
Synthesis of Pd nanorod arrays on Au nanoframes for excellent ethanol electrooxidation†
Qinru
Yun
a,
Juan
Xu
a,
Tingcha
Wei
ab,
Qifeng
Ruan
c,
Xingzhong
Zhu
 *ab and
Caixia
Kan
*ab
*ab and
Caixia
Kan
*ab
aCollege of Science, Nanjing University of Aeronautics and Astronautics, Nanjing 211106, China. E-mail: cxkan@nuaa.edu.cn; xzzhu@nuaa.edu.cn
bKey Laboratory of Aerospace Information Materials and Physics (NUAA), MIIT, Nanjing 211106, China
cEngineering Product Development, Singapore University of Technology and Design, Singapore 487372
First published on 6th December 2021
Abstract
Au–Pd hollow nanostructures have attracted a lot of attention because of their excellent ethanol electrooxidation performance. Herein, we report a facile preparation of Au nanoframe@Pd array electrocatalysts in the presence of cetylpyridinium chloride. The reduced Pd atoms were directed to mainly deposit on the surface of the Au nanoframes in the form of rods, leading to the formation of Au nanoframe@Pd arrays with a super-large specific surface area. The red shift and damping of the plasmon peak were ascribed to the deposition of the Pd arrays on the surface of the Au nanoframes and nanobipyramids, which was verified by electrodynamic simulations. Surfactants, temperature and reaction time determine the growth process and thereby the architecture of the obtained Au–Pd hollow nanostructures. Compared with the Au nanoframe@Pd nanostructures and Au nanobipyramid@Pd arrays, the Au nanoframe@Pd arrays exhibit an enhanced electrocatalytic performance towards ethanol electrooxidation due to an abundance of catalytic active sites. The Au NF@Pd arrays display 4.1 times higher specific activity and 13.7 times higher mass activity than the commercial Pd/C electrocatalyst. Moreover, the nanostructure shows improved stability towards the ethanol oxidation reaction. This study enriches the manufacturing technology to increase the active sites of noble metal nanocatalysts and promotes the development of direct ethanol fuel cells.
Introduction
Direct ethanol fuel cells (DEFCs) as a renewable and environmentally friendly energy source have attracted much attention due to the shortage of fossil fuels on earth.1–5 Noble metal nanostructures, especially Au–Pd nanostructures, have been recognized as important heterogeneous electrocatalysts for the ethanol oxidation reaction (EOR) due to their higher activity and better stability in alkaline media.6–16 However, the high cost and rarity of the Pd element prevent the large-scale commercial utilization of Au–Pd nanostructures. Increasing the specific surface area of the Au–Pd bimetallic nanoparticles has been demonstrated as a general strategy to enhance the atomic utilization and thus improve the ethanol oxidation performance.17–25Recently, numerous methods have been developed to synthesize controllable Au–Pd bimetallic nanoparticles with large specific surface areas.26–29 The transformation of solid Pd shells into discontinuous Pd shells is the most commonly used method to achieve this purpose. It should be noted that many Au–Pd nanoparticles have been successfully prepared by this method, in which the island-like or porous Pd shells were overgrown on the surface of solid Au cores.30 For example, Fang and co-workers demonstrated that the 2D/1D Au/Pd heterostructures with Pd nanorod arrays arranged in a very high order on two basal facets provide an abundance of active sites for the catalytic reaction.31 Yu et al. have successfully obtained distinctive types of Au/Pd NCs by selective deposition of satellite nanocrystals on the desired sites of a polyhedral central nanocrystal.32 In principle, when the inner atoms of the Au cores were further exposed, the added participating facets and nanoscale confined spaces of the obtained hollow nanostructures could provide more active sites and greater collision frequency to benefit the reaction's efficiency. Recently, Xia and co-workers prepared hollow Pd@Au@Pd nanocages by employing hollow Au nanocages as seeds.33 Pd shells were formed in both the inner and outer surfaces of the Au nanocages by controlling the deposition rate of the Pd atoms. It is worth noting that the precise control of the Pd growth process is challenging, which limited the large-scale production of these hollow Au–Pd nanostructures. In addition, the contribution of the dense Pd shells, in comparison with the porous ones, for improving the specific surface area of the nanostructure was clearly limited. However, there are a few reports on the Au–Pd nanostructures with both the Au core and Pd shell expanded to the limit. Therefore, the preparation of Au–Pd nanostructures with a hollow Au core and porous Pd shells to maximize the specific surface area is still an incipient area and in strong demand.
In this work, we demonstrated a general method to synthesize Au nanoframe@Pd (NF@Pd) arrays by employing hollow Au nanobipyramid (NBP)-embedded Au NFs as the seeds. The Pd arrays were uniformly distributed on the whole surface of the Au NFs, effectively enlarging the specific surface area. As the Pd atoms were deposited, the plasmon peak of the Au NF@Pd arrays was examined as a function of the Pd precursor amount, which was highly accordant with the electrodynamic simulation. Specifically, the surfactant was found to be crucial for the successful formation of the Pd arrays. In comparison with the other Au–Pd bimetallic nanoparticles, the advantage of the obtained Au NF@Pd arrays was the exposure of more active sites, which afforded them much higher electrocatalytic activity towards the EOR. Moreover, the Au NF@Pd arrays exhibited improved durability due to the existence of the Au.
