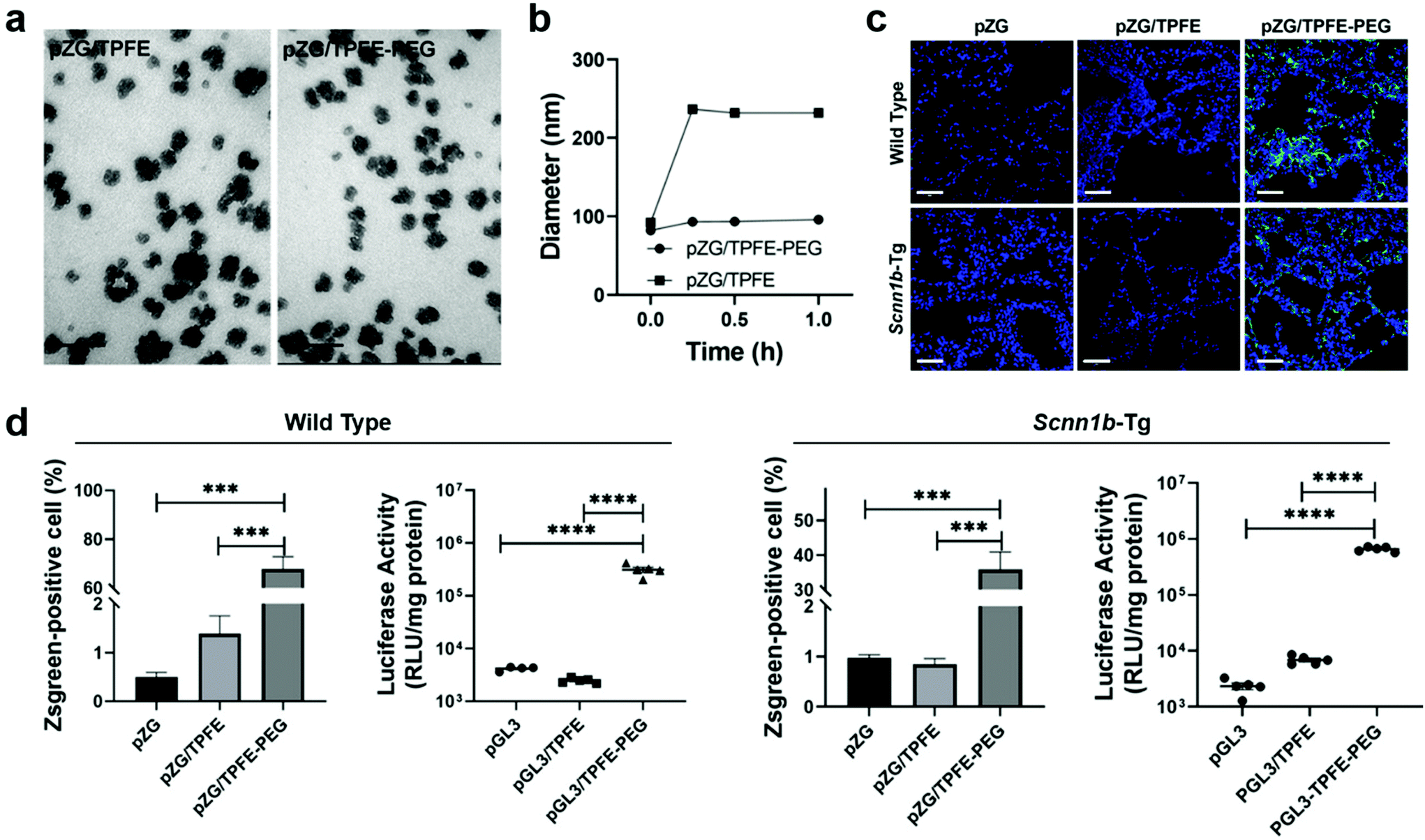 | ||
| Fig. 4 PEGylation strategy to facilitate overcoming of mucosal barriers to delivery vectors. (a) Representative TEM images of tetra(piperazino)fullerene epoxide (TPFE)-based delivery nanoparticles (scale bars = 100 nm). (b) Hydrodynamic diameter changes in a mucin solution at 37 °C over time (n = 3). (c) Representative confocal images demonstrating distribution of transgene expression mediated by naked pZG, pZG/TPFE and pZG/TPFE-PEG in the lungs of wild-type C57BL/6 and Scnn1b-Tg mice after intratracheal administration. Scale bar = 100 μm. (d) Percentages of cells with reporter transgene expression according to images in (c) (n = 3) and overall level of transgene expression determined by homogenate-based luciferase assay (n = 5) in the lungs of both wild-type C57BL/6 and Scnn1b-Tg mice. ****P < 0.001.47 Copyright 2021, WILEY-VCH GmbH. | ||
However, success in mucosal barrier crossing doesn't necessarily guarantee potent mucosal immune protection. Without proper targeting, the cargo antigens may not induce efficient cellular internalization and antigen cross-presentation. Undoubtedly, targeting APCs in the lungs is an attractive approach to address this problem. The target receptors that have been extensively explored in parenteral nanovaccines can apply to the design of mucosal nanovaccines, for example, the well-characterized dendritic cell (DC) receptor CD20550 and mannose receptor.51,52 In addition to APCs, pulmonary epithelial cells represent another important cell component for targeting mucosal nanovaccines. In particular, the neonatal Fc receptor (FcRn), which is overexpressed on some type I mucosal epithelial cells and involved in the bidirectionally transepithelial transport of IgG and proteins,53 providing great opportunities for targeted delivery of protein-based or protein hitchhiking-mediated nanovaccines.13,54 Moreover, M cell-targeted strategy can achieve highly effective mucosal immune responses.42,55,56 The most widely investigated M cell-targeting ligand is ulex europaeus agglutinin 1 (UEA-1),56,57 which specifically bind to α-L-fucose residues on the surfaces of murine intestinal M cells. Nevertheless, the targeting ligands of pulmonary M cells were seldomly reported. To identify an effective pulmonary M-cell targeting strategy, the M cell characteristics should be taken into consideration as highlighted by Longet et al. in 2018.55
Despite effective delivery, mucosal nanovaccines with antigens alone may not induce sufficiently strong immune responses for long-lasting protection against viruses associated with mucosal diseases.58,59 Co-delivery of adjuvants/immunostimulators with antigens could significantly potentiate mucosal immunization.54,60 This strategy has been verified by using polymeric nanoparticle delivery systems, such as poly-(D,L-lactide-co-glycolide) (PLGA), poly(lactic acid) (PLA), and chitosan, which could simultaneously act as mucosal adjuvants and delivery vectors in nanovaccines.56,61,62 In most cases, however, adjuvants need to be delivered separately.60 For example, a phosphatidylserine (PS)-coated and stimulator of interferon genes (STING) agonist cyclic guanosine monophosphate–adenosine monophosphate (cGAMP)-loaded liposome (NP-cGAMP) mucosal nanovaccine was able to target pulmonary APCs and boost anticancer immunity against lung metastases (Fig. 5a).63 Notably, in NP-cGAMP, the exposed PS functioned as the “eat-me” signal, which could be recognized and internalized by APCs through their PS receptors. The cGAMP was expected to release in cytosol, bind to STING in APCs, stimulate STING signaling and type I interferons production, and finally promote pro-inflammatory tumor microenvironment for increased immune protection. The NP-cGAMP and its counterparts had an average diameter of around 120 nm (Fig. 5b and c). After inhalation, the aerosolized NP-cGAMP predominately resided in lung tissues at a relatively high concentration for at least 48 hours, which was attributed to the PS-mediated APC targeting (Fig. 5d). Furthermore, pulmonary delivery of NP-cGAMP synergized with fractionated radiation (IR) could successfully activate anticancer immunity in a B16F10-OVA tumor metastasis model. In animal studies, fractionated IR was only delivered to the right lung of mice, which resulted in successful tumor inhibition in the right lung but little therapeutic efficacy against metastatic foci in the non-irradiated left lung. The mice treated with inhalable NP-cGAMP caused a moderate reduction in the number of tumor foci in both lungs. Impressively, combination of NP-cGAMP and fractionated IR (right lung only) induced almost complete tumor regression in both lungs (Fig. 5e and f). These data demonstrated that pulmonary delivery of immunostimulators, especially by using APC-targeting delivery vectors, could synergize with in situ tumor antigen release and enhance protective immunity against tumor development at the lung tissue. However, this study didn't take mucosal immune responses into careful considerations when exploring the mechanisms behind the synergistic therapeutic effects. A more comprehensive analysis of the protective immunity would be needed before applying the NP-cGAMP delivery strategy to other pulmonary nanovaccines. Although adjuvants could potentiate antigen-specific immune responses, an improper adjuvant may result in severe side effects. For example, an enterotoxin-adjuvanted nasal influenza vaccine caused a substantial number of Bell's palsy in a clinical trial, which was attributed to the high binding affinity between enterotoxin and GM1 ganglioside moieties of olfactory nerves.64–66 Pollen grains have been reported as an adjuvant to elicit allergic immune response and promote DC maturation,67 which therefore should be taken into consideration for the design of pulmonary vaccines. A pulmonary vaccine with too strong adjuvants may be at higher risks for triggering allergy, especially when it is administrated in pollen seasons.
