
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A bioinspired glycopolymer for capturing membrane proteins in native-like lipid-bilayer nanodiscs†
Bartholomäus
Danielczak
a,
Marie
Rasche
b,
Julia
Lenz
a,
Eugenio
Pérez Patallo
a,
Sophie
Weyrauch
a,
Florian
Mahler
a,
Michael Tope
Agbadaola
 ac,
Annette
Meister
d,
Jonathan Oyebamiji
Babalola
c,
Carolyn
Vargas
aefg,
Cenek
Kolar
b and
Sandro
Keller
*aefg
ac,
Annette
Meister
d,
Jonathan Oyebamiji
Babalola
c,
Carolyn
Vargas
aefg,
Cenek
Kolar
b and
Sandro
Keller
*aefg
aMolecular Biophysics, Technische Universität Kaiserslautern (TUK), Erwin-Schrödinger-Str. 13, 67663 Kaiserslautern, Germany
bGlycon Biochemicals GmbH, Im Biotechnologiepark TGZ 1, 14943 Luckenwalde, Germany
cDepartment of Chemistry, University of Ibadan, 200284, Ibadan, Nigeria
dInstitute of Biochemistry and Biotechnology, and ZIK HALOmem, Martin Luther University Halle–Wittenberg, Kurt-Mothes-Str. 3a, 06120 Halle (Saale), Germany
eBiophysics, Institute of Molecular Biosciences (IMB), NAWI Graz, University of Graz, Humboldtstr. 50/III, 8010 Graz, Austria. E-mail: sandro.keller@uni-graz.at
fField of Excellence BioHealth, University of Graz, Graz, Austria
gBioTechMed-Graz, Graz, Austria
First published on 23rd November 2021
Abstract
Amphiphilic copolymers that directly extract membrane proteins and lipids from cellular membranes to form nanodiscs combine the advantages of harsher membrane mimics with those of a native-like membrane environment. Among the few commercial polymers that are capable of forming nanodiscs, alternating diisobutylene/maleic acid (DIBMA) copolymers have gained considerable popularity as gentle and UV-transparent alternatives to aromatic polymers. However, their moderate hydrophobicities and high electric charge densities render all existing aliphatic copolymers rather inefficient under near-physiological conditions. Here, we introduce Glyco-DIBMA, a bioinspired glycopolymer that possesses increased hydrophobicity and reduced charge density but nevertheless retains excellent solubility in aqueous solutions. Glyco-DIBMA outperforms established aliphatic copolymers in that it solubilizes lipid vesicles of various compositions much more efficiently, thereby furnishing smaller, more narrowly distributed nanodiscs that preserve a bilayer architecture and exhibit rapid lipid exchange. We demonstrate the superior performance of Glyco-DIBMA in preparative and analytical applications by extracting a broad range of integral membrane proteins from cellular membranes and further by purifying a membrane-embedded voltage-gated K+ channel, which was fluorescently labeled and analyzed with the aid of microfluidic diffusional sizing (MDS) directly within native-like lipid-bilayer nanodiscs.
1. Introduction
The gentle isolation of membrane proteins from cellular membranes is a major challenge1 that is increasingly met by the use of nanodisc-forming amphiphilic polymers such as aliphatic diisobutylene/maleic acid (DIBMA, Fig. 1a)2–8 or aromatic styrene/maleic acid (SMA) copolymers.3,9–13 Unlike traditional head-and-tail detergents, which displace the native lipid environment of membrane proteins,14 amphiphilic copolymers co-extract a nanoscopic lipid patch from the cellular membrane to form polymer-bounded lipid-bilayer nanodiscs.12 Thus, these polymers allow encapsulated membrane proteins to remain embedded in a near-native lipid bilayer during isolation, purification, and investigation. The advantages offered by this approach manifest in superior membrane-protein stability6,10,13,15–22 and more native-like ligand interactions15 and other protein activities6,16,19 as compared with detergent micelles.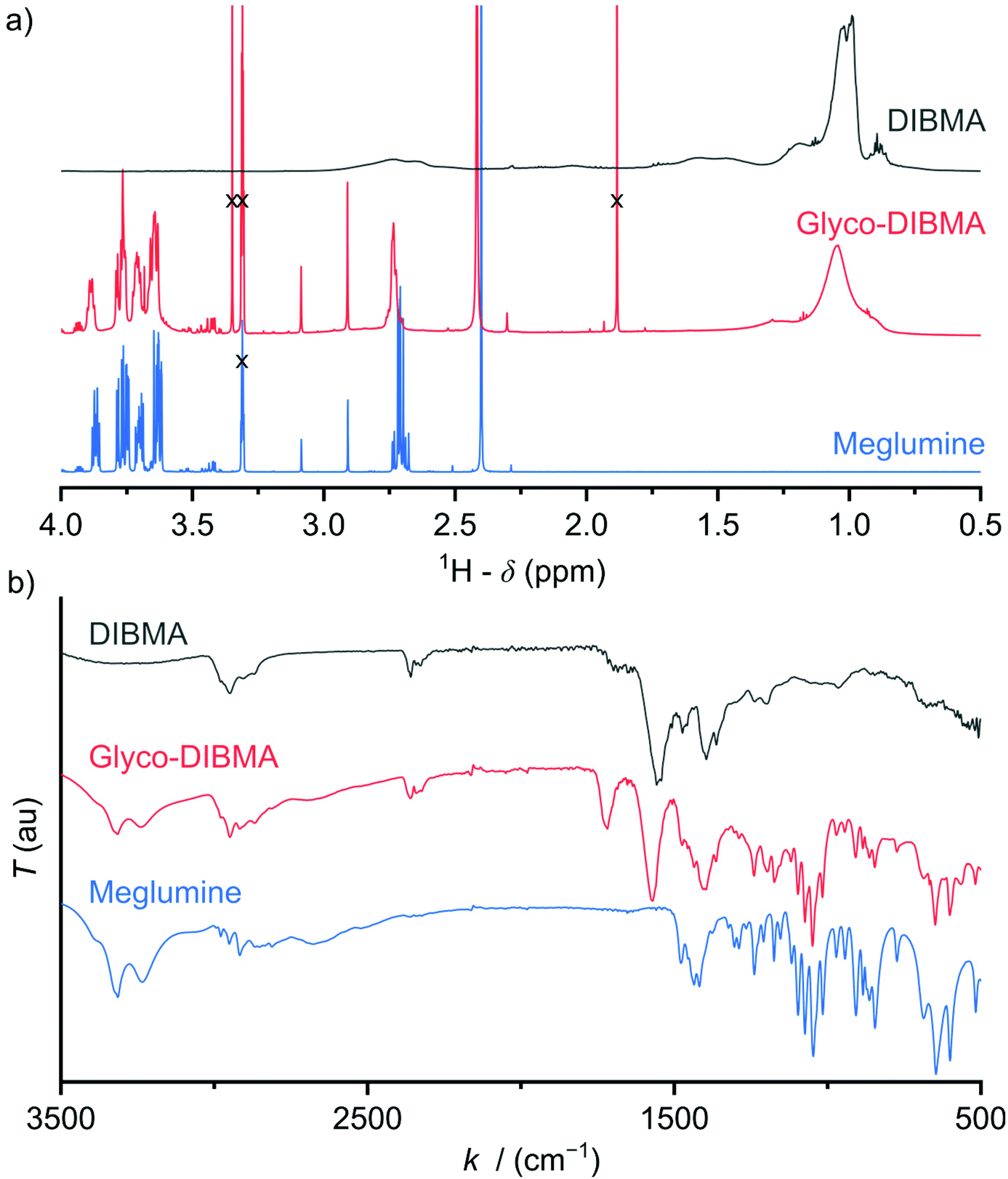 | ||
| Fig. 1 (a) 1H-NMR spectra of DIBMA, Glyco-DIBMA, and meglumine. The DIBMA spectrum was taken in D2O and referenced to the signal of 3-(trimethylsilyl)propane-1-sulfonate (DSS) at 0 ppm, whereas the spectra of Glyco-DIBMA and meglumine were taken in CD3OD and referenced to the signal of residual CD3OH at 3.31 ppm. Solvent signals are marked with a cross. (b) ATR-FTIR spectra of DIBMA, Glyco-DIBMA, and meglumine. | ||
Unlike most other polymers used for extracting membrane proteins,2,3,8 DIBMA copolymers contain no aromatic moieties and, thus, have substantially lower background absorption in the UV range. However, these alternating polymers are characterized by a high density of carboxylic acid groups and, consequently, relatively low hydrophobicity. Under physiological conditions of near-neutral pH, the high charge density of DIBMA results in strong coulombic repulsion between the polymer and anionic lipid membranes on the one hand and among polymer chains on the other.4,23,24
These adverse effects reduce the effective membrane affinity and, thereby, the solubilization efficiency of alternating aliphatic polymers. Moreover, coulombic repulsion is expected to limit the density of polymer chains in the nanodisc rim, which might be responsible for the broad size distributions observed for DIBMA/lipid particles (DIBMALPs).2,23 We and others have shown that coulombic repulsion can be reduced either by adjusting the buffer composition to increase the ionic strength of the solution2,11,25 or by reducing the effective charge on the polymer through partial protonation at low pH23,25 or through adsorption of divalent cations.2,4,8,26 Although effective, all of these remedies render the solution conditions quite unphysiological and, in doing so, partly offset the benefits of alternating aliphatic polymers for handling membrane proteins under conditions that ideally should be as native-like as possible.
On this premise, we sought to develop a new, powerful polymer that is intrinsically less charged and more hydrophobic, with the intention of rendering it an efficient solubilizer also under physiological conditions. At the same time, the new polymer should retain the essential favorable properties of existing aliphatic copolymers and should be synthetically accessible by post-polymerization functionalization of a commercial polymer backbone, thus avoiding the need for laborious and expensive de novo polymer synthesis. Herein, we present Glyco-DIBMA (Scheme 1) as a new glycopolymer that is partially amidated with the amino sugar N-methyl-D-glucamine (“meglumine”). Utilizing a range of biochemical, biophysical, and imaging techniques, we show that Glyco-DIBMA outperforms established aliphatic copolymers in that it gives rise to smaller and more narrowly distributed lipid-bilayer nanodiscs from both model and cellular membranes under a broad variety of solution conditions. Membrane proteins extracted with the aid of Glyco-DIBMA are amenable to protein purification by chromatography and to further manipulation and analysis under well-controlled yet native-like conditions.