Experimental
Materials
Hexadecyltrimethylammonium bromide (CTAB, 99%) and cetyltrimethylammonium chloride (CTAC, 99%) were obtained from Alfa Aesar. Hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O, 99%), silver nitrate (AgNO3, 99.8%), palladium chloride (PdCl2, 99%), sodium borohydride (NaBH4, 99%), L-ascorbic acid (AA, 99.7%) and hexadecylpyridinium chloride monohydrate (CPC, 99%) were purchased from Sigma-Aldrich. Deionized water with a resistivity of 18.2 MΩ-cm produced using a Direct-Q 5 ultraviolet water purification system was used in all of the experiments.Synthesis of Au NBPs
The Au NBPs were synthesized by the seed-mediated growth method. Briefly, a freshly prepared, ice-cold NaBH4 solution (0.15 mL, 0.01 M) was injected quickly into a pre-made aqueous solution of HAuCl4 (0.125 mL, 0.01 M) mixed with trisodium citrate (0.25 mL, 0.01 M) and water (9.625 mL). The resulting seed solution was kept at room temperature for 2 h. The seed solution (0.6 mL) was injected into the growth solution that was made in advance by mixing together the CTAB solution (40 mL, 0.1 M), HAuCl4 (2 mL, 0.01 M), AgNO3 (0.4 mL, 0.01 M), HCl (0.8 mL, 1 M) and AA (0.32 mL, 0.1 M). The reaction solution was kept at room temperature for the growth of the Au NBPs.Synthesis of the Au NBP-embedded Au NFs
The synthesis of the Au NBP-embedded Au NFs involves three steps: the overgrowth of Ag on Au NBPs, deposition of Au and etching of Ag. Per a typical synthesis, the purified Au NBPs (10 mL) were centrifuged at 7000 rpm for 10 min. The precipitate was redispersed into a CTAC solution (10 mL, 0.08 M), followed by the subsequent addition of AgNO3 (250 μL, 0.01 M) and AA (125 μL, 0.1 M) under gentle shaking. The mixture was placed in an air-bath shaker (65 °C) and kept for 4.5 h in order to form the Au NBP@Ag core–shell nanorods. The resulting sample was centrifuged and redispersed in a CTAB solution (4 mL, 0.05 M). The formation of the Au NBP-embedded Au NFs was carried out by the sequential addition of NaOH (1 mL, 0.2 M) and AA (1 mL, 0.1 M) under gentle shaking. Aqueous HAuCl4 (2.8 mL, 0.1 mM) was titrated into the solution using a syringe pump at a rate of 20 μL min−1 for the deposition of Au on the surface of the Au NBP@Ag nanorods. The resulting sample was centrifuged, followed by the addition of CTAB (300 μL, 0.1 M), NH3·H2O (200 μL, 28 wt%) and H2O2 (4 mL, 6 wt%) for the etching of Ag. The final product was centrifuged and redispersed in water for further use.Synthesis of four types of Au–Pd nanostructures
The obtained Au NFs (2 mL) were centrifuged at 6500 rpm for 10 min and redispersed in the CPC solution (2 mL, 0.01 M). The H2PdCl4 solution (90 μL, 1 mM) and AA (90 μL, 0.01 M) were added under gentle shaking. The reaction mixture was kept in an oven at 65 °C for 30 min. The resulting Au NF@Pd arrays were centrifuged at 6500 rpm for 10 min and stored in water for further use. For the synthesis of the Au NBP@Pd arrays, the procedure was the same as that used for the synthesis of the Au NF@Pd arrays except that the Au NFs were replaced with Au NBPs. For the synthesis of the Au NF@Pd nanostructures, the procedure was the same as that used for the synthesis of the Au NF@Pd arrays except that the CPC was replaced with CTAB or CTAC.Characterization
The extinction spectra were measured using a Shimadzu UV-3600 Plus ultraviolet/visible/near-infrared spectrophotometer with 0.5 cm optical path length plastic cuvettes. Transmission electron microscopy (TEM) imaging was carried out using a microscope (Hitachi HT 7700) operating at 120 kV. High-resolution transmission electron microscopy (HRTEM) imaging, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) characterization and elemental mapping were performed using an FEI Tecnai F20 microscope operating at 200 kV, which was equipped with an Oxford energy-dispersive X-ray (EDX) analysis system. X-ray diffraction (XRD) was performed using a Panalytical Empyrean diffractometer. X-ray photoelectron spectroscopy (XPS) was performed using a SHIMADZU AXIS Ultra DLD. Selected area electron diffraction (SAED) patterns were captured using a ThermoFisher Talos F200X. The electrocatalysts were digested using aqua regia to dissolve the metals and then analyzed by the inductively coupled plasma optical emission spectrometer (ICP-OES) (Agilent ICP-MS 7500a).Electrodynamic simulations