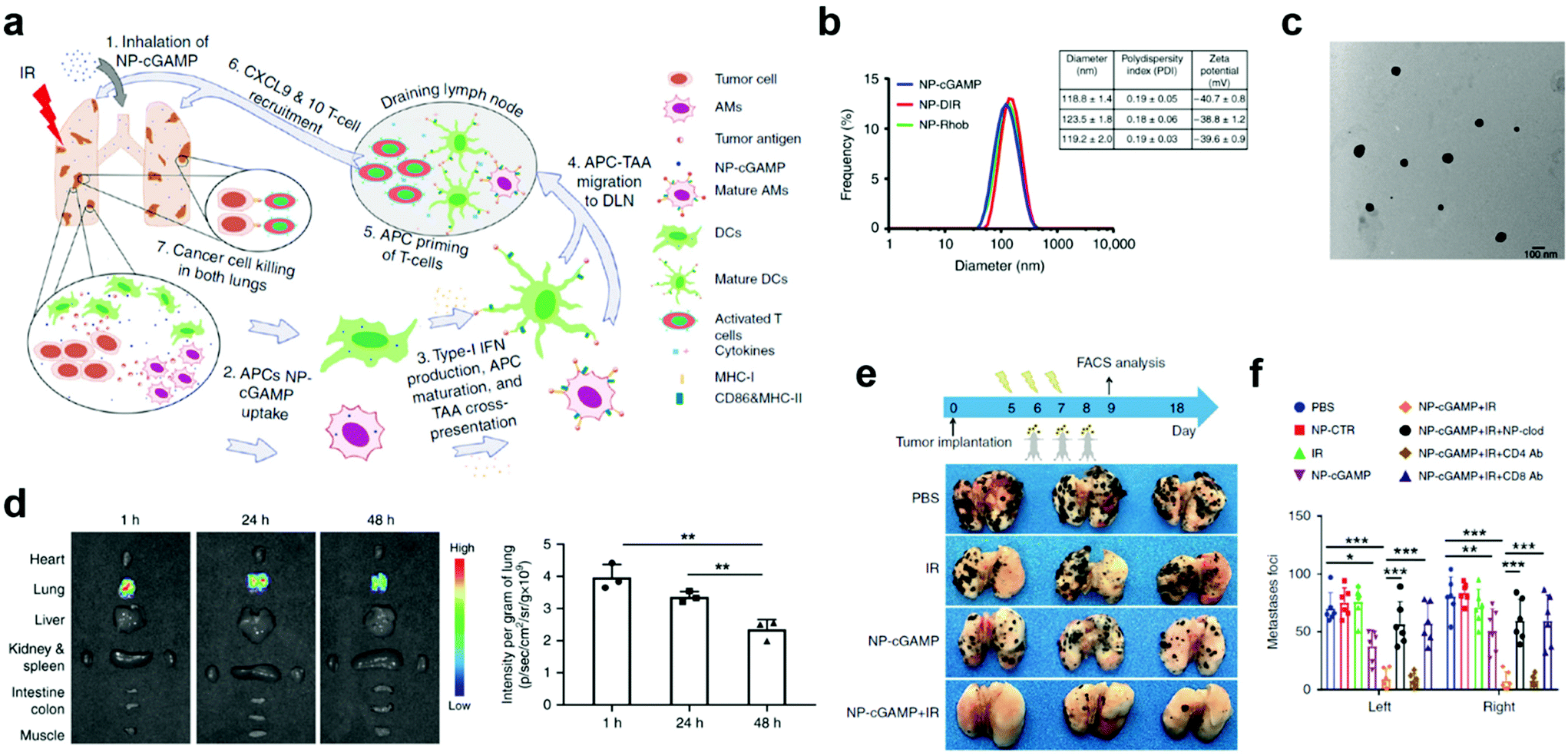 | ||
| Fig. 5 APC-targeting and immunostimulator co-delivery strategies to improve protective immunity at mucosal sites. (a) Schematic illustration of the inhalable phosphatidylserine-coated and STING agonist cyclic guanosine monophosphate-adenosine monophosphate (cGAMP)-loaded liposomes (NP-cGAMP) for enhancing antitumor immunity against lung metastases. APC, antigen presenting cell. DLN, draining lymph node. IR, irradiation. TA, tumor antigen. (b) Size distribution, polydispersity index, and surface charge of NP-cGAMP, NP labeled with DIR (NP-DIR), and NP labeled with Rhob (NP-Rhob) by dynamic light scattering (DLS). (c) TEM images of NP-cGAMP. (d) Representative ex vivo fluorescence imaging of major organs dissected from 4T1-luc lung metastases-bearing mice at 1, 24, and 48 h post inhalation of DiR-labeled PS-coated NPs. Light signals were exclusively from both lungs and the lung signals were quantified (n = 3 biologically independent mice/time; **P < 0.01, Student's t test). (e) NP-cGAMP inhalation synergized with radiotherapy by eliciting APC-mediated adaptive immunity in B16F10-OVA melanoma lung metastasis model. On day 5, after confirming development of multifocal metastases in both lungs, the mice were treated with fractionated radiation to the right lung (IR, 8 Gy × 3), inhalation of NP-cGAMP (24 h after each IR for three doses), or a combination of them. NP-CTR (2′5′-GpAp) served as a control of NP-cGAMP. To deplete pulmonary APCs, NP-clodronate was administered via inhalation 6 h before each of the three NP-cGAMP inhalations. To deplete CD4+ or CD8+ T cells, anti-CD4 Ab or anti-CD8α Ab was injected intraperitoneal (i.p.) (400 μg), respectively, one day before IR and repeated 7 days later. The mice (n = 6) were sacrificed on day 18 and representative lungs from the treatment groups were displayed. (f) Lungs after treatments in (e) were examined under a dissecting microscope and a total of metastatic lung foci on each lung were counted. Data are shown as mean ± SD of n = 6 biologically independent samples. *P < 0.05, **P < 0.01, and ***P < 0.001 by Student's t test. Reproduced with permission.63 Copyright 2019, Nature Publishing Group. | ||
4. Pulmonary nanovaccines for protective Immunity against mucosal diseases
As more extensively reviewed elsewhere, synthetic nanoparticles varying from LNPs, liposomes, polymeric nanoparticles, and hybrid nanoparticles, can be delivered via nasal, oral, sublingual, or colorectal routes for mucosal nanovaccines.42,68,69 These nanoparticles may also find broad applications in pulmonary mucosal nanovaccines. For example, aerosolized PLGA and PLA nanoparticles were used for pulmonary delivery of hepatitis B vaccine, which elicited enhanced humoral, mucosal and cytokine responses in comparison to the delivery of free antigens.70 In this section, inspired by the tremendous success of LNPs in parenteral nanovaccines for COVID-19, an infectious disease at pulmonary mucosal site, we will focus our discussion on lipid-based pulmonary nanovaccines.Liposome represents a paradigm of delivery vectors in the clinical translation of pulmonary mucosal nanovaccines. Very recently, Ming et al. prepared a bionic virus-like nanovaccine that comprised viral “capsid”-mimicking pulmonary surfactant liposomes as delivery vectors, viral genetic material-mimicking poly(I:C) (polyinosinic-polycytidylic acid) as adjuvants, and “spike”-mimicking receptor binding domain (RBD) proteins on the liposome surfaces as antigens. This inhalable mucosal vaccine elicited high titer of sIgA in respiratory secretions, effectively neutralized intranasally delivered pseudovirus, and demonstrated a much stronger mucosal protective immune responses than subcutaneous vaccination.71 It is worth noting that the lamellarity of liposomes can greatly impact the vaccine immunogenicity.19 Pulmonary immunization with interbilayer-crosslinked multilamellar liposome vesicles (ICMVs) were reported to activate high-frequency, long-lived, and antigen specific effector memory T cells (TEms) at multiple mucosal sites.19 The ICMVs were first reported by the Irvine group in 2011.72 They were composed of phospholipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), anionic 1,2-di-(9Z-octadecenoyl)-snglycero-3-phospho-(1′-rac-glycerol) (DOPG), and anionic maleimide-headgroup lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl) butyramide] (MPB) at a molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5. To synthesize them, simple liposomes were first prepared and fused to multilamellar vesicles (MLVs), in which functionalized lipid headgroups of adjacent bilayers were crosslinked to generate the ICMVs (Fig. 6a). The ICMVs allowed stable entrapment of protein antigens in the core and lipid-based immunostimulators in their multi-layer walls under extracellular conditions and rapid release of the cargoes in cell endosomes, therefore promoting antigen cross-presentation and eliciting robust humoral and cellular immune responses upon subcutaneous injection.72 In their following-up study, ICMVs were