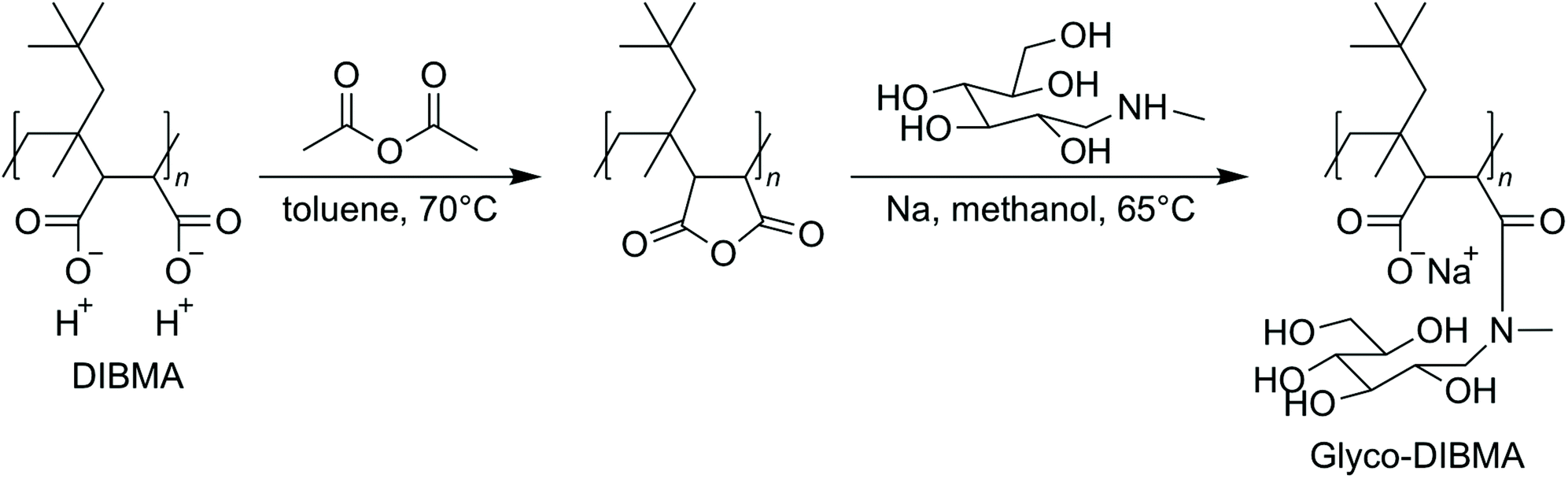 | ||
| Scheme 1 Synthesis of Glyco-DIBMA. | ||
2. Results and discussion
Glycosylation is nature's most powerful and versatile means of endowing biomacromolecules with enhanced water solubility without accumulating additional electric charge. Inspired by this, we reasoned that glycosylation might provide us with an opportunity to lower the charge density and, thereby, increase the hydrophobicity of a polyanionic polymer backbone without, however, compromising its excellent water solubility. A further crucial requirement was that the synthetic route should be reasonably straightforward and clean such as to afford the final product in sufficient quantity—that is, on the scale of hundreds of grams—and in high purity for compatibility with sensitive biomolecular specimens. In accordance with these considerations, we synthesized Glyco-DIBMA in a two-step process by first converting free DIBMA acid into the more reactive anhydride form and then amidating the latter with meglumine (Scheme 1).The expected chemical structure of Glyco-DIBMA was corroborated by 1H-NMR spectroscopy as well as attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectroscopy (Fig. 1) and its relatively narrow chain length distribution by analytical size exclusion chromatography (SEC) in aqueous phase (Fig. S1†). The 1H-NMR spectrum of Glyco-DIBMA displayed the broad polymer peaks also observed for DIBMA and, additionally, the considerably sharper multiplet peaks characteristic of meglumine (Fig. 1a). Nevertheless, all signals stemming from the sugar moiety became broader and shifted to lower magnetic field strengths in Glyco-DIBMA as compared with free meglumine (Fig. 1a), thus confirming successful coupling. This was further supported by ATR-FTIR (Fig. 1b), as the spectrum of Glyco-DIBMA again revealed both the peaks typical of DIBMA and those of meglumine. In SEC (Fig. S1†), Glyco-DIBMA eluted slightly earlier than DIBMA, as expected on the basis of its larger molar mass resulting from glycosylation. However, it should be noted that the elution behavior of polymers also depends on the degree of polymer compaction, that is, the distribution of effective polymer chain extensions in solution. The latter is notoriously difficult to account for quantitatively because it depends not only on intrinsic properties of the polymer such as the number of ionizable groups but also on solution conditions such as the pH value and the concentration of multivalent counterions.4,23 In summary, all three methods consistently attested to the high chemical purity and favorable solution behavior of Glyco-DIBMA, thus opening the way for a detailed investigation of its lipid solubilization and protein-extraction properties.
We examined the ability of Glyco-DIBMA to spontaneously form lipid-bilayer nanodiscs by mixing it with large unilamellar vesicles (LUVs) made from the zwitterionic phospholipid 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC). Dynamic light scattering (DLS) showed that addition of Glyco-DIBMA to DMPC at a mass ratio of polymer (P) to lipid (L) of mP/mL = 1.5 completely abolished the original vesicular structures, which gave way to nanoscopic particles having a z-average hydrodynamic diameter and an associated size distribution width of dz = (15 ± 6) nm (Fig. 2a). These Glyco-DIBMA/lipid particles (Glyco-DIBMALPs) were considerably smaller and more narrowly distributed than DIBMALPs produced at the same mass ratio, which were characterized by dz = (31 ± 13) nm. Upon titrating DMPC LUVs with Glyco-DIBMA (Fig. 2b), we observed an initial increase in dz at mP/mL ≤ 0.5 followed by a smooth decrease to reach dz = (12 ± 5) nm at mP/mL = 2.5. DIBMA exhibited a qualitatively similar pattern but, throughout the titration, gave rise to larger nanoparticles that displayed broader size distributions and that co-existed with “free” polymer not associated with nanodiscs. Both of these phenomena manifested particularly clearly in analytical SEC, which at mP/mL = 1.5 (Fig. 2c) revealed two broad peaks centered at elution volumes of VE ≈ 12 mL and 18 mL corresponding to nanodiscs and free polymer, respectively. In stark contrast with this, Glyco-DIBMALPs eluted in a single, sharp peak at VE ≈ 17 mL without a significant population of free polymer.
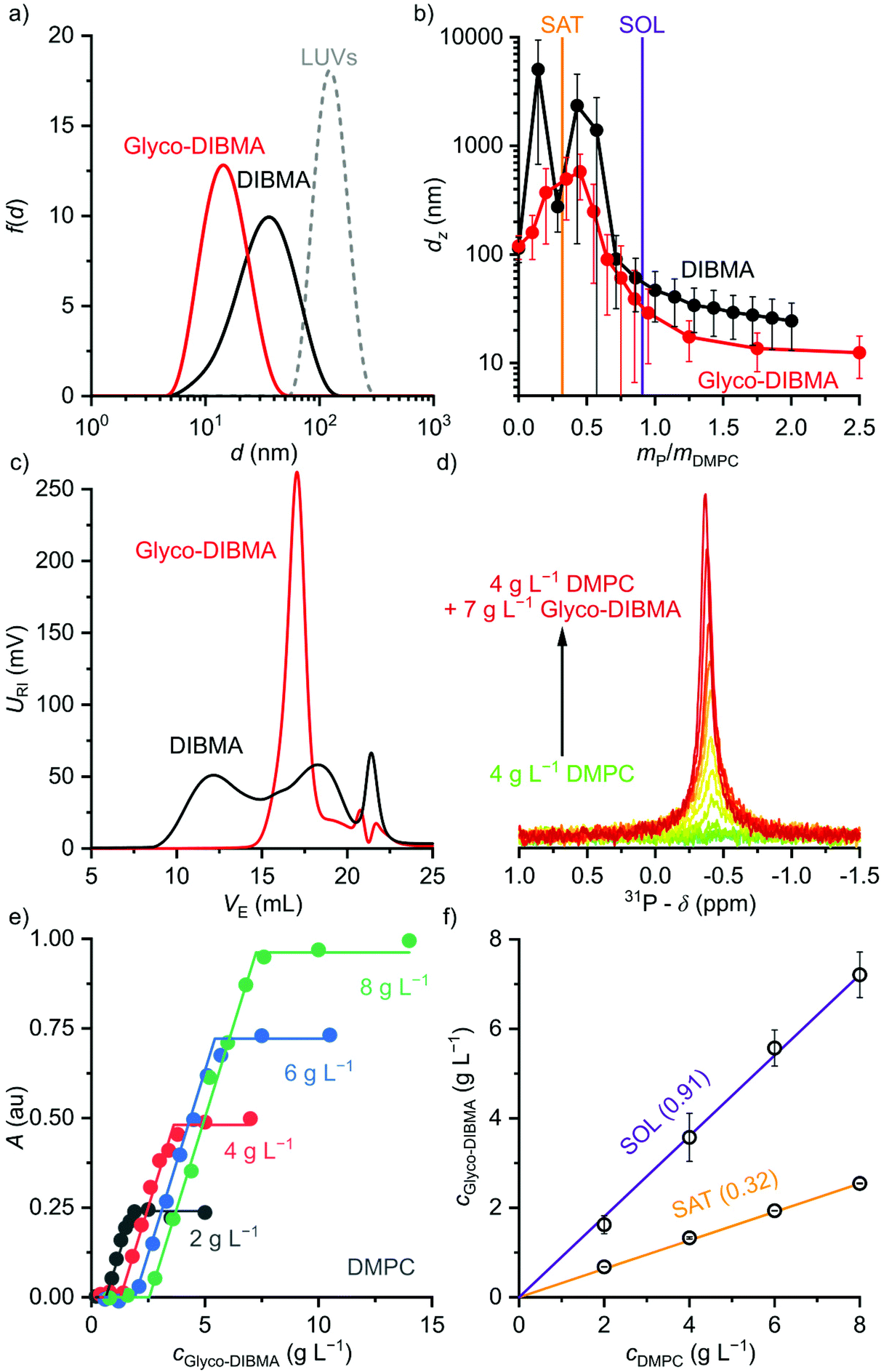 | ||
Fig. 2 Formation of polymer-encapsulated DMPC nanodiscs by Glyco-DIBMA and DIBMA in 50 mM Tris, 200 mM NaCl, pH 7.4, at 30 °C. (a) Intensity-weighted particle size distributions of 3.4 g L−1 (5 mM) DMPC initially present in the form of LUVs before and after nanodisc formation at a polymer/lipid mass ratio of mP/mL = 1.5. (b) z-Average hydrodynamic diameters (full circles) and size distribution widths (“error” bars) of 3.4 g L−1 (5 mM) DMPC at increasing mP/mL. Vertical lines indicate saturation (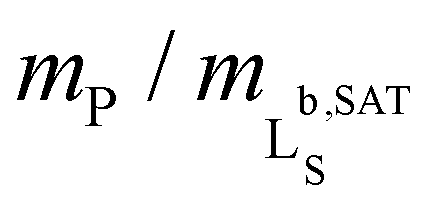 , SAT) and solubilization ( , SAT) and solubilization (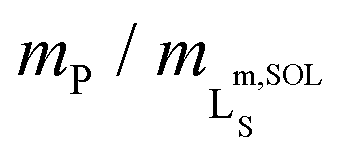 , SOL) thresholds as derived by 31P-NMR (see panels d–f). (c) SEC chromatograms showing the voltage of the refractive index detector, URI, as a function of elution volume, VE, for DMPC nanodiscs at mP/mL = 1.5. Minor peaks at ∼22 mL are due to NaCl and other solvent components. (d) 31P-NMR spectra of 4 g L−1 (5.9 mM) DMPC at increasing concentrations of Glyco-DIBMA. (e) NMR peak areas, A, at four different DMPC concentrations and increasing Glyco-DIBMA concentrations. Lines are from a global fit according to eqn (3)–(5) in the ESI.