Finite-difference time-domain (FDTD) simulations were performed to simulate the optical properties of the nanostructures using an FDTD Solution 8.24 (Lumerical Solution). A mesh size of 0.5 nm was employed in calculating the extinction spectra of the nanostructures. The dielectric function of Au was obtained by fitting the measured data of Johnson and Christy, and that of Pd was obtained by fitting Palik's data. The refractive index of water was set at 1.33. The shape and size of each nanostructure were taken to match their average values measured from the corresponding TEM results. Specifically, the Au NBP was modeled as two regular pentagonal frustums capped with two pentagonal pyramids at the ends, with a total length of 70 nm and a diameter of 24 nm. The thickness and length of the ridge of the Au nanoframe was 2 nm and 100 nm, respectively. For the models of the Au NF@Pd arrays, Pd cuboid arrays ranging in size from 1 × 1 × 4 nm3 to 3 × 3 × 9 nm3 were located at each ridge of the Au nanoframe with an interval of 5 nm.Electrochemical measurements
All of the electrochemical measurements were performed with a CHI 660E potentiostat using a three-electrode battery at room temperature. In this three-electrode workstation, the working electrode was a glassy carbon electrode (GCE, 3 mm in diameter), the reference electrode was Ag/AgCl, and the opposite electrode was a Pt electrode. The Au NF@Pd arrays (2 mL) were centrifuged and redispersed in 45 μL water and ultrasonically blended with 5 μL N,N-dimethylformamide/carbon black mixed solution and 5 μL 0.5 wt% Nafion solution for 30 min. Next, the 4 μL solution was dribbled onto the GCE and dried at room temperature. The electrochemically active surface area (ECSA) was calculated by the integration of the PdO reduction peaks during cyclic voltammetry (CV) in the N2-saturated KOH (1 M) solution to give a charge that was divided by the specific charge of 420 μC cm−2 for Pd. All of the other electrochemical measurements were performed in a solution of N2-saturated ethanol (0.5 M) and KOH (0.3 M). Chronoamperometric tests (CA) and CV tests were used to test the stability of the electrocatalyst at −0.2 V.Results and discussion
The first Au NBP samples were prepared by the seed-mediated method, followed by a depletion force-induced purification (Fig. S1a, ESI†).34,35 The overgrowth of the Ag atoms on the Au NBPs leads to the formation of the Au NBP@Ag nanorods (Fig. S1b, ESI†), which serve as a template to prepare the Au NBP-embedded Au NFs. The selective deposition of the Au atoms on the Au NBP@Ag nanorods followed the procedure described in our previous work.36 The samples possess uniform morphologies and narrow size distributions due to the high purity of the Au NBPs (Fig. 1a). The deposition of the Pd arrays on the Au NBP-embedded Au NFs was realized with the assistance of CPC. Particularly, the Au NFs with an open morphology and abundance of active sites were crucial for the anisotropic deposition of the Pd atoms. The TEM images shown in Fig. 1b–f reveal the detailed distribution of the Pd atoms on the Au NFs with the increased volume of Pd precursors. It is clearly shown that there are no obvious Pd nanoparticles on the Au NFs when the amount of added H2PdCl4 is less than 30 μL. As the volume is increased to 60 μL, significant Pd arrays with a uniform distribution occur on the edges of the Au NFs (Fig. 1d). Longer and coarser Pd arrays were obtained as the volume increased further to 160 μL, while the density of the Pd arrays show no distinct change (Fig. 1e and f). Taken together, the size of the Pd arrays could be controlled by the volume of the added Pd precursors. It seems that most of the reduced Pd atoms preferred to deposit on the outside of the Au NFs, while rare Pd atoms were seen on the surface of the Au NBPs. To further identify the deposition sites of the Pd atoms, HRTEM imaging and elemental mapping were performed as shown in Fig. 1g–i. The results clearly show the presence of Au, Ag and Pd atoms on the outside of the Au NFs, while the map shows that the majority of the Au atoms maintain their bipyramidal shape. Pd atoms were found to appear on the surface of both the Au NBPs and Au NFs, with most of them on the Au NFs in an array arrangement. | ||
| Fig. 1 (a–f) TEM images of the Au NBP-embedded Au NFs and Au NF@Pd arrays synthesized with 10, 30, 60, 90, and 160 μL H2PdCl4 (1 mM). (g) TEM image of a single Au NF@Pd array. (h) The HRTEM image recorded in the region indicated with a white box in (g). (i) HAADF-STEM and elemental mapping images of a single Au NF@Pd array. The rightmost image is the overlapped image of the Au, Ag, and Pd elemental maps. The elemental mapping images have the same size scale as the HAADF-STEM images. | ||