employed to pack antigen OVA and two TLR agonists, monophosphoryl lipid A (MPLA) and poly(I:C), for pulmonary vaccination. To investigate the distant mucosal immunity, luciferase-expressing OT-1 CD8+ T cells (OT-1-luc) were intravenousely transferred into mice twenty-four hours prior to intratracheal or subcutaneous administration of ICMV or soluble vaccines. According to bioluminescence imaging on day 3, T cell proliferation was only observed in draining lymph nodes (dLNs) in all vaccinated mice. In contrast, by day 5, higher T cell expansion was observed in the pulmonary ICMV group, showing bioluminescent signals in not only lymph nodes and spleen but also reproductive tracts and gut (Fig. 6b). These results indicated the unique mucosal homing property of T cell responses in mucosal immunization, which was attributed to the substantial expression of mucosal homing integrin α4β7 receptors in the tetramer+ peripheral blood OT-1 cells after pulmonary vaccination (Fig. 6c). Moreover, pulmonary ICMV vaccination biased T cell responses toward effector memory phenotype at both local and distal mucosal tissues. When tested in subcutaneous OVA-expressing B16F10 mouse melanoma tumor model, ICMV OVA pulmonary vaccination led to complete tumor rejection and long-term survival (over 18 weeks) of all animals. In contrast, soluble OVA pulmonary vaccination delayed tumor growth but failed to improve ultimate survival of mice (Fig. 6d). Similar results were observed in prophylactic mucosal viral challenge models (Fig. 6e). Collectively, pulmonary ICMV nanovaccine could prime TEMs that circulated to both local and distal mucosal tissues as well as systemic compartments, therefore providing robust protection in both therapeutic tumor and prophylactic viral infection settings. Further evaluation on the safety and efficacy of ICMV mucosal nanovaccines that carrying antigens of relevant mucosal disease, such as mucosal tumors and HIV, would provide significant guidance for their bench-to-bed translation.
:1:5. To synthesize them, simple liposomes were first prepared and fused to multilamellar vesicles (MLVs), in which functionalized lipid headgroups of adjacent bilayers were crosslinked to generate the ICMVs (Fig. 6a). The ICMVs allowed stable entrapment of protein antigens in the core and lipid-based immunostimulators in their multi-layer walls under extracellular conditions and rapid release of the cargoes in cell endosomes, therefore promoting antigen cross-presentation and eliciting robust humoral and cellular immune responses upon subcutaneous injection.72 In their following-up study, ICMVs were employed to pack antigen OVA and two TLR agonists, monophosphoryl lipid A (MPLA) and poly(I:C), for pulmonary vaccination. To investigate the distant mucosal immunity, luciferase-expressing OT-1 CD8+ T cells (OT-1-luc) were intravenousely transferred into mice twenty-four hours prior to intratracheal or subcutaneous administration of ICMV or soluble vaccines. According to bioluminescence imaging on day 3, T cell proliferation was only observed in draining lymph nodes (dLNs) in all vaccinated mice. In contrast, by day 5, higher T cell expansion was observed in the pulmonary ICMV group, showing bioluminescent signals in not only lymph nodes and spleen but also reproductive tracts and gut (Fig. 6b). These results indicated the unique mucosal homing property of T cell responses in mucosal immunization, which was attributed to the substantial expression of mucosal homing integrin α4β7 receptors in the tetramer+ peripheral blood OT-1 cells after pulmonary vaccination (Fig. 6c). Moreover, pulmonary ICMV vaccination biased T cell responses toward effector memory phenotype at both local and distal mucosal tissues. When tested in subcutaneous OVA-expressing B16F10 mouse melanoma tumor model, ICMV OVA pulmonary vaccination led to complete tumor rejection and long-term survival (over 18 weeks) of all animals. In contrast, soluble OVA pulmonary vaccination delayed tumor growth but failed to improve ultimate survival of mice (Fig. 6d). Similar results were observed in prophylactic mucosal viral challenge models (Fig. 6e). Collectively, pulmonary ICMV nanovaccine could prime TEMs that circulated to both local and distal mucosal tissues as well as systemic compartments, therefore providing robust protection in both therapeutic tumor and prophylactic viral infection settings. Further evaluation on the safety and efficacy of ICMV mucosal nanovaccines that carrying antigens of relevant mucosal disease, such as mucosal tumors and HIV, would provide significant guidance for their bench-to-bed translation.
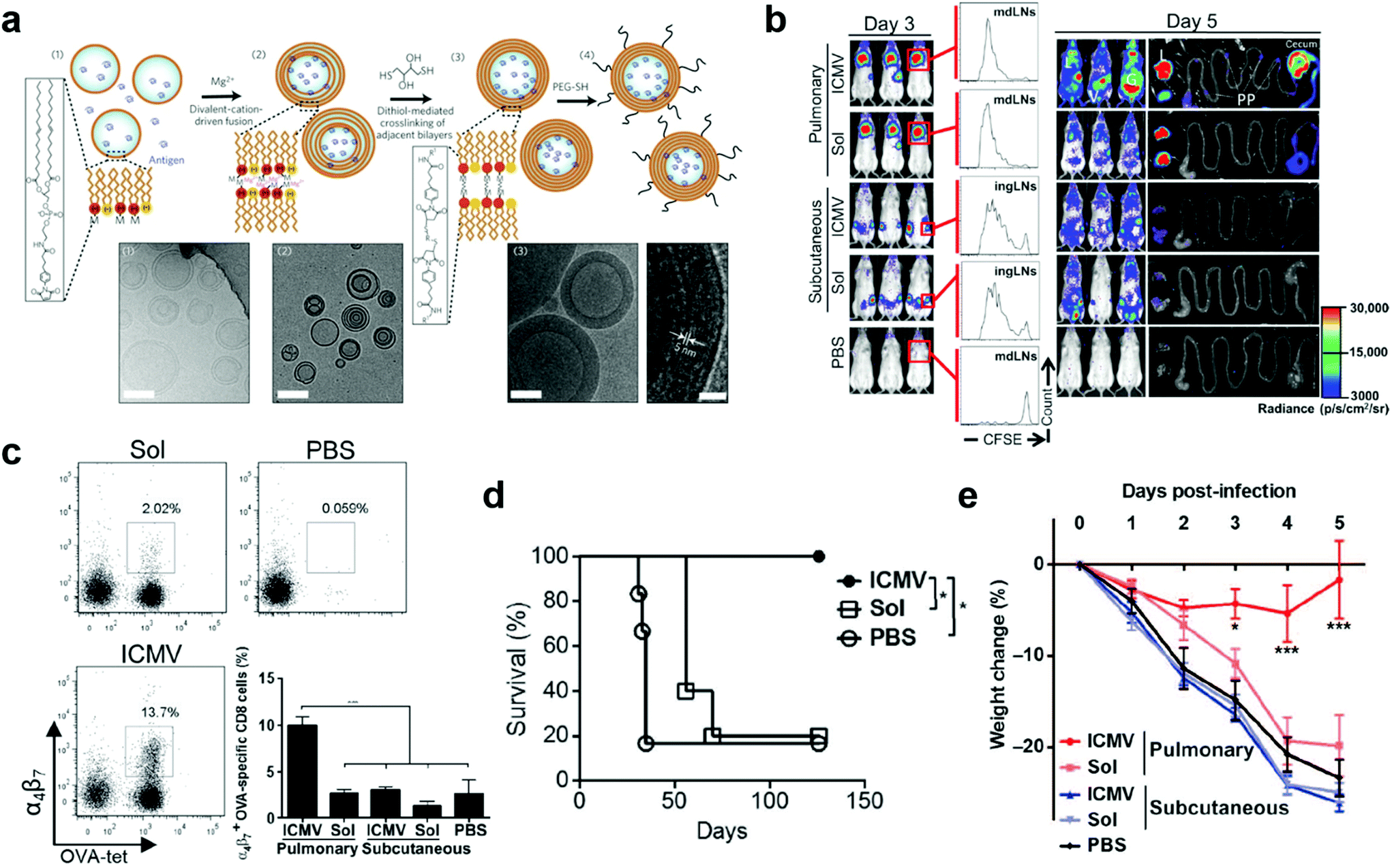 | ||