† (f) Glyco-DIBMA/DMPC pseudo-phase diagram as derived from peak areas in panel e. Shown are breakpoints from local fits (open circles) and corresponding 95% confidence intervals (error bars) as well as the result of a global fit (lines) according to eqn (3)–(5).† , SOL) thresholds as derived by 31P-NMR (see panels d–f). (c) SEC chromatograms showing the voltage of the refractive index detector, URI, as a function of elution volume, VE, for DMPC nanodiscs at mP/mL = 1.5. Minor peaks at ∼22 mL are due to NaCl and other solvent components. (d) 31P-NMR spectra of 4 g L−1 (5.9 mM) DMPC at increasing concentrations of Glyco-DIBMA. (e) NMR peak areas, A, at four different DMPC concentrations and increasing Glyco-DIBMA concentrations. Lines are from a global fit according to eqn (3)–(5) in the ESI.† (f) Glyco-DIBMA/DMPC pseudo-phase diagram as derived from peak areas in panel e. Shown are breakpoints from local fits (open circles) and corresponding 95% confidence intervals (error bars) as well as the result of a global fit (lines) according to eqn (3)–(5).† | ||
We further quantified the efficiency of Glyco-DIBMA in forming nanodiscs by monitoring the emergence of a 31P-NMR signal upon titration of DMPC LUVs with the polymer (Fig. 2d–f). As solution NMR spectroscopy is insensitive to nuclei residing in large, slowly tumbling particles such as LUVs, no peak was detectable in the absence or in the presence of low concentrations of Glyco-DIBMA (Fig. 2d). Increasing mP/mL beyond the so-called saturation (SAT) threshold resulted in the emergence of an isotropic peak, which indicated the formation of fast-tumbling Glyco-DIBMALPs. Further addition of Glyco-DIBMA caused a linear increase in the peak area until the latter reached a plateau at the so-called solubilization (SOL) boundary (Fig. 2d and e). Reading the SAT and SOL breakpoints from plots of 31P-NMR peak area vs. Glyco-DIBMA concentrations at several DMPC concentrations (Fig. 2e) furnished a pseudo-phase diagram27,28 (Fig. 2f) characterized by SAT and SOL boundaries and associated 95% confidence intervals of, respectively, 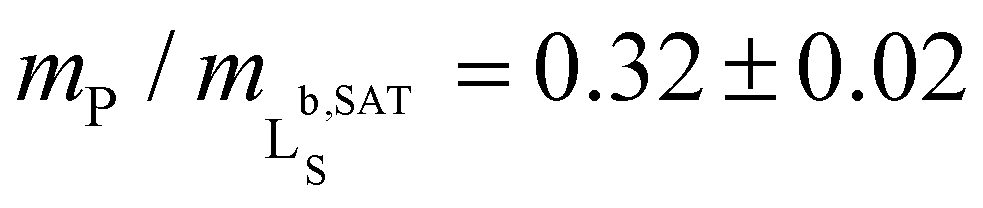 and
and 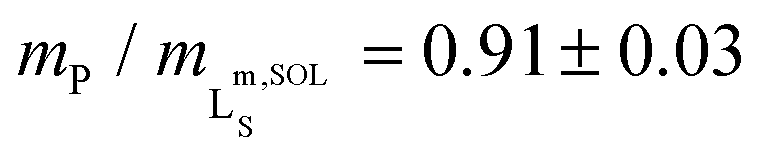 , which fall in the same range as the corresponding values determined for DIBMA.2 Hence, nanodisc formation by Glyco-DIBMA can be rationalized in terms of a simple three-stage model,27 as has been shown for other amphiphilic polymers.2,3,23,29–31
, which fall in the same range as the corresponding values determined for DIBMA.2 Hence, nanodisc formation by Glyco-DIBMA can be rationalized in terms of a simple three-stage model,27 as has been shown for other amphiphilic polymers.2,3,23,29–31
We further explored the efficiency of Glyco-DIBMA in forming lipid-bilayer nanodiscs under different solution conditions (Fig. 3a and b). To this end, we employed the unsaturated phospholipid 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), which is more challenging to accommodate in nanodiscs than DMPC, especially for DIBMA.3,4,23 Indeed, incubation of POPC LUVs with DIBMA under near-physiological conditions (i.e., 50 mM Tris, 200 mM NaCl, pH 7.4, 25 °C) produced large and broadly distributed nanoparticles with dz = (85 ± 45) nm even at a polymer/lipid mass ratio as high as mP/mL = 6 (Fig. 3a). By contrast, Glyco-DIBMA enabled the formation of narrowly distributed nanodiscs with dz = (15 ± 6) nm already at mP/mL = 2.2 under otherwise identical conditions (Fig. 3a). As is the case for DIBMA,23,24 the ionic strength of the solution substantially influenced the solubilization efficiency of Glyco-DIBMA (Fig. 3b). While nanodisc formation in the absence of added salt proved difficult, increasing cNaCl to 100 mM steadily increased the efficiency of Glyco-DIBMA to afford nanoparticles with dz ≈ (10 ± 4) nm already at mP/mL = 2. Although a further increase in cNaCl beyond 100 mM slightly reduced the solubilization efficiency and increased the size of the nanoparticles, this effect was suppressed by raising the pH value to 8.3, which afforded diameters of dz = (10 ± 4) nm already at mP/mL = 1 even at cNaCl = 200 mM (Fig. 3b). Under these conditions, the lipid-solubilization performance of Glyco-DIBMA was on par with that of the aromatic copolymer SMA(2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the current “gold standard” in the field (Fig. S2†). Moreover, Glyco-DIBMALPs were found to tolerate divalent cations up to 4.0 mM Mg2+ or 1.5 mM Ca2+ (Fig. S3†) and temperatures of up to 65 °C (Fig. S4†) at pH 8.3 and 200 mM NaCl. In summary, the efficiency of Glyco-DIBMA in facilitating the self-assembly of lipid-bilayer nanodiscs was greatest in the neutral to moderately alkaline pH range and at a reasonably physiological ionic strength of ∼100 mM. In contrast with DIBMA,4,23 neither acidic pH, nor high ionic strength, nor addition of divalent cations was required for optimal performance of Glyco-DIBMA, which thus proved to be a much more powerful nanodisc former.
:1), the current “gold standard” in the field (Fig. S2†). Moreover, Glyco-DIBMALPs were found to tolerate divalent cations up to 4.0 mM Mg2+ or 1.5 mM Ca2+ (Fig. S3†) and temperatures of up to 65 °C (Fig. S4†) at pH 8.3 and 200 mM NaCl. In summary, the efficiency of Glyco-DIBMA in facilitating the self-assembly of lipid-bilayer nanodiscs was greatest in the neutral to moderately alkaline pH range and at a reasonably physiological ionic strength of ∼100 mM. In contrast with DIBMA,4,23 neither acidic pH, nor high ionic strength, nor addition of divalent cations was required for optimal performance of Glyco-DIBMA, which thus proved to be a much more powerful nanodisc former.