Additionally, the detailed structure and composition of the Au NF@Pd arrays were identified by HRTEM, SAED patterns, XRD and XPS characterization. It is clearly shown that the Pd atoms preferred to selectively deposit on the surface of the Au NFs to form Pd arrays (Fig. 2a and Fig. S2, ESI†). The lattice fringe of the Pd array is 0.23 nm, which corresponds to the lattice spacing of the {111} plane of Pd. Meanwhile, the deposition of Pd arrays had little influence on the original structure of the Au NFs (Fig. S3, ESI†). The SAED of the Au NF@Pd arrays exhibit a series of concentric rings corresponding to the {111}, {200}, and {220} planes, respectively (Fig. 2b). However, the content of Pd was too low to be identified through an XRD pattern (Fig. S4, ESI†). The XPS measurement was performed to investigate the surface composition and metal valence of the Au NF@Pd arrays. The two symmetric peaks located at 83.1 and 86.8 eV in the high-resolution Au 4f spectrum are identified as the Au 4f7/2 and Au 4f5/2 signals, respectively, and therefore demonstrate the metallic Au0 state (Fig. 2c). In addition, the peaks at 334.3 eV (Pd 3d5/2) and 339.4 eV (Pd 3d3/2) are attributed to the metallic state of Pd, whereas those at 335.2 and 340.1 eV are assigned to the Pd2+ species. The percentage of Pd0 is about 74.3%, implying that its metallic state is predominant in the Au NF@Pd arrays.
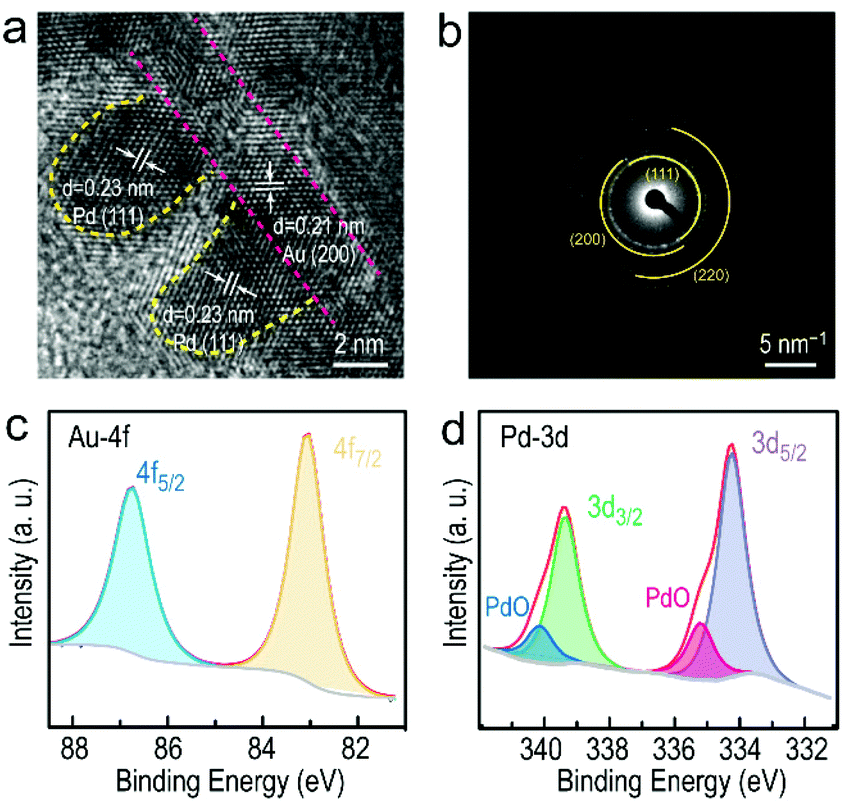 | ||
| Fig. 2 (a) HRTEM image of the Pd arrays on Au NFs. (b) SAED image of the corresponding Au NF@Pd arrays. (c) Au 4f and (d) Pd 3d XPS spectra for the Au NF@Pd arrays. | ||
Apart from the morphology evolution, the increase of the Pd precursors leads to the gradual red shift of the longitudinal plasmon peak of the Au NF@Pd arrays in comparison with that of the starting Au NBP-embedded Au NFs, which is accompanied by peak broadening (Fig. 3a). In contrast, the transverse plasmon peak shows a slight shift. As shown in Fig. 3b, the peak's position and intensity were plotted as a function of the added volume of the Pd precursors. The peak intensity changes dramatically and becomes saturated as the Pd precursors continuously increase. The red shift of the wavelength and the reduction of the peak intensity arise mainly from the increase in the size distribution of the Au NF@Pd arrays and the plasmon damping caused by the large imaginary part of the dielectric function of Pd.37,38
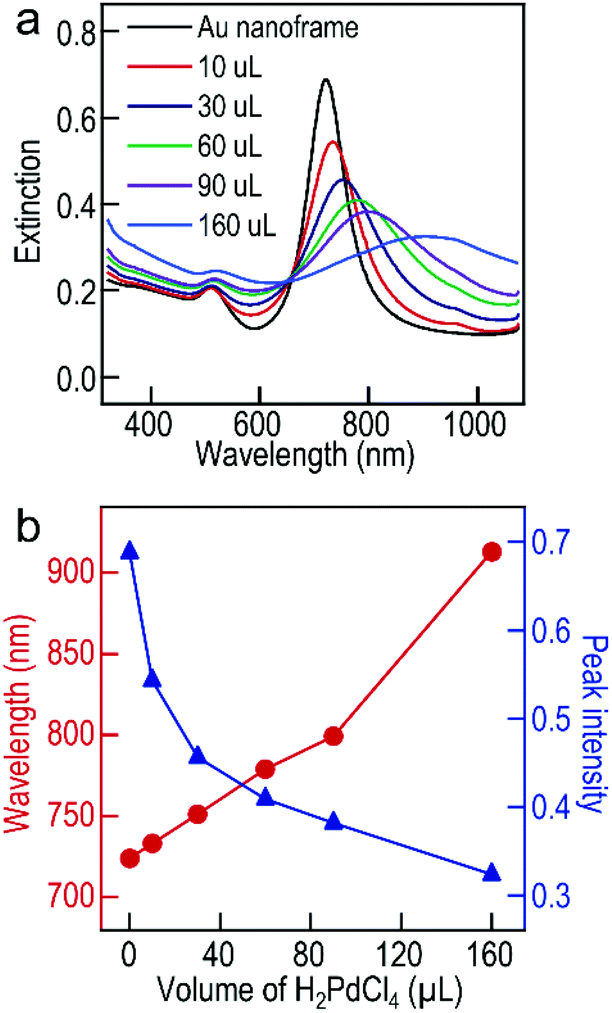 | ||
| Fig. 3 (a) Extinction spectra of the Au NBP-embedded Au NF and Au NF@Pd array samples produced with different amounts of H2PdCl4. (b) Variations of the longitudinal plasmon wavelength and peak intensity as a function of the volume of H2PdCl4. | ||
In order to obtain a further understanding of the red shift and damping of the plasmon peaks caused by the growth of the Pd arrays, FDTD simulations were performed to ascertain the plasmonic properties of the Au NF@Pd arrays (Fig. 4). The geometrical models used in the simulations were set according to the relevant parameters obtained from the TEM images. The length and waist diameter of the Au NBPs were set at 70 nm and 24 nm, respectively. The thickness and height of the Au nanoframes were set at 2 nm and 100 nm, respectively. To simplify the model, the Pd nanorod arrays on the Au nanoframe were set as cuboids. The length, width and height of the Pd cuboids were varied from 1 × 1 × 4 to 3 × 3 × 9 nm3 at an interval of 0.5, 0.5, and 1 nm (Fig. 4a). The diameter of the Pd nanospheres on the surface of the Au NBP was varied with the width of the Pd cuboids. In these models, the direction of the excitation light was perpendicular to the length axis. Thus, only the longitudinal plasmon mode is excited. The peak intensity of the longitudinal plasmon peak greatly decreased as the volume of the Pd nanocrystal increased (Fig. 4b), which is in agreement with the experimental results in Fig. 3a. When comparing the extinction spectra of the models in Fig. 4 with those cases depositing Pd arrays only on the outside of the Au NFs (Fig. S5, ESI†), we can conclude that the presence of the Pd nanoparticles on the surface of the Au NBPs also contributes to the damping of the plasmon peak.
 | ||
| Fig. 4 FDTD simulations. (a) Schematic models used in the simulations. (b) Simulated extinction spectra for the nanostructures. | ||
Based on the above results, we proposed the growth process of the Pd arrays on the Au NBP-embedded Au NFs in Fig. 5. Initially, Pd atoms would deposit at the Au NFs preferentially due to the abundant active sites. The subsequently reduced Pd atoms would continuously deposit at the same sites to form Pd arrays in the presence of CPC. CPC is found to be crucial for directing the formation of the Pd arrays via the self-organization of the surfactant molecules on the surfaces of the Au NFs, which originates from the stronger polarizability and additional aromatic π–π stacking interactions of CP+.30,39 The control experiments using other surfactants achieved different phenomena: the product obtained using CTAB shows a continuous Pd shell on the surface of the Au NFs while CTAC causes random island growth of the Pd atoms (Fig. S6, ESI†). Additionally, it is worth noting that an appropriate reaction temperature is also an important factor for the uniform growth of the Pd arrays by tailoring the surface diffusion rate of the Pd atoms (Fig. S7, ESI†). The reaction time should be controlled within half an hour to ensure the integrity of the Pd arrays (Fig. S8, ESI†).