| Fig. 6 Liposome nanoparticle-based pulmonary vaccine. (a) Schematic illustration of interbilayer-crosslinked multilamellar vesicles (ICMVs) synthesis and cryoelectron microscope images: (i) anionic, maleimide-functionalized liposomes are prepared from dried lipid films, (ii) divalent cations are added to induce fusion of liposomes and the formation of multilamellar vesicles (MLVs), (iii) membrane-permeable dithiols are added, which crosslink maleimide-lipids on apposed lipid bilayers in the vesicle walls, and (iv) the resulting lipid particles are PEGylated with thiol-terminated PEG. Cryo-EM images from each step of the synthesis show (i) initial liposomes, (ii) MLVs, and (iii) ICMVs with thick lipid walls. Scale bars = 100 nm. Right-hand image of (iii) shows a zoomed image of an ICMV wall, where stacked bilayers are resolved as electron-dense striations; scale bar = 20 nm. Reproduced with permission.49 Copyright 2011, Nature Publishing Group. (b) OT-I-luc CD8+ T cells were adoptively transferred into C57BL/6 mice (n = 5) 1 day before ICMV or soluble OVA vaccine immunization via intratracheal or subcutaneous routes. Trafficking and proliferation of OT-I-luc T cells were monitored by flow cytometry and bioluminescence imaging on days 3 and 5 after immunization. Flow cytometry histograms show representative CFSE dilutions in transferred T cells on day 3 after immunization at draining lymph nodes (dLNs) (mdLNs for intratracheal vaccines and ingLNs for subcutaneous vaccinations). Lungs (L) and gastrointestinal tracts (G) were dissected on day 5 and imaged to identify T cell localization. V, vaginal tract; PP, Peyer's patches. (c) Flow cytometry analyses of integrin a4b7+ OT-I T cells in blood on day 5. (d) C57BL/6 mice (n = 5 to 6 per group) inoculated with 5 × 105 B16F10-OVA cells subcutaneously in the flank on day 0 were treated on days 3 and 10 with intratracheal administration of ICMVs or soluble OVA vaccines. Survival of tumor-bearing mice was tracked for 18 weeks. *P < 0.05, log-rank (Mantel-Cox) test. (e) C57BL/6 mice were immunized subcutaneously or intratracheally on days 0 and 28 with antigen AL11 (AAVKNWMTQTL) and the universal CD4+ T cell helper epitope PADRE peptide ICMV or soluble vaccines, and then challenged by intratracheal administration of simian immunodeficiency virus (SIV) gag-expressing vaccinia virus (1 × 106 PFU) on day 42. Body weight changes were recorded over time. Dots show titers from individual animals. Data are means ± SEM with n = 3 to 7 animals per group. *P < 0.5, **P < 0.01, ***P < 0.001, by one-way ANOVA (c) or two-way ANOVA (e). Reproduced with permission.19 Copyright 2013, American Association for the Advancement of Science. | ||
In addition to liposome nanoparticles, lipid conjugates with “albumin hitching” property have demonstrated to effectively navigate amphiphilic phospholipid-coupling peptide antigens or molecular adjuvants (amph-vaccines) to dLNs.33,73,74 In this strategy, the lipid tail of the amph-vaccines served as an albumin-binding domain. Upon injection, endogenous albumin was bound to and chaperoned amph-vaccines to dLNs, and then transferred the lipid tails from albumin to insert in cell membranes.33,74 The Irvine group explored this amph-vaccine approach in pulmonary vaccination on the basis that (1) albumin is abundant in the bronchoalveolar lavage (BAL) fluid of naïve mice and its concentration can be rapidly increased from approximate 200 μg mL−1 to a few mg mL−1 after pulmonary administration of the TLR9 agonist CpG; (2) FcRn is highly expressed on pulmonary epithelial cells, which can facilitate the transportation of albumin across the airway epithelium.13 The amph-vaccines comprised a PEG spacer linked with a melanoma peptide antigen gp10020–39 and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) phospholipid tail as well as a similar C18 lipid tail-coupled adjuvant CpG DNA (Fig. 7a). The amph-gp100 conjugates led to about 10 times higher accumulation in the lung parenchyma than free gp100 peptide 24 h after intratracheal administration, and remained easily detectable in lungs and mediastinal lymph nodes (MLNs) at 48 h (Fig. 7b–d). Similar results were found in the amph-CpG conjugates. Importantly, pulmonary vaccination with amph-vaccines stimulated prolonged antigen presentation in MLNs for over two weeks, high levels of antigen-specific lung TRMs, and enhanced protection against viral and tumor challenge. In a prophylactic viral infection model, mice were first intratracheally immunized with amph-AL11/amph-CpG vaccines, in which AL11 is a murine gag peptide. After five months, the mice were challenged with a lethal dose of gag epitope-expressing Vaccinia virus (Fig. 7e). All mice immunized with pulmonary amph-vaccines survived. In control groups, mice kept losing body weight and succumbed by day 7 (Fig. 7f and g). To test the protective capacity in tumor models, the antigens of the amph-vaccines in Fig. 7e were replaced with a combination of three melanoma peptides (Trp1455–463, Trp2180–188, and gp10020–39) and mice were challenged 30 days after full vaccination by intravenously injecting B16F10 melanoma cells. According to survive curves in Fig. 7h, 80% mice in the pulmonary vaccine group survived at 100 days after the tumor inoculation, while only 40% mice were protected by subcutaneous vaccination. Taken together, pulmonary amph-vaccines primed long-lived antigen-specific TRMs in the lungs, generated robust mucosal immunity, and effectively protected against viral or tumor challenge.