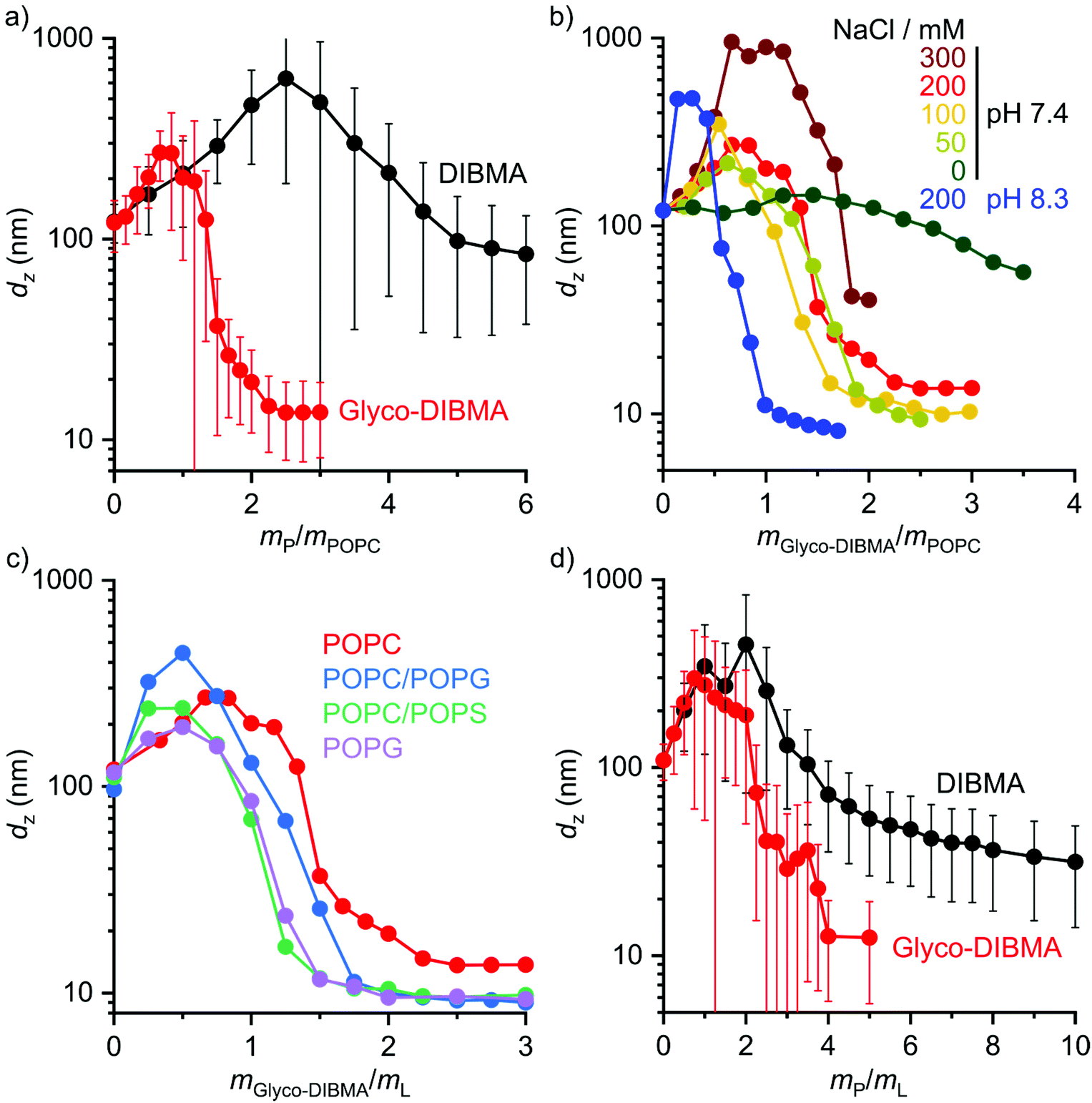 | ||
| Fig. 3 Formation of polymer-encapsulated nanodiscs by Glyco-DIBMA and DIBMA using different buffer compositions and various lipid species. (a and b) z-Average hydrodynamic diameters of 3.8 g L−1 (5 mM) POPC LUVs at increasing polymer/lipid mass ratios in the presence of (a) 50 mM Tris, 200 mM NaCl, pH 7.4 or (b) various NaCl concentrations and pH values (for Glyco-DIBMA only). (c) z-Average hydrodynamic diameters of LUVs harboring POPC, POPC/POPG (1:1 mol mol−1), POPC/POPS (7:3 mol mol−1), or POPG, each at ∼4 g L−1 total lipid at increasing Glyco-DIBMA/lipid mass ratios. (d) z-Average hydrodynamic diameters of 4 g L−1 POPC/cholesterol (3.55:1 mol mol−1) at increasing polymer/lipid mass ratios. Data shown in panels c and d were collected in 50 mM Tris, 200 mM NaCl, pH 7.4. | ||
In addition to varying the solution conditions, we tested the performance of Glyco-DIBMA in harboring more challenging lipid mixtures in nanodiscs that more closely mimic the complex compositions of eukaryotic and prokaryotic membranes (Fig. 3c and d). To this end, we incubated Glyco-DIBMA with LUVs made either from the anionic phospholipid 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) or from mixtures of POPC with POPG, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), or cholesterol. Addition of Glyco-DIBMA to all of the anionic LUVs produced nanoparticles with dz ≈ (10 ± 4) nm already at mP/mL = 2. By contrast, highly negatively charged DIBMA failed to solubilize any of the anionic membranes, even at ratios as high as mP/mL = 10 (Fig. S5†). These contrasting findings again reflect the reduced charge density on Glyco-DIBMA, which results in lower coulombic repulsion between the polymer and anionic membranes. Finally, we turned our attention to cholesterol, which does not affect the charge state of a zwitterionic phosphocholine-based lipid-bilayer membrane but increases both its hydrophobic thickness and the lateral pressure within its acyl chain region.32,33 Both of these effects can, in principle, be counterbalanced by an increase in the polymer density in the nanodisc rim. This, however, is limited by coulombic repulsion among the polymer chains in the rim, especially for highly charged DIBMA. Indeed, the solubilization of LUVs containing 30 mol% cholesterol by DIBMA turned out very difficult, as nanoparticle sizes of dz = (31 ± 17) nm were attained only at an exceedingly high polymer/lipid mass ratio of mP/mL = 10. By contrast, Glyco-DIBMA yielded much more narrowly sized nanoparticles with dz = (13 ± 7) nm already at mP/mL = 4.
To assess the morphology and lipid-bilayer architecture of Glyco-DIBMALPs, we turned to negative-stain transmission electron microscopy (TEM) and differential scanning calorimetry (DSC) (Fig. 4). TEM of Glyco-DIBMALPs encapsulating DMPC (Fig. 4a) or POPC (Fig. 4b) corroborated the formation of nanoscopic discs having diameters in excellent agreement with those determined by DLS (Fig. 2a and 3a). Of note, we observed numerous “rouleaux”, that is, assemblies of several nanodiscs stacking on top of one another. These artifacts are most likely caused by the staining procedure,2,34,35 as DLS (Fig. 2a, b; 3; 7d), SEC (Fig. 2c), as well as microfluidic diffusional sizing (MDS; see Fig. 7f below) provide no evidence of nanodisc stacking in solution. Nevertheless, the nanodisc stacks observed by TEM were helpful in that they enabled many side views of Glyco-DIBMALPs and, thus, clearly confirmed a nanodisc thickness typical of a phospholipid bilayer. With the aid of DSC, we probed the thermotropic gel-to-fluid phase transition of DMPC in Glyco-DIBMALPs at various polymer/lipid mass ratios (Fig. 4c). We observed a broadening of the phase transition in Glyco-DIBMALPs as compared with polymer-free LUVs, which is indicative of nanoscopic lipid-bilayer patches displaying a reduced size of the so-called cooperative unit.2–4,36 Upon addition of Glyco-DIBMA, the melting temperature of DMPC slightly increased from Tm = 24.0 °C in the absence of polymer to Tm = 24.8 °C at 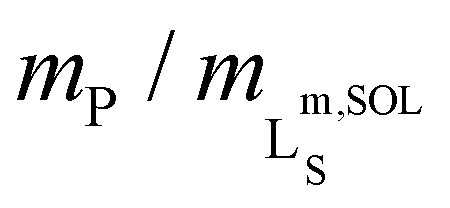 , similar to what has been observed for DIBMA2 and SMA(2:1).3 Upon complete nanodisc formation at mP/mL = 1, a value of Tm = 22 °C indicated that the lipid-bilayer core encapsulated by Glyco-DIBMA was only slightly affected by the polymer. However, further addition of excess polymer caused Tm to decline to ∼13 °C at mP/mL = 4, which is similar to the effect of other polymers such as SMA(2:1).3
, similar to what has been observed for DIBMA2 and SMA(2:1).3 Upon complete nanodisc formation at mP/mL = 1, a value of Tm = 22 °C indicated that the lipid-bilayer core encapsulated by Glyco-DIBMA was only slightly affected by the polymer. However, further addition of excess polymer caused Tm to decline to ∼13 °C at mP/mL = 4, which is similar to the effect of other polymers such as SMA(2:1).3
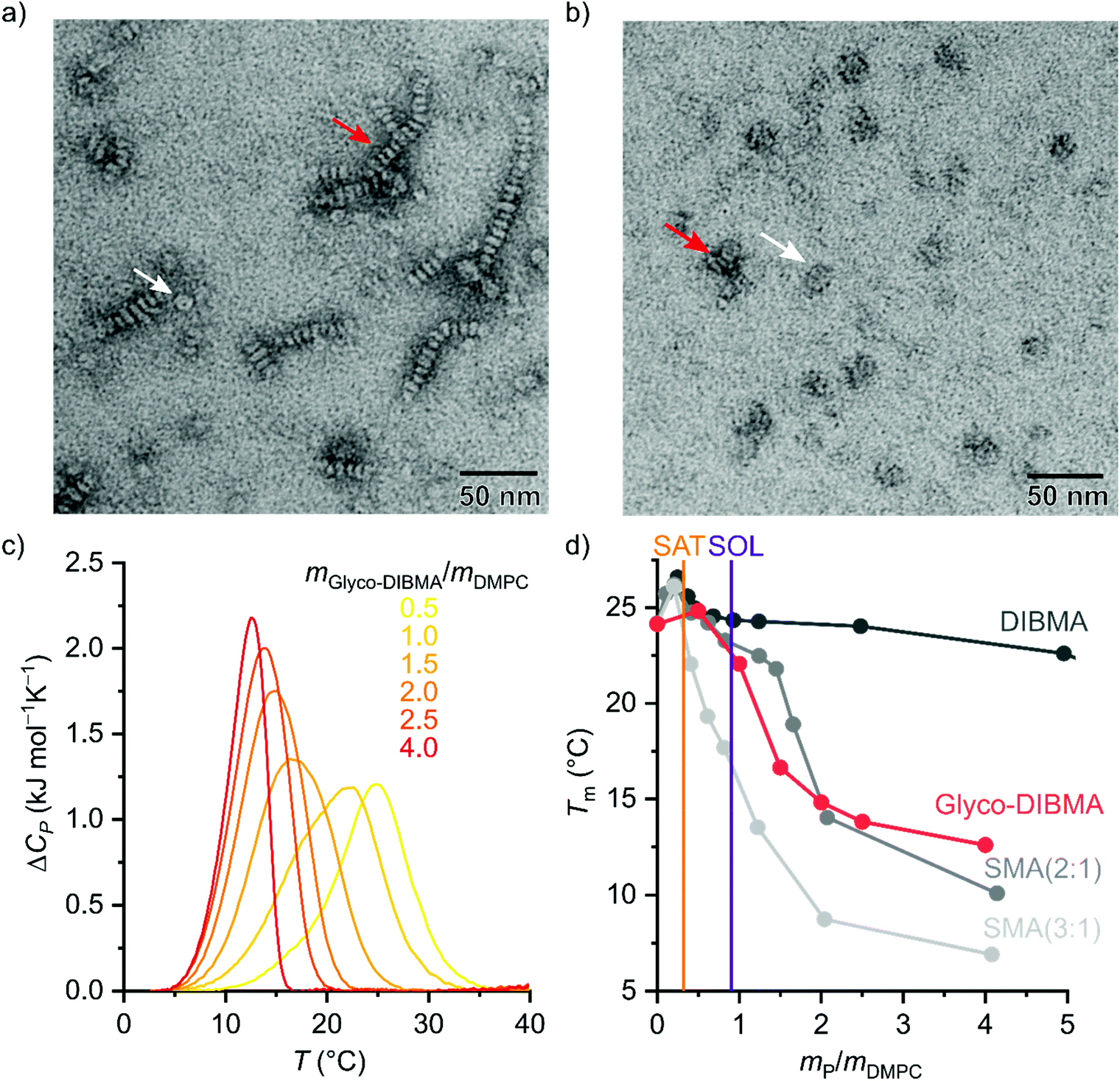 | ||
Fig. 4 Morphology and lipid-bilayer architecture of Glyco-DIBMALPs. (a and b) TEM images of negatively stained Glyco-DIBMALPs harboring (a) DMPC or (b) POPC at a polymer/lipid mass ratio of 1.5 or 2, respectively. Red and white arrows indicate nanodiscs in edge-on and face-on views, respectively. (c) DSC thermograms showing excess molar isobaric heat capacities, ΔCP, of 3.8 g L−1 (5.6 mM) DMPC at increasing Glyco-DIBMA/DMPC mass ratios. (d) Gel-to-fluid phase transition temperatures, Tm, of DMPC as functions of polymer/DMPC mass ratio for Glyco-DIBMA (this work), DIBMA,2 SMA(2:1),3 and SMA(3:1).2 Vertical lines indicate saturation ( , SAT) and solubilization ( , SAT) and solubilization ( , SOL) thresholds for Glyco-DIBMA as obtained from 31P-NMR (see Fig. 2d–f). , SOL) thresholds for Glyco-DIBMA as obtained from 31P-NMR (see Fig. 2d–f). | ||
The stronger impact on Tm of Glyco-DIBMA and the aromatic SMA copolymers as compared with DIBMA is readily explained by the smaller sizes of Glyco-DIBMALPs (Fig. 2b) and SMALPs,3,30 respectively, which lead to a more pronounced exposure of encapsulated phospholipid molecules to the polymer rim. Conversely, these observations suggest that the chemical nature of the polymer moieties in direct contact with the lipid core (i.e., diisobutylene or styrene) plays only a minor role in determining a polymer's effect on the thermotropic phase transition of the encapsulated lipid bilayer. Furthermore, it should be kept in mind that virtually all Glyco-DIBMA chains but only a fraction of DIBMA chains participate in nanodisc formation (Fig. 2c), so that the effective polymer/lipid ratios in DIBMALPs are lower than the nominal values. Together, TEM and DSC confirmed that the solubilization of vesicular membranes by Glyco-DIBMA resulted in the formation of lipid-bilayer nanodiscs, as desired, rather than mixed micellar structures, even at high mass ratios.