 | ||
| Fig. 5 Schematic illustration of the growth process of the Au NF@Pd arrays. | ||
In the past few years, Au–Pd nanostructures have been considered as important electrocatalysts towards the ethanol oxidation reaction in DEFCs due to their excellent activity and stability.40 The architectures of the Au NF@Pd arrays provide a large specific surface area and abundant catalytically active sites, which play a vital role in the determination of the performance during the catalytic process. We therefore evaluated the Au NF@Pd arrays as electrocatalysts for the oxidation of ethanol in an alkaline environment and benchmarked them against the commercial Pd/C, Au NBP@Pd array (Fig. 6) and Au NF@Pd nanostructures (Fig. S6b, ESI†).
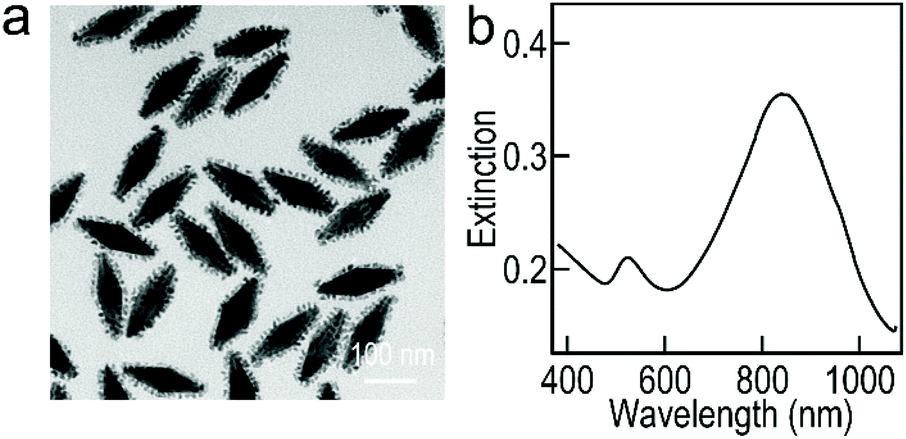 | ||
| Fig. 6 Characterization of the Au NBP@Pd array electrocatalysts. (a) TEM image of the Au NBP@Pd arrays. (b) Extinction spectrum of the Au NBP@Pd arrays. | ||
Prior to the electrochemical catalysis, all of the catalysts should be tested by CV in a N2-saturated KOH solution (Fig. 7a). The amount of Pd in each catalyst was determined to be 0.3, 0.278, 0.305, and 0.3 μg, respectively, by ICP-OES. In an alkaline environment, the electrochemical surface area (ECSA) can be calculated by the following formula:41

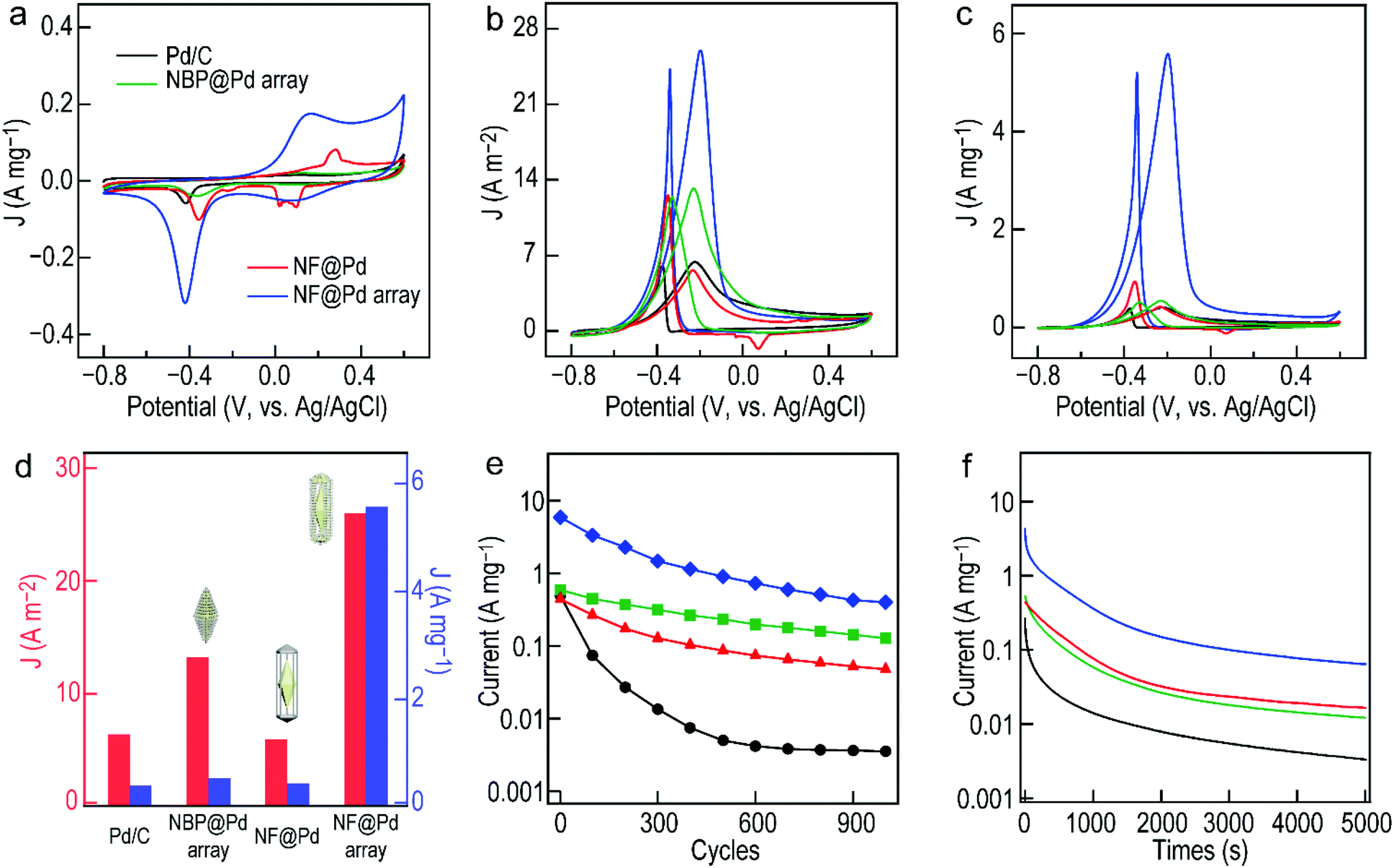 | ||
| Fig. 7 CV curves of the commercial Pd/C, Au NBP@Pd array, Au NF@Pd and Au NF@Pd array in a N2-saturated (a) aqueous KOH and (b and c) mixture of ethanol and KOH. The currents shown in (a and c) and (b) are normalized by the Pd mass loaded and ECSA values, respectively. (d) Specific and mass activities towards the EOR. (e) Cycling measurements and (f) CA curves using the commercial Pd/C, Au NBP@Pd array, Au NF@Pd and Au NF@Pd array as electrocatalysts. The scan rate was fixed at 50 mV s−1. | ||
The CV curves recorded in the presence of the above electrocatalysts in a mixture of ethanol and KOH depict two characteristic oxidation peaks observed for the EOR (Fig. 7b). The peak in the forward scan around −0.2 V can be ascribed to the oxidation of the freshly chemisorbed ethanol, while the peak in the backward scan around −0.35 V comes primarily from the further oxidation of the carbonaceous materials generated during ethanol oxidation. The peak value of the forward scan therefore represents the catalytic activity of the corresponding electrocatalyst. The specific activity calculated by normalizing the peak intensity to its ECSA is 25.99 A m−2 for the Au NF@Pd array catalyst, which is 4.1 times higher than that of the commercial Pd/C catalyst (6.39 A m−2). The Au NBP@Pd array and Au NF@Pd electrocatalysts exhibit specific activities of 13.25 and 5.96 A m−2, respectively. The mass activity based on the Pd mass loading on a working electrode made of the Au NF@Pd arrays is 5.58 A mg−1, while those of the commercial Pd/C, Au NBP@Pd arrays, and Au NF@Pd nanostructures are 0.41, 0.54, and 0.45 A mg−1, respectively (Fig. 7c). The change of the potential to a reversible hydrogen electrode (RHE) shows similar results (Fig. S9, ESI†). The specific and mass activities of