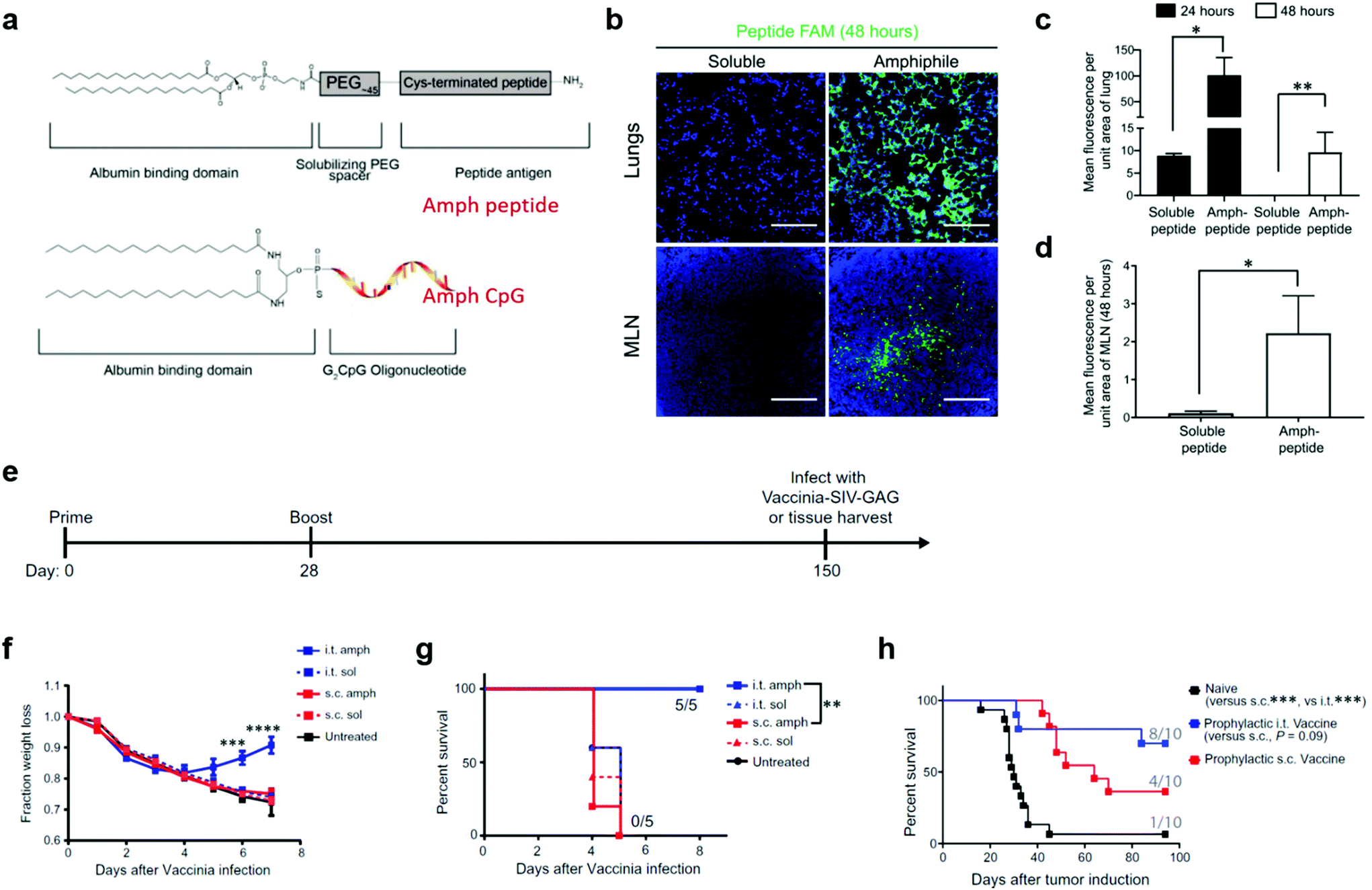 | ||
| Fig. 7 Albumin-binding lipid-based pulmonary vaccine. (a) Structure of amph-vaccines. Upper: schematic of 1,2-distearoyl-sn-glycero-3phosphoethanolamine (DSPE) conjugated to cysteine(Cys)-terminated peptide via a polyethylene glycol (PEG) spacer. Lower: schematic of diacyl lipid tail conjugated to CpG. (b) Representative immunofluorescence images of the lungs and mediastinal lymph nodes (MLNs) harvested from C57BL/6 mice (n = 3) at 48 hours after immunization with 20 μg of fluorescein amidite (FAM)-labeled (green) gp100 peptide or an equimolar amount of labeled amph-gp100. Scale bars, 100 μm. DAPI (blue) was used to stain nuclei. (c) Quantification of peptide signals from the lungs at 24 and 48 hours after the immunization in (b). (d) Quantification of peptide signals from the MLNs at 48 hours after the immunization in (b). (e) C57BL/6 mice (n = 5) were vaccinated with 5 μg of AL11 peptide combined with 8 μg of CpG or equimolar doses of amph-AL11 and amph-CpG administered intratracheally or subcutaneously on days 0 and 28 and then challenged with a lethal dose of SIV-Gag Vaccinia virus (106 PFU) intratracheally on day 150. (f) Animal weights over time after Vaccinia virus challenge in (e). (g) Animal survival after Vaccinia virus challenge in (e). (h) C57BL/6 mice (n = 10) were left naïve or vaccinated with 10 μg of gp100, 10 μg of Trp1, and 10 μg of Trp2 peptide antigens combined with 8 μg of CpG or vaccinated with equimolar doses of amph-peptides and amph-CpG on days 0 and 14. Thirty days after boost, mice were challenged with 4 × 105 B16F10 melanoma cells intravenously, and survival over time is plotted. In each cohort, the number of mice surviving at 80 days was indicated. Data are represented as means ± SEM from three independent experiments (e–g) and two independent experiments (b–d and h); *P < 0.05, **P < 0.001, and ****P < 0.0001 as determined by one-way ANOVA. Survival data were analyzed using a log-rank test. Reproduced with permission.13 Copyright 2021, American Association for the Advancement of Science. | ||
LNPs are another important class of lipid-based nanoparticles. LNPs are usually constructed by four lipid components: (1) ionizable/cationic lipids that protect the cargoes and facilitate endosomal escape, (2) PEG-lipids that stabilize the LNPs and protect them from opsonization, (3) cholesterol that promotes cellular endocytosis, and (4) other helper lipids such as phospholipids that support the LNP structure.75,76 Each component and their molar ratio can be carefully tuned to efficiently deliver nucleic acid therapeutics to hepatocytes.77–79 Recently, the Siegwart research group engineered LNPs with a fifth lipid component and achieved liver-, spleen-, or lung-specific delivery after intravenous injection of the LNPs.75 In brief, addition of cationic lipid 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP) was identified to be a key regulator of the selective organ targeting (SORT), which shifted the accumulation of LNPs from liver to spleen to lung by increasing its molar percentages from 0 to 100% in the LNPs. Anionic lipid 1,2-dioleoyl-sn-glycero-3-phosphate (18PA) (at a molar ratio of 10–40%) was screened for spleen-targeted delivery. In addition, they found that ionizable cationic lipids such as 1,2-dioleoyl-3-dimethylammonium-propane (DODAP) (at a molar ratio of 20%) did not affect biodistribution but could increase mRNA delivery to the liver.75 LNPs have been used in the intramuscular delivery of COVID-19 mRNA vaccines and are exerting a broad impact on the activation of potent systemic immune protection against SARS-CoV-2. They can also be applied to the delivery of mucosal nanovaccines. For example, rectal delivery of LNPs containing Hepatitis B surface antigen (HBsAg) and MPLA adjuvant induced strong systemic and robust mucosal immunity.80 Further encapsulation of this HBsAg nanovaccine into enteric-coated minicapsules converted it to an oral nanovaccine, which can target the colonic region upon oral delivery and induce significant immune protection against hepatitis B.81 Few studies have been reported for using LNPs in pulmonary vaccination thus far. Notably, upon pulmonary delivery, LNPs would interact with proteins, biomolecules, cells, and physical barriers in the airway, which are different from those they would interact with in systemic delivery.9 In addition, the shearing stress from nebulizers or inhalers may disrupt the LNP structures.82 Therefore, LNP formulations that succeed in parenteral administration may need further optimization for pulmonary delivery. Very recently, Lokugamage et al. established an in vivo workflow to systematically study how different lipid components in LNPs affected their nebulized delivery of mRNA therapeutics.82 They constructed oligomer-lipid conjugate 7C1-baed LNPs by varying the molar ratio and structure of PEG-lipids, the charge of phospholipids, and the presence or absence of cholesterol. They found that PEG-lipids, rather than cholesterol and helper lipids, were the key regulators of the formation of stable 7C1-based LNPs for nebulized delivery. A higher PEG content and cationic helper lipids in LNPs could improve mRNA delivery. The optimized LNPs (NLD1) were composed of 7C1, cholesterol, C14-PEG2000, and 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) at a molar ratio of 35:5:55:5, which could efficiently deliver mRNA-encoding neutralizing antibody to protect mice from influenza A viruses. Moreover, nebulized delivery of NLD1 demonstrated excellent biosafety. Compared with untreated controls, NLD1 caused only 6 of 547 inflammatory genes in the lung tissues that increased by more than two-fold at 4 h after the exposure, whereas that in the mild-dose lipopolysaccharides (LPS) control group was 86 out of 547 inflammatory genes. In addition, nebulized delivery of NLD1 didn't induce significant loss of mouse body weights within 24 hours, further supporting excellent tolerability even at early timepoints.82 Taken together, the NLD1 LNPs hold great potential to be safe and effective delivery nanocarriers for pulmonary vaccines.