It has been shown that polymer-encapsulated nanodiscs can exchange their contents with each other4,24,37,38 or with other lipidic systems such as vesicles,30 lipid monolayers,39 planar lipid bilayers,40 and cubic phases.11 This fast exchange of contents is enabled by the flexible, soft nature of the polymer rim, which itself exchanges rapidly among nanodiscs.41 Importantly, DIBMALPs and SMALPs therefore represent equilibrium nanocomposites rather than kinetically trapped lipid assemblies such as LUVs42 or nanodiscs bounded by membrane scaffold proteins (MSPs).43 Under typical in vitro conditions (i.e., millimolar lipid concentrations), lipid exchange occurs predominantly through nanodisc collisions4,24,37,38 and, consequently, slows down with increasing charge density in the polymer rim.4,24,38 Following this rationale, we expected lipid exchange among Glyco-DIBMALPs to be faster than among nanodiscs suffering from high charge densities such as DIBMALPs. To test this prediction, we quantified the kinetics and unraveled the mechanisms of lipid transfer by time-resolved Förster resonance energy transfer (TR-FRET). To this end, we produced fluorescently labeled nanodiscs consisting of a DMPC matrix hosting 1 mol% of each N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (NBD-PE) and N-(lissamine rhodamine B sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Rh-PE), which served as donor and acceptor fluorophores, respectively. When co-localized within the same nanodisc, NBD-PE and Rh–PE form an efficient FRET pair, but redistribution of the fluorescent lipids in a larger lipid pool will dequench the donor and cause an increase in NBD-PE fluorescence.
Indeed, mixing labeled and unlabeled Glyco-DIBMALPs at various lipid concentrations resulted in a strong increase in the emission intensity of the donor dye NBD-PE in a time- and concentration-dependent manner (Fig. 5a). Local fits (eqn (9) in the ESI†) yielded transfer rate constants, kobs, similar to those retrieved from a global fit (eqn (10)†) across all lipid concentrations tested, showing a linear increase in kobs except at the lowest lipid concentrations (Fig. 5b). These are the hallmarks of two independent lipid-exchange mechanisms, with diffusional lipid transfer playing a significant role only at low lipid concentrations <1 mM and collisional lipid transfer dominating at higher concentrations (Fig. 5c). We found the diffusional and collisional transfer rate constants and their associated 95% confidence intervals to amount to, respectively, kdif = (0.048 ± 0.002) s−1 and kcol = (43.1 ± 0.5) M−1 s−1. Thus, the second-order collisional lipid transfer among Glyco-DIBMALPs is ∼30-fold faster than among DIBMALPs,24 which again can be ascribed to a substantial reduction in the charge density on the polymer chain upon glycosylation. In agreement with this interpretation, the overall lipid-exchange rate constant, kobs, among nanodiscs encapsulated by Glyco-DIBMA falls between those bounded by SMA(2:1) and SMA(3:1) (Fig. 5d), which have considerably lower charge densities than DIBMA. Of note, the slow diffusional transfer of lipid monomers among Glyco-DIBMALPs confirms the above findings by DSC and EM that these nanodiscs harbor a well-preserved lipid-bilayer core.
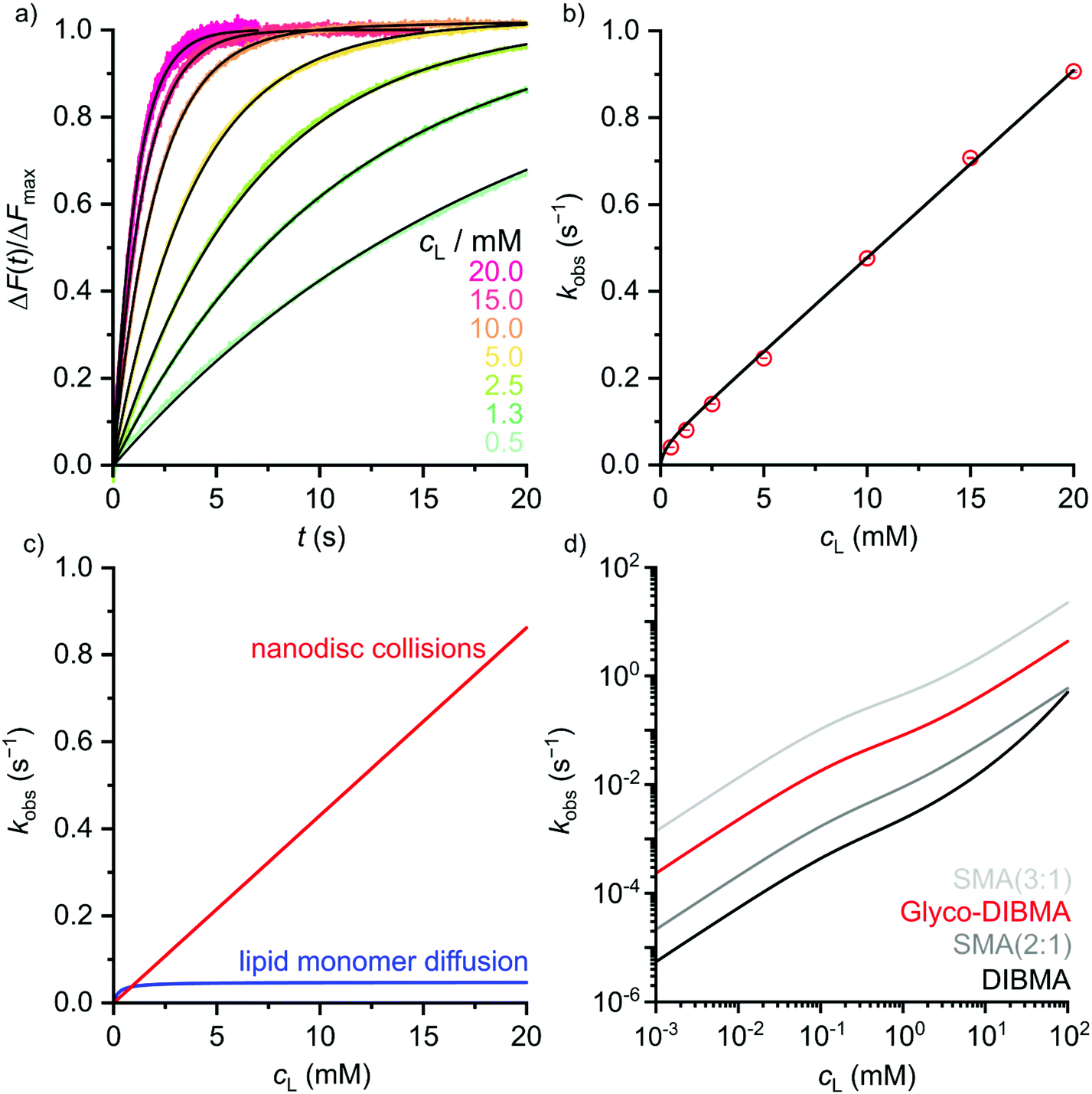 | ||
| Fig. 5 Lipid transfer among fluorescently labeled and unlabeled DMPC nanodiscs bounded by Glyco-DIBMA. (a) Normalized fluorescence emission intensity of NBD-PE, ΔF(t)/ΔFmax, upon rapid mixing of labeled and unlabeled nanodiscs at various lipid concentrations. Shown are experimental data points (colored dots) and a global fit (black lines) based on eqn (10).† (b) Observed lipid-transfer rate constants, kobs, as functions of lipid concentration, cL. Shown are local fits (empty circles) and a global fit (black line) based on eqn (9) and (10),† respectively. Error bars (within circles) correspond to 95% confidence intervals from local fits. (c) Contributions of diffusional and collisional lipid-transfer mechanisms to kobs as derived from eqn (6) and (7),† respectively. (d) kobs values as functions of cL as determined for nanodiscs bounded by Glyco-DIBMA (this work), DIBMA,24 SMA(2:1),38 or SMA(3:1).37 | ||