the above Pd-based electrocatalysts are summarized and shown in Fig. 7d. It is clear that the Au NF@Pd arrays show the highest electrocatalytic activity among the four electrocatalysts, with mass and specific activity 13.7 and 4.1 times higher than those of the commercial Pd/C, respectively. Compared to the state-of-the-art Au–Pd bimetallic electrocatalysts, this result is at the same tier of the best performance reported in the literature (Table 1). We believe that the improvement of the catalytic performance is due to its unique structure. First, Pd arrays on the surface of the electrocatalysts provide more catalytic sites. Second, a charge transfer between two dissimilar surface atoms (Au and Pd) alters their electronic structure and activity, which change the redox properties of the PdO formation/reduction.31,52,53
| Name | ECSA (m2 g−1) | MA (A mg−1) | Timesa |
|---|---|---|---|
| a Time is the ratio of MA between the catalysts and commercial Pd/C. | |||
| Au NF@Pd array [this work] | 214.6 | 5.58 | 13.7 |
| PdNN41 | 85.6 | 2.04 | 3.5 |
| Au@AuPd NPs43 | 125.8 | 11.9 | 31.3 |
| PdAgPt nanoalloys44 | 64.5 | 3.33 | 7.02 |
| Au/PdAg NFs45 | 42.6 | 1.1 | 5.5 |
| AuPd NWs46 | 56.99 | 7.09 | 10.30 |
| Au8Pd3 NWs47 | 169.28 | 10.57 | 11.74 |
| Au@PdNi48 | 38.5 | 5.89 | 9.48 |
| TOH Au@Pd NPs49 | 100.9 | 13.2 | 66 |
| CuPd/Ir@Au NSs22 | 83.45 | 7.78 | 17.3 |
| Pd1Au3 NSFs50 | 4.89 | 0.365 | 1.4 |
| PdAu NNWs51 | 18 | 10.8 | 7.2 |
In addition to the activity, the durability of the catalyst is another important factor to assess the catalytic performance of the electrocatalysts. First of all, the cycling durability of the catalysts was studied by CV for 1000 cycles between −0.8 and 0.6 V at a fixed scan rate (Fig. 7e). The plot of the mass activity against cycle number for the four electrocatalysts shows that the mass activity of the commercial Pd/C almost drops to 0 (0.003 A mg−1) after 1000 cycles. For the Au NF@Pd arrays, the mass activity is retained by 0.399 A mg−1, still much higher than those of the NBP@Pd array (0.127 A mg−1) and NF@Pd nanostructures (0.048 A mg−1). Next, the electrochemical stability of the catalyst was tested by the timing current method. The chronoamperometric (CA) tests were conducted at −0.2 V for 5000 s (Fig. 7f). All of the electrocatalysts suffer an unavoidable activity loss at the first stage due to the concentration difference caused by the diffusion of ethanol on the anode surface and the PdO formation (Fig. S10, ESI†). Although the Au NF@Pd arrays show a drop in activity, they still retain the highest mass activity (0.064 A mg−1) towards the EOR in comparison with the other Au–Pd nanostructures. Moreover, the TEM images of the Au NF@Pd arrays after CA measurement verify their structural stability during electrocatalytic evaluation (Fig. S11, ESI†). In addition, the relative contents of Au, Pd, and Ag were all constant after the cycling durability process (Table S1, ESI†). The increase in the durability of the Au–Pd hollow nanostructures with a large specific surface area and excellent electrocatalytic activity certainly warrants further study.