5. Concluding remarks
Mucosal vaccination offers significant benefits over parenteral immunization by eliciting immune defenses at the principal sites of infection. The sterilizing immunity is mainly achieved by local secretory antibody responses and tissue-resident T cells. A comprehensive understanding of the lung anatomy as well as the lung immunobiology is required for the rational design of pulmonary mucosal nanovaccines that induce adaptive and innate immune responses, both of which are pivotal for vaccines. Meanwhile, current pulmonary delivery technologies (i.e., devices and formulations) provide important guidance for the development of pulmonary vaccines. A major challenge of mucosal vaccine is to effectively deliver antigen/adjuvant agents to the desired mucosal sites. Nanovaccines can facilitate immunogens to cross mucosal barriers and target immune cells, thus promoting mucosal immune responses. Moreover, nanovaccines provide a facile method to co-deliver adjuvants with antigens, whereby promoting the immunogenicity of antigens and hence improving the resulting adaptive immune responses.In this review, we summarized recent design strategies and progresses of pulmonary mucosal nanovaccines. We mainly discussed the improvement in the design of nanocarriers for vaccines. Other critical parameters, such as antigen selection and expression, antigen/adjuvant stability in mucosal environment, administration doses and timing of antigens/adjuvants, mucosal immunity measurement, and prime-boost combination with parenteral vaccination are also critical for the pulmonary vaccines to induce the optimal immune responses. Furthermore, the accessibility of storage, transportation, and massive immunization should also be considered for the successful clinical translation and commercialization of pulmonary mucosal nanovaccines. Notably, the crosstalk between mucosal compartments allows pulmonary nanovaccines to provide protection against not only lung infections but also reproductive or gastrointestinal tract pathogens, for instance, HIV/AIDS.6 Moreover, the pulmonary delivery strategies can also be expanded to the delivery of therapeutic agents other than vaccines for alternative approaches of mucosal disease treatment. For example, nebulized delivery of lung spheroid cell exosome-based nanodecoys accelerated the clearance of SARS-CoV-2 mimics from the lungs without apparent toxicity.83 In another study, nebulized delivery of PBAE polymer formulated Cas13a mRNA efficiently degraded influenza or SARS-CoV-2 RNA in lung tissue, mitigated influenza virus A or SARS-CoV-2 replication, and reduced disease symptoms in rodents.27 Besides, intratracheal administration of multiplexing activity-based nanosensors achieved sensitive and specific lung cancer detection.84 Combination of pulmonary nanovaccines with these applications may provide new solutions for the management of mucosal diseases. Taken together, pulmonary mucosal nanovaccines will provide powerful tools for preventing infection and transmission of mucosal pathogens, as well as for the treatment of pre-existing diseases such as infections and cancer at mucosal sites and other distant tissues.
Conflicts of interest
G. Z. is a co-founder and CSO of AmpedRNA Biosciences, LLC, which develops novel vaccine technologies for cancer and other diseases.Acknowledgements
The work was supported by the National University of Singapore Start-Up funding (W. T.). G. Z. acknowledges support from NIH-NIGMS (R35GM143014), NIH-NINDS (R21NS114455), NIH-NCATS KL2 Scholarship (KL2TR002648) and Endowment Fund from VCU C. Kenneth and Dianne Wright Center for Clinical and Translational Research (UL1TR002649), and start-up fund from the Center for Pharmaceutical Engineering and Sciences-VCU School of Pharmacy.References
- L. R. Baden, H. M. El Sahly, B. Essink, K. Kotloff, S. Frey, R. Novak, D. Diemert, S. A. Spector, N. Rouphael, C. B. Creech, J. McGettigan, S. Khetan, N. Segall, J. Solis, A. Brosz, C. Fierro, H. Schwartz, K. Neuzil, L. Corey, P. Gilbert, H. Janes, D. Follmann, M. Marovich, J. Mascola, L. Polakowski, J. Ledgerwood, B. S. Graham, H. Bennett, R. Pajon, C. Knightly, B. Leav, W. Deng, H. Zhou, S. Han, M. Ivarsson, J. Miller, T. Zaks and C. S. Group, N. Engl. J. Med., 2021, 384, 403 CrossRef CAS PubMed.
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. P. Marc, E. D. Moreira, C. Zerbini, R. Bailey, K. A. Swanson, S. Roychoudhury, K. Koury, P. Li, W. V. Kalina, D. Cooper, R. W. Frenck Jr., L. L. Hammitt, O. Tureci, H. Nell, A. Schaefer, S. Unal, D. B. Tresnan, S. Mather, P. R. Dormitzer, U. Sahin, K. U. Jansen, W. C. Gruber and C. C. T. Group, N. Engl. J. Med., 2020, 383, 2603 CrossRef CAS PubMed.
- E. J. Anderson, N. G. Rouphael, A. T. Widge, L. A. Jackson, P. C. Roberts, M. Makhene, J. D. Chappell, M. R. Denison, L. J. Stevens, A. J. Pruijssers, A. B. McDermott, B. Flach, B. C. Lin, N. A. Doria-Rose, S. O'Dell, S. D. Schmidt, K. S. Corbett, P. A. Swanson, M. Padilla, K. M. Neuzil, H. Bennett, B. Leav, M. Makowski, J. Albert, K. Cross, V. V. Edara, K. Floyd, M. S. Suthar, D. R. Martinez, R. Baric, W. Buchanan, C. J. Luke, V. K. Phadke, C. A. Rostad, J. E. Ledgerwood, B. S. Graham and J. H. Beigel, N. Engl. J. Med., 2020, 383, 2427 CrossRef CAS PubMed.
- E. C. Lavelle and R. W. Ward, Nat. Rev. Immunol., 2021 DOI:10.1038/s41577-021-00583-2.
- J. Holmgren and C. Czerkinsky, Nat. Med., 2005, 11, S45–S53 CrossRef CAS PubMed.
- M. R. Neutra and P. A. Kozlowski, Nat. Rev. Immunol., 2006, 6, 148 CrossRef CAS PubMed.
- C. J. Wei, M. C. Crank, J. Shiver, B. S. Graham, J. R. Mascola and G. J. Nabel, Nat. Rev. Drug Discovery, 2020, 19, 239 CrossRef CAS PubMed.
- S. Nambulli, Y. Xiang, N. L. Tilston-Lunel, L. J. Rennick, Z. Sang, W. B. Klimstra, D. S. Reed, N. A. Crossland, Y. Shi and W. P. Duprex, Sci. Adv., 2021, 7, eabh0319 CrossRef CAS PubMed.
- J. A. Whitsett and T. Alenghat, Nat. Immunol., 2015, 16, 27 CrossRef CAS PubMed.
- Z. Zuo, Y. Zou, Q. Li, Y. Guo, T. Zhang, J. Wu, C. He and F. O. Eko, Sci. Rep., 2021, 11, 10389 CrossRef CAS PubMed.
- D. Sterlin, A. Mathian, M. Miyara, A. Mohr, F. Anna, L. Claer, P. Quentric, J. Fadlallah, H. Devilliers, P. Ghillani, C. Gunn, R. Hockett, S. Mudumba, A. Guihot, C. E. Luyt, J. Mayaux, A. Beurton, S. Fourati, T. Bruel, O. Schwartz, J. M. Lacorte, H. Yssel, C. Parizot, K. Dorgham, P. Charneau, Z. Amoura and G. Gorochov, Sci. Transl. Med., 2021, 13, eabd2223 CrossRef CAS PubMed.
- G. Stary, A. Olive, A. F. Radovic-Moreno, D. Gondek, D. Alvarez, P. A. Basto, M. Perro, V. D. Vrbanac, A. M. Tager, J. Shi, J. A. Yethon, O. C. Farokhzad, R. Langer, M. N. Starnbach and U. H. Von Andrian, Science, 2015, 348, aaa8205 CrossRef PubMed.
- K. Rakhra, W. Abraham, C. Wang, K. D. Moynihan, N. Li, N. Donahue, A. D. Baldeon and D. J. Irvine, Sci. Immunol., 2021, 6, eabd8003 CrossRef CAS PubMed.