After testing Glyco-DIBMA with respect to the encapsulation of well-defined model lipid bilayers within nanodiscs, we explored its usefulness for extracting and accommodating integral membrane proteins from complex, cellular membranes in nanodiscs. For this purpose, we first exposed Escherichia coli membranes to increasing concentrations of Glyco-DIBMA and quantified the total amounts of extracted membrane proteins by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 6). Across all polymer concentrations, Glyco-DIBMALPs were able to encapsulate a large variety of membrane proteins from both the inner and the outer bacterial membranes,44,45 revealing an SDS-PAGE band pattern similar to that found for DIBMA (Fig. 6a), other, aromatic polymers such as SMA(2:1), and conventional detergents such as DDM.2,3 At low polymer concentrations of 0.3% (w/v), both aliphatic copolymers performed roughly on par, but Glyco-DIBMA outperformed DIBMA by more than 2.5-fold at more commonly used polymer concentrations of 0.5% and 1.0% (w/v) (Fig. 6b). As E. coli membranes are highly anionic,46 nanodisc-forming polymers need to overcome strong coulombic repulsion in order to interact with and fragment such membranes. This explains why, for optimal performance, partial charge compensation by divalent cations is required for highly charged DIBMA4 but not for Glyco-DIBMA, which revealed a much higher protein-extraction efficiency under mild, near-physiological conditions. Under such conditions, Glyco-DIBMA enabled overall membrane-protein yields similar to those obtained using SMA(2:1).3
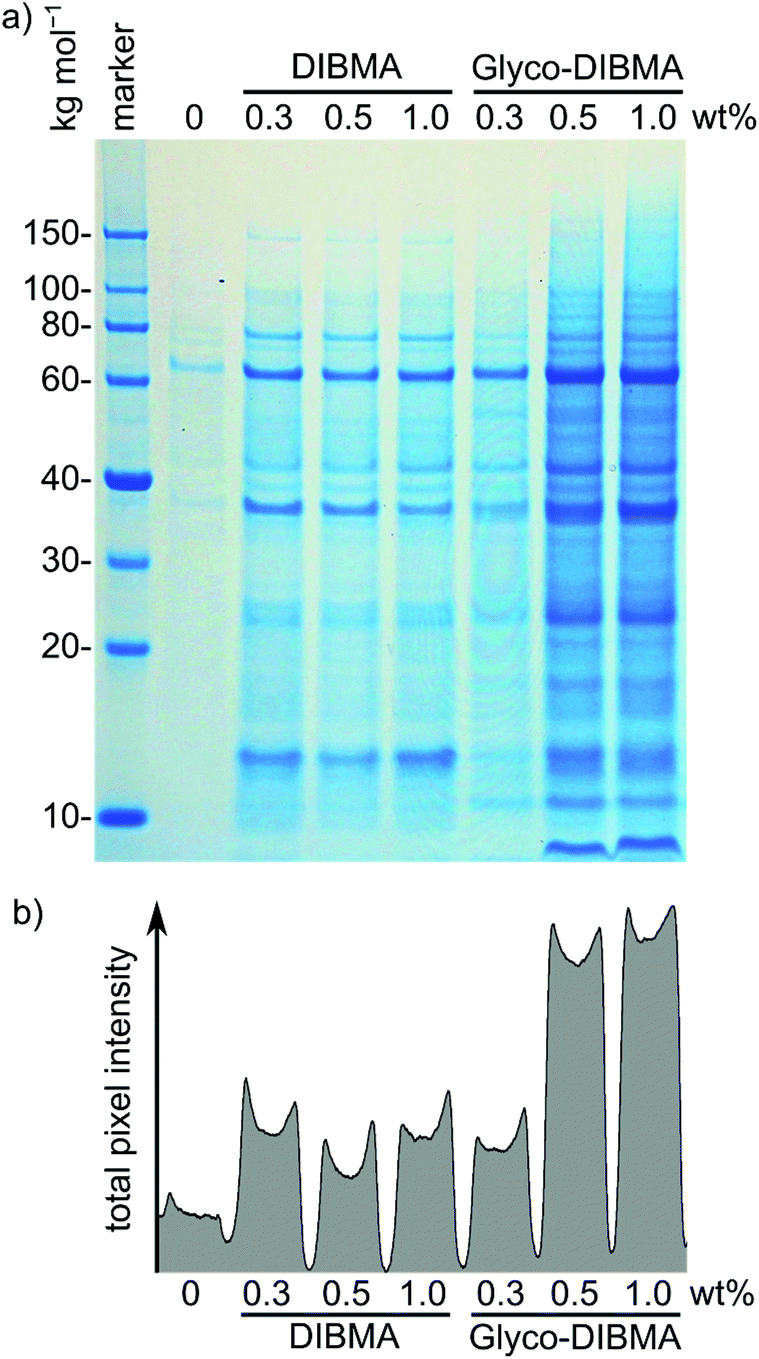 | ||
| Fig. 6 Extraction of the membrane proteome of E. coli cells into polymer-encapsulated nanodiscs mediated by Glyco-DIBMA and DIBMA. Shown are (a) a Coomassie-stained gel after SDS-PAGE of polymer-solubilized fractions and (b) a projection of the total pixel intensity across all lanes in the gel. Cell debris and water-soluble proteins were removed by serial ultracentrifugation, and samples were gently agitated overnight at 23 °C in the presence of Glyco-DIBMA or DIBMA at a constant E. coli membrane concentration of 5% (w/v). Prior to electrophoresis, unsolubilized material and polymer were removed by ultracentrifugation and organic solvent extraction, respectively. A control without polymer was produced under otherwise identical conditions. | ||
Mass spectrometry confirmed that the dominant bands were, as expected, due to the most abundant membrane proteins such as OmpF (∼60 kg mol−1), OmpA (∼37 kg mol−1), and Lpp (<10 kg mol−1). The latter two proteins provide interesting examples of preferential extraction: the integral outer membrane protein OmpA has been found to be extracted well by SMA(2:1) and even better by DDM but worse by DIBMA, while the membrane-anchored lipoprotein Lpp is extracted by both SMA(2:1) and DDM but is lost upon treatment of E. coli membranes with DIBMA.3 Interestingly, Glyco-DIBMA (Fig. 6a) not only improved the yield of extracted OmpA over that afforded by DIBMA but also retained Lpp at levels similar to those found for SMA(2:1) and DDM.3
Next, we sought to demonstrate that Glyco-DIBMA not only extracts membrane proteins from cellular membranes to accommodate them in nanodiscs but can also be used to purify and chemically manipulate a particular target protein directly within Glyco-DIBMALPs (Fig. 7). Hence, we extracted a His6-tagged variant of the voltage-gated K+ channel KvAP47 from E. coli membranes and purified it by Co2+ affinity chromatography and SEC. This two-step purification resulted in a single strong band at the expected molar mass of ∼25 kg mol−1 on SDS-PAGE, both in a Coomassie stain (Fig. 7a) and in a western blot relying on an antibody directed against the His6-tag (Fig. 7b). Mass spectrometry confirmed that this band corresponded to KvAP. Of note, SEC (Fig. 7c) allowed neat separation of KvAP from SlyD (Fig. 7a, fraction 5), a histidine-rich protein that is a common contaminant in the affinity purification of His-tagged proteins,48,49 to afford excellent purity of KvAP in Glyco-DIBMALPs (Fig. 7a and b). The yield and purity of KvAP purified with the aid of Glyco-DIBMA compared favorably with those obtained using 2% (w/v) n-decyl-β-D-maltopyranoside (DM; Fig. S6†), the conventional, micelle-forming detergent previously used for this membrane protein.47 Fraction 3 of the SEC run was concentrated and subjected to further analysis by DLS (Fig. 7d) and TEM (Fig. 7e) to confirm that it contained discoidal nanoparticles with an average diameter of ∼15 nm.