Conclusions
In summary, we have successfully synthesized a kind of Au–Pd hollow nanostructure, named Au NF@Pd arrays, by using a Au NBP-embedded Au NF as a template. In the presence of CPC, most of the reduced Pd atoms were preferentially deposited on the surface of the Au NF in an array arrangement. The resulting Au NF@Pd arrays have a large specific surface area and thus more catalytic active sites. The deposition of the Pd atoms led to the red shift and damping of the longitudinal plasmon peaks, which has been verified by FDTD simulation. The type of surfactant, temperature and reaction time were found to be the critical factors in the formation of the Au NF@Pd arrays. Ethanol electrooxidation in an alkaline environment was carried out to test the electrocatalytic performance of the Au NF@Pd arrays. In comparison with the Au NBP@Pd arrays and Au NF@Pd nanostructures, the combination of the Au NF and Pd array provides our Au NF@Pd arrays with 4.1 times higher specific activity and 13.7 times higher mass activity than the commercial Pd/C electrocatalyst. Meanwhile, the Au NF@Pd arrays also show better stability than the other three electrocatalysts. Our work provides a versatile strategy for the construction of Au–Pd hollow nanostructures with a super-large special surface area and superior EOR, which can be extended to other metal electrocatalysts and promote the development of DEFCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21805137, 11774171, 11874220 and 11974182) and the Natural Science Foundation of Jiangsu Province (BK20180444).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- V. M. Barragán and A. Heinzel, J. Power Sources, 2002, 104, 66–72 CrossRef.
- C. W. Xu, P. K. Shen and Y. L. Liu, J. Power Sources, 2007, 164, 527–531 CrossRef CAS.
- Y. S. Li, T. S. Zhao and Z. X. Liang, J. Power Sources, 2009, 187, 387–392 CrossRef CAS.
- F. Vigier, C. Coutanceau, A. Perrard, E. M. Belgsir and C. Lamy, J. Appl. Electrochem., 2004, 34, 439–446 CrossRef CAS.
- Z. X. Liang, T. S. Zhao, J. B. Xu and L. D. Zhu, Electrochim. Acta, 2009, 54, 2203–2208 CrossRef CAS.
- M. S. Ahmed, M.-S. Kim and S. Jeon, J. Nanosci. Nanotechnol., 2016, 16, 8294–8301 CrossRef CAS.
- Z. Y. Zhang, L. Xin, K. Sun and W. Z. Li, Int. J. Hydrogen Energy, 2011, 36, 12686–12697 CrossRef CAS.
- M. S. Ahmed and S. Jeon, ACS Catal., 2014, 4, 1830–1837 CrossRef CAS.
- C. N. Brodsky, A. P. Young, K. C. Ng, C.-H. Kuo and C.-K. Tsung, ACS Nano, 2014, 8, 9368–9378 CrossRef CAS PubMed.
- J. C. Guo, R. Huang, Y. Li, Z. Y. Yu, L. Y. Wan, L. Huang, B. B. Xu, J. Y. Ye and S. G. Sun, J. Phys. Chem. C, 2019, 123, 23554–23562 CrossRef CAS.
- Z. J. Li, M. Li, X. Wang, G. T. Fu and Y. W. Tang, Nanoscale Adv., 2021, 3, 1813–1829 RSC.
- M. Li, Z. J. Li, G. T. Fu and Y. W. Tang, Small, 2021, 17, 2007179 CrossRef CAS PubMed.
- W. Q. Yao, X. Jiang, M. Li, Y. L. Li, Y. Y. Liu, X. Zhan, G. T. Fu and Y. W. Tang, Appl. Catal., B, 2021, 282, 119595 CrossRef CAS.
- Z. S. Teng, M. Li, Z. J. Li, Z. Y. Liu, G. T. Fu and Y. W. Tang, Mater. Today Energy, 2021, 19, 100596 CrossRef CAS.
- J. H. Hu, R. B. Jiang, H. Zhang, Y. Z. Guo, J. Wang and J. F. Wang, Nanoscale, 2018, 10, 18473–18481 RSC.
- B. Lim and Y. N. Xia, Angew. Chem., Int. Ed., 2011, 50, 76–85 CrossRef CAS PubMed.
- F. Godínez-Salomón, R. Mendoza-Cruz, M. J. Arellano-Jimenez, M. Jose-Yacaman and C. P. Rhodes, ACS Appl. Mater. Interfaces, 2017, 9, 18660–18674 CrossRef PubMed.
- T. Asset, R. Chattot, M. Fontana, B. Mercier-Guyon, N. Job, L. Dubau and F. Maillard, ChemPhysChem, 2018, 19, 1552–1567 CrossRef CAS PubMed.
- Y. X. Gao and Y. Ding, Chem. – Eur. J., 2020, 26, 8845–8856 CrossRef CAS PubMed.
- T. Kwon, M. Jun, G. J. Bang, H. Yang, J. Joo, T. Kim, J. Kim, J. M. Kim, H. Baik, Y. Jung, J. Y. Kim and K. Lee, Cell Rep. Phys. Sci., 2020, 1, 100260 CrossRef.
- L. X. Luo, C. H. Fu, X. H. Yan, S. Y. Shen, F. Yang, Y. G. Guo, F. J. Zhu, L. J. Yang and J. L. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 25961–25971 CrossRef CAS PubMed.
- T. Wu, Y. Ma, Z. B. Qu, J. C. Fan, Q. X. Li, P. H. Shi, Q. J. Xu and Y. L. Min, ACS Appl. Mater. Interfaces, 2019, 11, 5136–5145 CrossRef CAS PubMed.
- X. B. Yang, Z. P. Liang, S. Chen, M. J. Ma, Q. Wang, X. L. Tong, Q. H. Zhang, J. Y. Ye, L. Gu and N. J. Yang, Small, 2020, 16, 2004727 CrossRef CAS PubMed.
- N. S. Marinkovic, M. Li and R. R. Adzic, Top. Curr. Chem., 2019, 377, 11 CrossRef PubMed.
- B. Karimi, H. Barzegar and H. Vali, Chem. Commun., 2018, 54, 7155–7158 RSC.
- H. Jing and H. Wang, CrystEngComm, 2014, 16, 9469–9477 RSC.
- G.-R. Zhang, J. Wu and B.-Q. Xu, J. Phys. Chem. C, 2012, 116, 20839–20847 CrossRef CAS.
- R. G. Weiner and S. E. Skrabalak, Chem. Mater., 2016, 28, 4139–4142 CrossRef CAS.
- J. Guo, Y. Zhang, L. Shi, Y. F. Zhu, M. F. Mideksa, K. Hou, W. S. Zhao, D. W. Wang, M. T. Zhao, X. F. Zhang, J. W. Lv, J. Q. Zhang, X. L. Wang and Z. Y. Tang, J. Am. Chem. Soc., 2017, 139, 17964–17972 CrossRef CAS PubMed.
- C. H. Fang, T. Bi, Q. Ding, Z. Q. Cui, N. Yu, X. X. Xu and B. Y. Geng, ACS Appl. Mater. Interfaces, 2019, 11, 20117–20124 CrossRef CAS PubMed.
- Y. Yu, Q. B. Zhang, J. P. Xie and J. Y. Lee, Nat. Commun., 2013, 4, 1454 CrossRef PubMed.
- M. X. Yang, W. X. Wang, K. D. Gilroy and Y. N. Xia, Nano Lett., 2017, 17, 5682–5687 CrossRef CAS PubMed.
- Q. Li, X. L. Zhuo, S. Li, Q. F. Ruan, Q.-H. Xu and J. F. Wang, Adv. Opt. Mater., 2015, 3, 801–812 CrossRef CAS.
- X. Z. Zhu, X. L. Zhuo, Q. Li, Z. Yang and J. F. Wang, Adv. Funct. Mater., 2016, 26, 341–352 CrossRef CAS.
- X. Z. Zhu, J. Xu, H. Zhang, X. M. Cui, Y. Z. Guo, S. Cheng, C. X. Kan and J. F. Wang, Chem. Sci., 2020, 11, 3198–3207 RSC.
- Y. W. Lee, M. Kim, Z. H. Kim and S. W. Han, J. Am. Chem. Soc., 2009, 131, 17036–17037 CrossRef CAS PubMed.
- H. J. Chen, F. Wang, K. Li, K. C. Woo, J. F. Wang, Q. Li, L.-D. Sun, X. X. Zhang, H.-Q. Lin and C.-H. Yan, ACS Nano, 2012, 6, 7162–7171 CrossRef CAS PubMed.
- D. Kuang, T. Brezesinski and B. Smarsly, J. Am. Chem. Soc., 2004, 126, 10534–10535 CrossRef CAS PubMed.
- J. Li, H.-M. Yin, X.-B. Li, E. Okunishi, Y.-L. Shen, J. He, Z.-K. Tang, W.-X. Wang, E. Yücelen, C. Li, Y. Gong, L. Gu, S. Miao, L.-M. Liu, J. Luo and Y. Ding, Nat. Energy, 2017, 2, 17111 CrossRef CAS.
- H. Begum, M. S. Ahmed and S. Jeon, ACS Appl. Mater. Interfaces, 2017, 9, 39303–39311 CrossRef CAS PubMed.
- X. H. Xie, Y. Nie, S. G. Chen, W. Ding, X. Q. Qi, L. Li and Z. D. Wei, J. Mater. Chem. A, 2015, 3, 13962–13969 RSC.
- C. X. Bi, Y. H. Song, H. P. He, C. S. Wu, W. Du, L. H. Huang, H. Moehwald and H. B. Xia, J. Mater. Chem. A, 2018, 6, 7675–7685 RSC.
- W. Y. Lu, X. Y. Xia, X. X. Wei, M. M. Li, M. Zeng, J. Guo and S. Cheng, ACS Appl. Mater. Interfaces, 2020, 12, 21569–21578 CrossRef CAS PubMed.
- C. H. Fang, G. L. Zhao, Z. J. Zhang, J. P. Zheng, Q. Ding, X. X. Xu, L. Shao and B. Y. Geng, Part. Part. Syst. Charact., 2018, 35, 1800258 CrossRef.
- L. L. Zhou, X. L. Xie, R. G. Xie, H. Guo, M. H. Wang and L. J. Wang, Int. J. Hydrogen Energy, 2019, 44, 25589–25598 CrossRef CAS.
- L. K. Wang, Z. Liu, S. Z. Zhang, M. Y. Li, Y. Y. Zhang, Z. F. Li and Z. H. Tang, Int. J. Hydrogen Energy, 2021, 46, 8549–8556 CrossRef CAS.
- N. Sui, T. Wang, Q. Bai, R. P. Yue, H. R. Jiang, H. L. Xiao, M. H. Liu, L. N. Wang, Z. L. Zhu and W. W. Yu, J. Alloys Compd., 2019, 817, 153335 CrossRef.
- Y. H. Song, C. S. Xiang, C. X. Bi, C. S. Wu, H. P. He, W. Du, L. H. Huang, H. Tian and H. B. Xia, Nanoscale, 2018, 10, 22302–22311 RSC.
- Z.-Z. Yang, L. Liu, A.-J. Wang, J. H. Yuan, J.-J. Feng and Q.-Q. Xu, Int. J. Hydrogen Energy, 2017, 42, 2034–2044 CrossRef CAS.
- W. Hong, J. Wang and E. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 9481–9487 CrossRef CAS PubMed.
- C. X. Bi, C. Feng, T. T. Miao, Y. H. Song, D. Y. Wang and H. B. Xia, Nanoscale, 2015, 7, 20105–20116 RSC.
- T. Ma and F. Liang, J. Phys. Chem. C, 2020, 124, 7812–7822 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images, extinction spectra and simulations. See DOI: 10.1039/d1nr05987d |
| This journal is © The Royal Society of Chemistry 2022 |