- A. O. Hassan, N. M. Kafai, I. P. Dmitriev, J. M. Fox, B. K. Smith, I. B. Harvey, R. E. Chen, E. S. Winkler, A. W. Wessel, J. B. Case, E. Kashentseva, B. T. McCune, A. L. Bailey, H. Zhao, L. A. VanBlargan, Y. N. Dai, M. Ma, L. J. Adams, S. Shrihari, J. E. Danis, L. E. Gralinski, Y. J. Hou, A. Schafer, A. S. Kim, S. P. Keeler, D. Weiskopf, R. S. Baric, M. J. Holtzman, D. H. Fremont, D. T. Curiel and M. S. Diamond, Cell, 2020, 183, 169 CrossRef CAS PubMed.
- D. Hartl, R. Tirouvanziam, J. Laval, C. M. Greene, D. Habiel, L. Sharma, A. O. Yildirim, C. S. Dela Cruz and C. M. Hogaboam, J. Innate Immun., 2018, 10, 487 CrossRef CAS PubMed.
- S. U. Islam, A. Shehzad, M. B. Ahmed and Y. S. Lee, Molecules, 2020, 25, 1929 CrossRef CAS PubMed.
- L. A. Keller, O. Merkel and A. Popp, Drug Delivery Transl. Res., 2021, 11, 1 CrossRef PubMed.
- D. Hartl, R. Tirouvanziam, J. Laval, C. M. Greene, D. Habiel, L. Sharma, A. O. Yildirim, C. S. Dela Cruz and C. M. Hogaboam, J. Innate Immun., 2018, 10, 487 CrossRef CAS PubMed.
- A. V. Li, J. J. Moon, W. Abraham, H. Suh, J. Elkhader, M. A. Seidman, M. Yen, E. J. Im, M. H. Foley, D. H. Barouch and D. J. Irvine, Sci. Transl. Med., 2013, 5, 204ra130 Search PubMed.
- M. Hellfritzsch and R. Scherließ, Pharmaceutics, 2019, 11, 375 CrossRef CAS PubMed.
- W. Liang, H. W. Pan, D. Vllasaliu and J. K. W. Lam, Pharmaceutics, 2020, 12, 1025 CrossRef CAS PubMed.
- C. A. Ruge, J. Kirch and C. M. Lehr, Lancet Respir. Med., 2013, 1, 402 CrossRef CAS PubMed.
- T. E. Rajapaksa, K. M. Bennett, M. Hamer, C. Lytle, V. G. Rodgers and D. D. Lo, J. Biol. Chem., 2010, 285, 23739 CrossRef CAS PubMed.
- W. M. Colwell, D. G. Simmons, J. R. Harris, T. G. Fulp, J. H. Carrozza and T. A. Maag, Avian Dis., 1975, 19, 781 CrossRef CAS PubMed.
- D. T. Brandau, L. S. Jones, C. M. Wiethoff, J. Rexroad and C. R. Middaugh, J. Pharm. Sci., 2003, 92, 218 CrossRef CAS PubMed.
- A. K. Patel, J. C. Kaczmarek, S. Bose, K. J. Kauffman, F. Mir, M. W. Heartlein, F. DeRosa, R. Langer and D. G. Anderson, Adv. Mater., 2019, 31, e1805116 CrossRef PubMed.
- E. L. Blanchard, D. Vanover, S. S. Bawage, P. M. Tiwari, L. Rotolo, J. Beyersdorf, H. E. Peck, N. C. Bruno, R. Hincapie, F. Michel, J. Murray, H. Sadhwani, B. Vanderheyden, M. G. Finn, M. A. Brinton, E. R. Lafontaine, R. J. Hogan, C. Zurla and P. J. Santangelo, Nat. Biotechnol., 2021, 39, 717 CrossRef CAS PubMed.
- Z. Li, Y. Wang, Y. Shen, C. Qian, D. Oupicky and M. Sun, Sci. Adv., 2020, 6, eaaz9240 CrossRef CAS PubMed.
- A. Minne, H. Boireau, M. J. Horta and R. Vanbever, Eur. J. Pharm. Biopharm., 2008, 70, 839 CrossRef CAS PubMed.
- J. Raula, F. Thielmann, M. Naderi, V. P. Lehto and E. I. Kauppinen, Int. J. Pharm., 2010, 385, 79 CrossRef CAS PubMed.
- C. Weiler, M. Egen, M. Trunk and P. Langguth, J. Pharm. Sci., 2010, 99, 303 CrossRef CAS PubMed.
- J. L. Wang, M. S. Hanafy, H. Xu, J. Leal, Y. Zhai, D. Ghosh, R. O. Williams III, H. D. C. Smyth and Z. Cui, Int. J. Pharm., 2021, 596, 120215 CrossRef CAS PubMed.
- L. Ma, T. Dichwalkar, J. Y. H. Chang, B. Cossette, D. Garafola, A. Q. Zhang, M. Fichter, C. Wang, S. Liang, M. Silva, S. Kumari, N. K. Mehta, W. Abraham, N. Thai, N. Li, K. D. Wittrup and D. J. Irvine, Science, 2019, 365, 162 CrossRef CAS PubMed.
- G. Zhu, L. Mei, H. D. Vishwasrao, O. Jacobson, Z. Wang, Y. Liu, B. C. Yung, X. Fu, A. Jin, G. Niu, Q. Wang, F. Zhang and X. Chen, Nat. Commun., 2017, 8, 1482 CrossRef PubMed.
- G. Zhu, G. M. Lynn, O. Jacobson, K. Chen, Y. Liu, H. Zhang, Y. Ma, F. Zhang, R. Tian, Q. Ni, S. Cheng, Z. Wang, N. Lu, B. C. Yung, Z. Wang, L. Lang, X. Fu, A. Jin, I. D. Weiss, H. Vishwasrao, G. Niu, H. Shroff, D. M. Klinman, R. A. Seder and X. Chen, Nat. Commun., 2017, 8, 1954 CrossRef PubMed.
- M. E. Aikins, C. Xu and J. J. Moon, Acc. Chem. Res., 2020, 53, 2094 CrossRef CAS PubMed.
- S. Yan, Z. Luo, Z. Li, Y. Wang, J. Tao, C. Gong and X. Liu, Angew. Chem., Int. Ed., 2020, 59, 17332 CrossRef CAS PubMed.
- C. Zhang and K. Pu, Chem. Soc. Rev., 2020, 49, 4234 RSC.
- W. Sang, Z. Zhang, Y. Dai and X. Chen, Chem. Soc. Rev., 2019, 48, 3771 RSC.
- Y. Zhang, S. Lin, X. Y. Wang and G. Zhu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, 1559 Search PubMed.
- W. Tang, W. Fan, J. Lau, L. Deng, Z. Shen and X. Chen, Chem. Soc. Rev., 2019, 48, 2967 RSC.
- K. A. Woodrow, K. M. Bennett and D. D. Lo, Annu. Rev. Biomed. Eng., 2012, 14, 17 CrossRef CAS PubMed.
- S. K. Lai, Y.-Y. Wang and J. Hanes, Adv. Drug Delivery Rev., 2009, 61, 158 CrossRef CAS PubMed.
- J. K. Sheehan, K. Oates and I. Carlstedt, Biochem. J., 1986, 239, 147 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28 CrossRef CAS PubMed.
- Q. Xu, L. M. Ensign, N. J. Boylan, A. Schön, X. Gong, J. C. Yang, N. W. Lamb, S. Cai, T. Yu, E. Freire and J. Hanes, ACS Nano, 2015, 9, 9217 CrossRef CAS PubMed.
- D. Chen, S. Liu, D. Chen, J. Liu, J. Wu, H. Wang, Y. Su, G. Kwak, X. Zuo, D. Rao, H. Cui, C. Shu and J. S. Suk, Angew. Chem., Int. Ed., 2021, 60, 15225 CrossRef CAS PubMed.
- E. Nakamura and H. Isobe, Chem. Rec., 2010, 10, 260–270 CrossRef CAS PubMed.
- H. Isobe, S. Sugiyama, K. I. Fukui Ki, Y. Iwasawa and E. Nakamura, Angew. Chem., Int. Ed., 2001, 40, 3364 CrossRef CAS PubMed.
- C. Macri, C. Dumont, A. P. Johnston and J. D. Mintern, Clin. Transl. Immunol., 2016, 5, 66 CrossRef PubMed.
- A. R. Poh and M. Ernst, Front. Oncol., 2018, 8, 49 CrossRef PubMed.