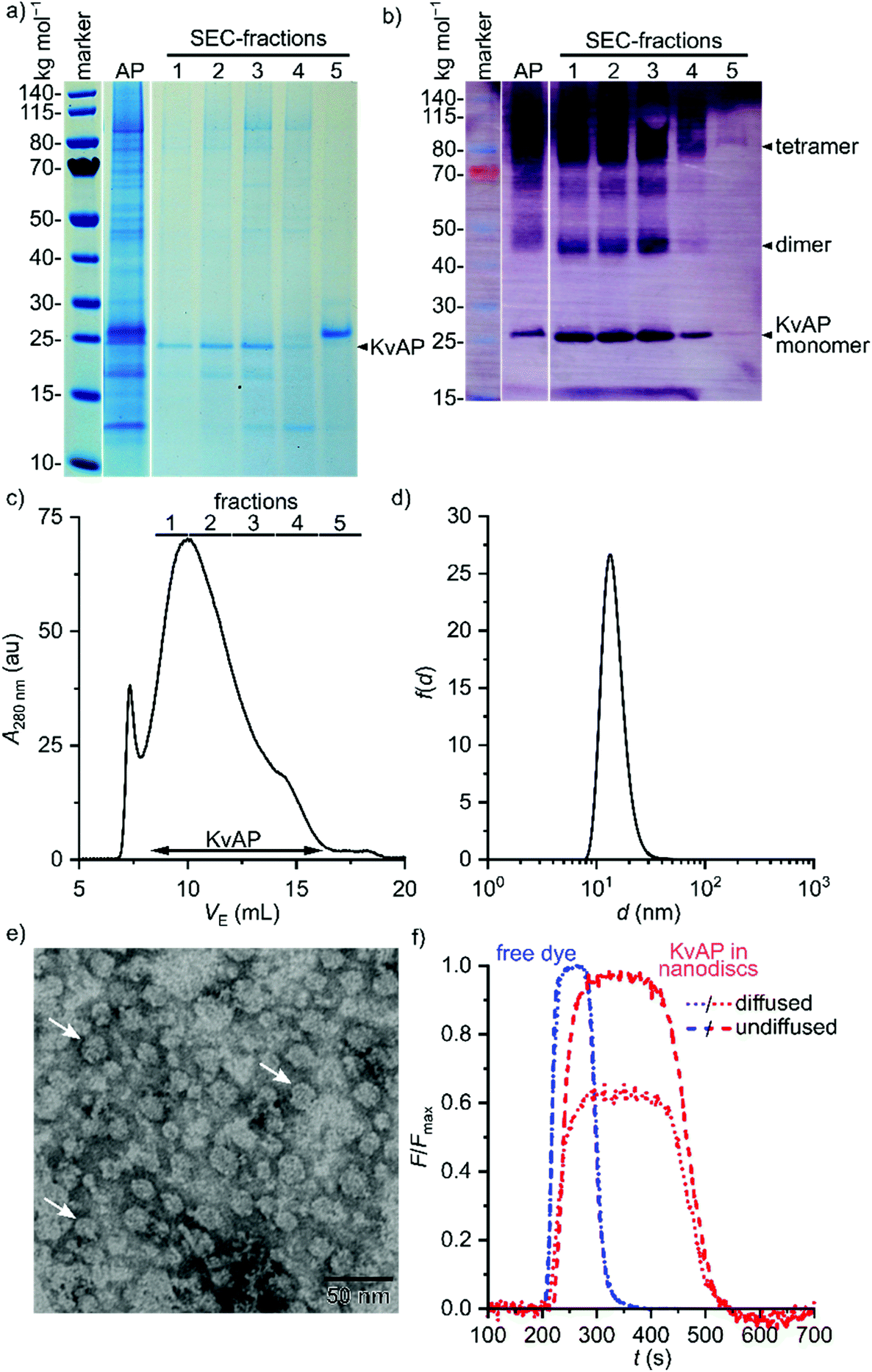 | ||
| Fig. 7 Extraction and purification of the voltage-gated K+ channel KvAP embedded in Glyco-DIBMA nanodiscs. (a and b) SDS-PAGE of KvAP extracted and purified with 5% (w/v) Glyco-DIBMA as visualized by (a) Coomassie stain and (b) western blot of two gels run with the same sample. Shown are samples after His6-tag affinity purification and concentration (AP) and after SEC as indicated in panel c. (c) Representative SEC elution profile of the concentrated sample (AP) shown in panels a and b on a Superose 6 Increase column. Shown is the absorption at 280 nm as a function of elution volume, VE. (d) Number-weighted particle size distribution of Glyco-DIBMALPs harboring purified KvAP, as concentrated from SEC fraction 3 shown in panels a–c. (e) TEM image of KvAP-containing Glyco-DIBMALPs (same sample as shown in panel d). White arrows indicate nanodiscs in face-on views. (f) Normalized fluorescence emission intensity at ∼520 nm of labeled KvAP (from SEC fraction 3) and unconjugated ATTO-488 maleimide after injection into a microfluidic laminar flow chamber and separation into two detection channels corresponding to diffused (dotted lines) and undiffused fluorophores (dashed lines). | ||
As a final proof of concept, microfluidic diffusional sizing (MDS)50 was used to show that protein-containing Glyco-DIBMALPs are amenable to protein-conjugation techniques and further in vitro scrutiny. In contrast with DLS and TEM, MDS selectively reports on the hydrodynamic size of fluorescently labeled nanoparticles only. Hence, we labeled affinity-purified KvAP embedded in Glyco-DIBMALPs by conjugation of the thiol-reactive fluorophore ATTO-488 maleimide to the protein's sole cysteine residue at position 243. After removal of unconjugated dye by SEC, labeled KvAP incorporated in Glyco-DIBMALPs was subjected to MDS and compared with the free, unconjugated dye. The MDS raw data revealed that the fraction of diffused KvAP was much smaller than that of the free dye (Fig. 7f), as expected from the large increase in size of the fluorescent species upon conjugation to a nanodisc-embedded membrane protein. Quantitative analysis of the MDS data furnished a diffusion coefficient of 38 μm2 s−1 and a corresponding hydrodynamic diameter of 11 nm for KvAP nanodiscs, whereas free ATTO-488 maleimide displayed a diffusion coefficient of 300 μm2 s−1 and a hydrodynamic diameter of 1.5 nm. These values are in excellent agreement with structural considerations and, thus, demonstrate that nanodiscs encapsulsated by Glyco-DIBMA are compatible with chemical protein-conjugation techniques, state-of-the-art microfluidic methods, and sensitive fluorescence detection without ever removing the target protein from its native-like lipid-bilayer background.
3. Conclusions
We have introduced Glyco-DIBMA as a more hydrophobic, less charged, and more powerful nanodisc-forming polymer than established aliphatic membrane-solubilizing copolymers. Compared with DIBMA, this new, bioinspired glycopolymer fragments and accommodates model and cellular membranes with substantially higher efficiency—in particular, under near-physiological conditions—to form smaller and more narrowly distributed lipid-bilayer nanodiscs. Glyco-DIBMALPs harbor a native-like, dynamic lipid-bilayer core and are stable across a wide range of buffer compositions and temperatures. Glyco-DIBMALPs are able to accommodate within their lipid-bilayer core a broad range of membrane proteins, which thus become amenable to chromatographic purification as well as further in vitro manipulation and analysis while remaining embedded in a nanoscale membrane patch at all times.Author contributions
BD: conceptualization, investigation, writing–original draft, writing–review & editing; MR: investigation, writing–review & editing; JL: investigation, writing–review & editing; EPP: investigation, supervision, writing–review & editing; SW: investigation, writing–review & editing; FM: investigation, writing–review & editing; MTA: investigation, writing–review & editing; AM: conceptualization, investigation, writing–review & editing; JOB: conceptualization, funding acquisition, supervision, writing–review & editing; CV: conceptualization, investigation, funding acquisition, supervision, writing–review & editing; CK: conceptualization, investigation, supervision, writing–review & editing; SK: conceptualization, investigation, funding acquisition, supervision, writing–review & editing.Conflicts of interest
CK is the founder and owner of Glycon Biochemicals GmbH, which is offering Glyco-DIBMA for sale. MR is an employee of Glycon Biochemicals GmbH.Acknowledgements
We thank Dr Harald Kelm, Lisa Hamsch, Christiane Müller, Ann-Cathrin Schlapp, Dr Frederik Sommer, and Kai Zwara (all TUK) for excellent technical assistance and Prof. Peter Pohl (University of Linz) for providing the E. coli strain used for producing KvAP. This work was supported by the Carl Zeiss Foundation through the Centre for Lipidomics (CZSLip). M.T.A. acknowledges the Deutsche Akademische Austauschdienst (DAAD) for a Ph.D. scholarship.References
- N. Bordag and S. Keller, Alpha-helical transmembrane peptides: a “divide and conquer” approach to membrane proteins, Chem. Phys. Lipids, 2010, 163, 1–26 CrossRef CAS PubMed
.
- A. O. Oluwole, B. Danielczak, A. Meister, J. O. Babalola, C. Vargas and S. Keller, Solubilization of Membrane Proteins into Functional Lipid-Bilayer Nanodiscs Using a Diisobutylene/Maleic Acid Copolymer, Angew. Chem., Int. Ed., 2017, 56, 1919–1924 CrossRef CAS PubMed
- A. Grethen, A. O. Oluwole, B. Danielczak, C. Vargas and S. Keller, Thermodynamics of nanodisc formation mediated by styrene/maleic acid (2:1) copolymer, Sci. Rep., 2017, 7, 11517 CrossRef PubMed
- B. Danielczak, A. Meister and S. Keller, Influence of Mg2+ and Ca2+ on Nanodisc Formation by Diisobutylene/Maleic Acid (DIBMA) Copolymer, Chem. Phys. Lipids, 2019, 221, 30–38 CrossRef CAS PubMed
- G. A. Pellowe, H. E. Findlay, K. Lee, T. M. Gemeinhardt, L. R. Blackholly, E. Reading and P. J. Booth, Capturing Membrane Protein Ribosome Nascent Chain Complexes in a Native-like Environment for Co-translational Studies, Biochemistry, 2020, 59, 2764–2775 CrossRef CAS PubMed
- M. Barniol-Xicota and S. H. L. Verhelst, Stable and Functional Rhomboid Proteases in Lipid Nanodiscs by Using Diisobutylene/Maleic Acid Copolymers, J. Am. Chem. Soc., 2018, 140, 14557–14561 CrossRef CAS PubMed
- V. J. Flegler, A. Rasmussen, S. Rao, N. Wu, R. Zenobi, M. S. P. Sansom, R. Hedrich, T. Rasmussen and B. Böttcher, The MscS-like channel YnaI has a gating mechanism based on flexible pore helices, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28754–28762 CrossRef CAS PubMed
- A. A. Gulamhussein, R. Uddin, B. J. Tighe, D. R. Poyner and A. J. Rothnie, A comparison of SMA (styrene maleic acid) and DIBMA (di-isobutylene maleic acid) for membrane protein purification, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183281 CrossRef CAS PubMed
- S. R. Tonge and B. J. Tighe, Responsive hydrophobically associating polymers: a review of structure and properties, Adv. Drug Delivery Rev., 2001, 53, 109–122 CrossRef CAS PubMed
- T. J. Knowles, R. Finka, C. Smith, Y.-P. Lin, T. Dafforn and M. Overduin, Membrane proteins solubilized intact in lipid containing nanoparticles bounded by styrene maleic acid copolymer, J. Am. Chem. Soc., 2009, 131, 7484–7485 CrossRef CAS PubMed
- J. Broecker, B. T. Eger and O. P. Ernst, Crystallogenesis of Membrane Proteins Mediated by Polymer-Bounded Lipid Nanodiscs, Structure, 2017, 25, 384–392 CrossRef CAS PubMed
- S. C. Lee, T. J. Knowles, V. L. G. Postis, M. Jamshad, R. A. Parslow, Y.-P. Lin, A. Goldman, P. Sridhar, M. Overduin, S. P. Muench and T. R. Dafforn, A method for detergent-free isolation of membrane proteins in their local lipid environment, Nat. Protoc., 2016, 11, 1149–1162 CrossRef CAS PubMed
- C. Sun, S. Benlekbir, P. Venkatakrishnan, Y. Wang, S. Hong, J. Hosler, E. Tajkhorshid, J. L. Rubinstein and R. B. Gennis, Structure of the alternative complex III in a supercomplex with cytochrome oxidase, Nature, 2018, 557, 123–126 CrossRef CAS PubMed
- A. M. Seddon, P. Curnow and P. J. Booth, Membrane proteins, lipids and detergents: not just a soap opera, Biochim. Biophys. Acta, 2004, 1666, 105–117 CrossRef CAS
- M. Jamshad, J. Charlton, Y.-P. Lin, S. J. Routledge, Z. Bawa, T. J. Knowles, M. Overduin, N. Dekker, T. R. Dafforn, R. M. Bill, D. R. Poyner and M. Wheatley, G-protein coupled receptor solubilization and purification for biophysical analysis and functional studies, in the total absence of detergent, Biosci. Rep., 2015, 35, 1–10 CrossRef CAS PubMed
- D. J. K. Swainsbury, S. Scheidelaar, R. van Grondelle, J. A. Killian and M. R. Jones, Bacterial Reaction Centers Purified with Styrene Maleic Acid Copolymer Retain Native Membrane Functional Properties and Display Enhanced Stability, Angew. Chem., Int. Ed., 2014, 126, 11997–12001 CrossRef
- S. Rehan, V. O. Paavilainen and V.-P. Jaakola, Functional reconstitution of human equilibrative nucleoside transporter-1 into styrene maleic acid co-polymer lipid particles, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1059–1065 CrossRef CAS PubMed
- V. M. Luna, M. Vazir, A. Vaish, S. Chong, I. Chen and H. K. Yamane, Generation of membrane proteins in polymer-based lipoparticles as flow cytometry antigens, Eur. Polym. J., 2018, 109, 483–488 CrossRef CAS
- E. Reading, Z. Hall, C. Martens, T. Haghighi, H. Findlay, Z. Ahdash, A. Politis and P. J. Booth, Interrogating Membrane Protein Conformational Dynamics within Native Lipid Compositions, Angew. Chem., Int. Ed., 2017, 56, 15654–15657 CrossRef CAS PubMed
- S. Gulati, M. Jamshad, T. J. Knowles, K. A. Morrison, R. Downing, N. Cant, R. Collins, J. B. Koenderink, R. C. Ford, M. Overduin, I. D. Kerr, T. R. Dafforn and A. J. Rothnie, Detergent-free purification of ABC (ATP-binding-cassette) transporters, Biochem. J., 2014, 461, 269–278 CrossRef CAS PubMed
- C. Logez, M. Damian, C. Legros, C. Dupré, M. Guéry, S. Mary, R. Wagner, C. M'Kadmi, O. Nosjean, B. Fould, J. Marie, J.-A. Fehrentz, J. Martinez, G. Ferry, J. A. Boutin and J.-L. Banères, Detergent-free Isolation of Functional G Protein-Coupled Receptors into Nanometric Lipid Particles, Biochemistry, 2016, 55, 38–48 CrossRef CAS PubMed
- D. Hardy, R. M. Bill, A. J. Rothnie and A. Jawhari, Stabilization of Human Multidrug Resistance Protein 4 (MRP4/ABCC4) Using Novel Solubilization Agents, SLAS Discovery, 2019, 24, 1009–1017 CAS
- A. O. Oluwole, J. Klingler, B. Danielczak, J. O. Babalola, C. Vargas, G. Pabst and S. Keller, Formation of Lipid-Bilayer Nanodiscs by Diisobutylene/Maleic Acid (DIBMA) Copolymer, Langmuir, 2017, 33, 14378–14388 CrossRef CAS PubMed
- B. Danielczak and S. Keller, Collisional lipid exchange among DIBMA-encapsulated nanodiscs (DIBMALPs), Eur. Polym. J., 2018, 109, 206–213 CrossRef CAS
- S. Scheidelaar, M. C. Koorengevel, C. A. van Walree, J. J. Dominguez, J. M. Dörr and J. A. Killian, Effect of Polymer Composition and pH on Membrane Solubilization by Styrene-Maleic Acid Copolymers, Biophys. J., 2016, 1974–1986 CrossRef CAS PubMed
- A. A. Gulamhussein, D. Meah, D. D. Soja, S. Fenner, Z. Saidani, A. Akram, S. Lallie, A. Mathews, C. Painter, M. K. Liddar, Z. Mohammed, L. K. Chiu, S. S. Sumar, H. Healy, N. Hussain, J. H. Patel, S. C. L. Hall, T. R. Dafforn and A. J. Rothnie, Examining the stability of membrane proteins within SMALPs, Eur. Polym. J., 2019, 112, 120–125 CrossRef CAS
- D. Lichtenberg, E. Opatowski and M. M. Kozlov, Phase boundaries in mixtures of membrane-forming amphiphiles and micelle-forming amphiphiles, Biochim. Biophys. Acta, Biomembr., 2000, 1508, 1–19 CrossRef CAS
- H. Heerklotz, Interactions of surfactants with lipid membranes, Q. Rev. Biophys., 2008, 41, 205–264 CrossRef CAS PubMed
- C. Vargas, R. C. Arenas, E. Frotscher and S. Keller, Nanoparticle self-assembly in mixtures of phospholipids with styrene/maleic acid copolymers or fluorinated surfactants, Nanoscale, 2015, 7, 20685–20696 RSC
- R. Cuevas Arenas, J. Klingler, C. Vargas and S. Keller, Influence of lipid bilayer properties on nanodisc formation mediated by styrene/maleic acid copolymers, Nanoscale, 2016, 8, 15016–15026 RSC
- S. C. L. Hall, C. Tognoloni, J. Charlton, É. C. Bragginton, A. J. Rothnie, P. Sridhar, M. Wheatley, T. J. Knowles, T. Arnold, K. J. Edler and T. R. Dafforn, An acid-compatible co-polymer for the solubilization of membranes and proteins into lipid bilayer-containing nanoparticles, Nanoscale, 2018, 10, 10609–10619 RSC
- O. H. S. Ollila, T. Róg, M. Karttunen and I. Vattulainen, Role of sterol type on lateral pressure profiles of lipid membranes affecting membrane protein functionality: Comparison between cholesterol, desmosterol, 7-dehydrocholesterol and ketosterol, J. Struct. Biol., 2007, 159, 311–323 CrossRef
- J. Gallová, D. Uhríková, N. Kucerka, J. Teixeira and P. Balgavý, Hydrophobic thickness, lipid surface area and polar region hydration in monounsaturated diacylphosphatidylcholine bilayers: SANS study of effects of cholesterol and beta-sitosterol in unilamellar vesicles, Biochim. Biophys. Acta, 2008, 1778, 2627–2632 CrossRef PubMed
- S. Bibow, Y. Polyhach, C. Eichmann, C. N. Chi, J. Kowal, S. Albiez, R. A. McLeod, H. Stahlberg, G. Jeschke, P. Güntert and R. Riek, Solution structure of discoidal high-density lipoprotein particles with a shortened apolipoprotein A-I, Nat. Struct. Mol. Biol., 2017, 24, 187–193 CrossRef CAS PubMed
- D. Martinez, M. Decossas, J. Kowal, L. Frey, H. Stahlberg, E. J. Dufourc, R. Riek, B. Habenstein, S. Bibow and A. Loquet, Lipid Internal Dynamics Probed in Nanodiscs, ChemPhysChem, 2017, 18, 2651–2657 CrossRef CAS PubMed
- M. C. Orwick, P. J. Judge, J. Procek, L. Lindholm, A. Graziadei, A. Engel, G. Gröbner and A. Watts, Detergent-free formation and physicochemical characterization of nanosized lipid-polymer complexes: Lipodisq, Angew. Chem., Int. Ed., 2012, 51, 4653–4657 CrossRef CAS PubMed
- R. Cuevas Arenas, B. Danielczak, A. Martel, L. Porcar, C. Breyton, C. Ebel and S. Keller, Fast Collisional Lipid Transfer Among Polymer-Bounded Nanodiscs, Sci. Rep., 2017, 7, 45875 CrossRef CAS PubMed
- A. Grethen, D. Glueck and S. Keller, Role of Coulombic Repulsion in Collisional Lipid Transfer Among SMA(2:1)-Bounded Nanodiscs, J. Membr. Biol., 2018, 251, 443–451 CrossRef CAS PubMed
- G. Hazell, T. Arnold, R. D. Barker, L. A. Clifton, N.-J. Steinke, C. Tognoloni and K. J. Edler, Evidence of Lipid Exchange in Styrene Maleic Acid Lipid Particle (SMALP) Nanodisc Systems, Langmuir, 2016, 32, 11845–11853 CrossRef CAS PubMed
- J. M. Dörr, M. C. Koorengevel, M. Schäfer, A. V. Prokofyev, S. Scheidelaar, E. A. W. van der Cruijsen, T. R. Dafforn, M. Baldus and J. A. Killian, Detergent-free isolation, characterization, and functional reconstitution of a tetrameric K+channel: the power of native nanodiscs, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18607–18612 CrossRef PubMed
- V. Schmidt and J. N. Sturgis, Modifying styrene-maleic acid co-polymer for studying lipid nanodiscs, Biochim. Biophys. Acta, 2018, 1860, 777–783 CrossRef CAS PubMed
- M. Nakano, M. Fukuda, T. Kudo, H. Endo and T. Handa, Determination of interbilayer and transbilayer lipid transfers by time-resolved small-angle neutron scattering, Phys. Rev. Lett., 2007, 98, 238101 CrossRef PubMed
- M. Nakano, M. Fukuda, T. Kudo, M. Miyazaki, Y. Wada, N. Matsuzaki, H. Endo and T. Handa, Static and dynamic properties of phospholipid bilayer nanodiscs, J. Am. Chem. Soc., 2009, 131, 8308–8312 CrossRef CAS PubMed
- K. Duquesne and J. N. Sturgis, Membrane protein solubilization, Methods Mol. Biol., 2010, 601, 205–217 CrossRef CAS PubMed
- B. T. Arachea, Z. Sun, N. Potente, R. Malik, D. Isailovic and R. E. Viola, Detergent selection for enhanced extraction of membrane proteins, Protein Expression Purif., 2012, 86, 12–20 CrossRef CAS PubMed
- C. R. Raetz and W. Dowhan, Biosynthesis and function of phospholipids in Escherichia coli, J. Biol. Chem., 1990, 265, 1235–1238 CrossRef CAS
- V. Ruta, Y. Jiang, A. Lee, J. Chen and R. MacKinnon, Functional analysis of an archaebacterial voltage-dependent K+ channel, Nature, 2003, 422, 180–185 CrossRef CAS PubMed
- S. Hottenrott, T. Schumann, A. Plückthun, G. Fischer and J. U. Rahfeld, The Escherichia coli SlyD is a metal ion-regulated peptidyl-prolyl cis/trans-isomerase, J. Biol. Chem., 1997, 272, 15697–15701 CrossRef CAS PubMed
- V. M. Bolanos-Garcia and O. R. Davies, Structural analysis and classification of native proteins from E. coli commonly co-purified by immobilised metal affinity chromatography, Biochim. Biophys. Acta, 2006, 1760, 1304–1313 CrossRef CAS PubMed
- M. Azouz, M. Gonin, S. Fiedler, J. Faherty, M. Decossas, C. Cullin, S. Villette, M. Lafleur, I. D. Alves, S. Lecomte and A. Ciaccafava, Microfluidic diffusional sizing probes lipid nanodiscs formation, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183215 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr03811g |
| This journal is © The Royal Society of Chemistry 2022 |