- D. S. Wilson, S. Hirosue, M. M. Raczy, L. Bonilla-Ramirez, L. Jeanbart, R. Wang, M. Kwissa, J. F. Franetich, M. A. S. Broggi, G. Diaceri, X. Quaglia-Thermes, D. Mazier, M. A. Swartz and J. A. Hubbell, Nat. Mater., 2019, 18, 175 CrossRef CAS PubMed.
- M. Yoshida, S. M. Claypool, J. S. Wagner, E. Mizoguchi, A. Mizoguchi, D. C. Roopenian, W. I. Lencer and R. S. Blumberg, Immunity, 2004, 20, 769 CrossRef CAS PubMed.
- S. Jangra, J. De Vrieze, A. Choi, R. Rathnasinghe, G. Laghlali, A. Uvyn, S. Van Herck, L. Nuhn, K. Deswarte, Z. Zhong, N. Sanders, S. Lienenklaus, S. David, S. Strohmeier, F. Amanat, F. Krammer, H. Hammad, B. N. Lambrecht, L. Coughlan, A. Garcia-Sastre, B. de Geest and M. Schotsaert, Angew. Chem., Int. Ed., 2021, 60, 9467 CrossRef CAS PubMed.
- S. Longet, M. L. E. Lundahl and E. C. Lavelle, Bioconjugate Chem., 2018, 29, 613 CrossRef CAS PubMed.
- M. Li, Y. Wang, Y. Sun, H. Cui, S. J. Zhu and H.-J. Qiu, Immunol. Lett., 2020, 217, 116 CrossRef CAS PubMed.
- L. Du, Z. Yu, F. Pang, X. Xu, A. Mao, W. Yuan, K. He and B. Li, Front. Cell. Infect. Microbiol., 2018, 8, 7 CrossRef PubMed.
- T. Azegami, Y. Yuki, R. Nakahashi, H. Itoh and H. Kiyono, Mol. Immunol., 2018, 98, 19 CrossRef CAS PubMed.
- F. C. Knight, P. Gilchuk, A. Kumar, K. W. Becker, S. Sevimli, M. E. Jacobson, N. Suryadevara, L. Wang-Bishop, K. L. Boyd, J. E. Crowe Jr., S. Joyce and J. T. Wilson, ACS Nano, 2019, 13, 10939 CrossRef CAS PubMed.
- N. Lycke, Nat. Rev. Immunol., 2012, 12, 592 CrossRef CAS PubMed.
- A. Muralidharan, M. S. Russell, L. Larocque, C. Gravel, S. Sauvé, Z. Chen, C. Li, W. Chen, T. Cyr, M. Rosu-Myles, L. Wang and X. Li, Vaccine, 2019, 37, 4031 CrossRef CAS PubMed.
- L. Feng, X. R. Qi, X. J. Zhou, Y. Maitani, S. C. Wang, Y. Jiang and T. Nagai, J. Controlled Release, 2006, 112, 35 CrossRef CAS PubMed.
- Y. Liu, W. N. Crowe, L. Wang, Y. Lu, W. J. Petty, A. A. Habib and D. Zhao, Nat. Commun., 2019, 10, 5108 CrossRef CAS PubMed.
- M. Mutsch, W. Zhou, P. Rhodes, M. Bopp, R. T. Chen, T. Linder, C. Spyr and R. Steffen, N. Engl. J. Med., 2004, 350, 896 CrossRef CAS PubMed.
- F. W. van Ginkel, R. J. Jackson, N. Yoshino, Y. Hagiwara, D. J. Metzger, T. D. Connell, H. L. Vu, M. Martin, K. Fujihashi and J. R. McGhee, Infect. Immun., 2005, 73, 6892 CrossRef CAS PubMed.
- J. H. Rhee, S. E. Lee and S. Y. Kim, Clin. Exp. Vaccine Res., 2012, 1, 50 CrossRef CAS PubMed.
- Z. Allakhverdi, S. Bouguermouh, M. Rubio and G. Delespesse, Allergy, 2005, 60, 1157 CrossRef CAS PubMed.
- A. Thakur and C. Foged, Nanoengineered Biomaterials for Advanced Drug Delivery, Elsevier, 2020, pp. 603–646, DOI:10.1016/b978-0-08-102985-5.00025-5.
- V. Bernasconi, K. Norling, M. Bally, F. Höök and N. Y. Lycke, J. Immunol. Res., 2016, 2016, 1 CrossRef PubMed.
- C. Thomas, A. Rawat, L. Hope-Weeks and F. Ahsan, Mol. Pharm., 2011, 8, 405 CrossRef CAS PubMed.
- B. Zheng, W. Peng, M. Guo, M. Huang, Y. Gu, T. Wang, G. Ni and D. Ming, Chem. Eng. J., 2021, 418, 129392 CrossRef CAS PubMed.
- J. J. Moon, H. Suh, A. Bershteyn, M. T. Stephan, H. Liu, B. Huang, M. Sohail, S. Luo, S. H. Um, H. Khant, J. T. Goodwin, J. Ramos, W. Chiu and D. J. Irvine, Nat. Mater., 2011, 10, 243 CrossRef CAS PubMed.
- H. Liu, K. D. Moynihan, Y. Zheng, G. L. Szeto, A. V. Li, B. Huang, D. S. Van Egeren, C. Park and D. J. Irvine, Nature, 2014, 507, 519 CrossRef CAS PubMed.
- K. D. Moynihan, R. L. Holden, N. K. Mehta, C. Wang, M. R. Karver, J. Dinter, S. Liang, W. Abraham, M. B. Melo, A. Q. Zhang, N. Li, S. L. Gall, B. L. Pentelute and D. J. Irvine, Cancer Immunol. Res., 2018, 6, 1025 CrossRef CAS PubMed.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313 CrossRef CAS PubMed.
- M. D. Buschmann, M. J. Carrasco, S. Alishetty, M. Paige, M. G. Alameh and D. Weissman, Vaccines, 2021, 9, 65 CrossRef CAS PubMed.
- A. Akinc, A. Zumbuehl, M. Goldberg, E. S. Leshchiner, V. Busini, N. Hossain, S. A. Bacallado, D. N. Nguyen, J. Fuller, R. Alvarez, A. Borodovsky, T. Borland, R. Constien, A. de Fougerolles, J. R. Dorkin, K. Narayanannair Jayaprakash, M. Jayaraman, M. John, V. Koteliansky, M. Manoharan, L. Nechev, J. Qin, T. Racie, D. Raitcheva, K. G. Rajeev, D. W. Sah, J. Soutschek, I. Toudjarska, H. P. Vornlocher, T. S. Zimmermann, R. Langer and D. G. Anderson, Nat. Biotechnol., 2008, 26, 561 CrossRef CAS PubMed.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. de Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Nat. Biotechnol., 2010, 28, 172 CrossRef CAS PubMed.
- J. A. Kulkarni, D. Witzigmann, S. Chen, P. R. Cullis and R. van der Meel, Acc. Chem. Res., 2019, 52, 2435 CrossRef CAS PubMed.
- K. K. Sahu and R. S. Pandey, Int. Immunopharmacol., 2016, 39, 343 CrossRef CAS PubMed.
- K. K. Sahu, M. Kaurav and R. S. Pandey, Int. Immunopharmacol., 2019, 66, 317 CrossRef CAS PubMed.
- M. P. Lokugamage, D. Vanover, J. Beyersdorf, M. Z. C. Hatit, L. Rotolo, E. S. Echeverri, H. E. Peck, H. Ni, J.-K. Yoon, Y. Kim, P. J. Santangelo and J. E. Dahlman, Nat. Biomed. Eng., 2021, 5, 1059 CrossRef PubMed.
- Z. Li, Z. Wang, P.-U. C. Dinh, D. Zhu, K. D. Popowski, H. Lutz, S. Hu, M. G. Lewis, A. Cook, H. Andersen, J. Greenhouse, L. Pessaint, L. J. Lobo and K. Cheng, Nat. Nanotechnol., 2021, 16, 942 CrossRef CAS PubMed.
- J. D. Kirkpatrick, A. D. Warren, A. P. Soleimany, P. M. K. Westcott, J. C. Voog, C. Martin-Alonso, H. E. Fleming, T. Tammela, T. Jacks and S. N. Bhatia, Sci. Transl. Med., 2020, 12, eaaw0262 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |