
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioactive natural products from Bacteroidetes
Stephan
Brinkmann
 a,
Marius S.
Spohn
a and
Till F.
Schäberle
*abc
a,
Marius S.
Spohn
a and
Till F.
Schäberle
*abc
aFraunhofer Institute for Molecular Biology and Applied Ecology (IME), Branch for Bioresources, 35392 Giessen, Germany. E-mail: stephan.brinkmann@ime.fraunhofer.de; marius.spohn@ime.fraunhofer.de; till.f.schaeberle@agrar.uni-giessen.de
bInstitute for Insect Biotechnology, Justus Liebig University of Giessen, 35392 Giessen, Germany
cGerman Centre for Infection Research (DZIF), Partner Site Giessen-Marburg-Langen, Giessen, Germany
First published on 22nd March 2022
Abstract
Covering: up to end of January 2022
Bacteria representing the phylum Bacteroidetes produce a diverse range of natural products, including polyketides, peptides and lactams. Here, we discuss unique aspects of the bioactive compounds discovered thus far, and the corresponding biosynthetic pathways if known, providing a comprehensive overview of the Bacteroidetes as a natural product reservoir.
 Stephan Brinkmann | Stephan Brinkmann received his bachelor's degree in Biotechnology and Instrumentation Engineering from the Bielefeld University of Applied Sciences in 2014 and his master's degree in Molecular Biotechnology from the University Bielefeld in 2017. In his following PhD thesis, he developed microfluidic-based high-throughput approaches for the cultivation and screening of microorganisms for their potential to produce bioactive natural products and also analyzed the Bacteroidetes phylum for its potential to biosynthesize natural products using omics technologies at the Fraunhofer Institute for Molecular Biology and Applied Ecology (IME), Branch for Bioresources, in Giessen, Germany. |
 Marius S. Spohn | Marius Spohn received his master's degree in Microbiology in 2012 from the University of Tübingen. After his PhD studies focusing on the investigation of regulatory circuit's controlling microbial natural products biosynthesis and a short postdoctoral position he joined in 2016 the Fraunhofer Institute for Molecular Biology and Applied Ecology (IME). Working as the group leader for molecular microbiology, he continues his research on microbial natural products in joint projects with partners from pharma-, agro- and veterinary industry. |
 Till F. Schäberle | Till F. Schäberle studied biology and at the age of 29, he received his PhD from the University of Tübingen, Germany at the Microbiology/Biotechnology department. In 2009, he moved to the University of Bonn, Germany where he worked as a postdoc and subsequently was heading his own working group within the Institute for Pharmaceutical Biology. Currently, he is professor at the Justus-Liebig-University Giessen and department head at the Fraunhofer Institute for Molecular Biology and Applied Ecology (IME), Branch Bioresources. His natural product (NP) research groups applies genetic, biochemical, bioinformatic and chemical analytical methods for NP biosynthesis/discovery. |
1 Introduction
Antimicrobial resistance (AMR) is a serious threat to modern medicine and agriculture.1 Bacteria and fungi resistant to many or most antimicrobials have dire socioeconomic consequences for human health and global food security.2 The threat of AMR is exacerbated by the limited number of candidate antibiotics entering the development pipeline and thus the declining number of approvals for new anti-infective drugs.3The AMR crisis is being addressed by efforts to discover natural products that can serve as new anti-infective leads, and microbes are a rich source of such compounds.4 However, natural product research is hindered by high rediscovery rates, especially in classical producers such as Actinobacteria, Firmicutes and Myxobacteria – despite the strong genetic potential for the biosynthesis of novel molecules.5 This limitation can be circumvented by focusing on underexplored bacterial taxa.
Members of the phylum Bacteroidetes are well-known, abundant and widely distributed sources of natural products, and many members are easy to cultivate. They colonize all types of habitats, including soil, ocean, freshwater, and the gastrointestinal tracts of animals.6 The phylum comprises six classes of gram-negative, non-spore forming, chemo-organotrophic bacteria. Gastrointestinal tracts harbor species from the mostly anaerobic class Bacteroidia, whereas classes Chitinophagia, Cytophagia, Flavobacteriia, Saprospiria and Sphingobacteriia are mainly found in the environment.7 All of them are non-motile (or motile by gliding)8 bacteria that can degrade polymeric organic matter.6 A few are pathogenic9 or endosymbiotic.10 Their promoter structures feature a unique consensus sequence11 recognized by the core RNA polymerase and an unusual primary sigma factor.11,12 The strong genetic potential for the biosynthesis of natural products13,14 is enriched in a few genera (e.g., Chitinophaga and Pedobacter) and less abundant in the anaerobic class Bacteroidia.14 However, the number of compounds isolated thus far is rather low. Known products are structurally diverse and some are potential antibiotic leads, such as the (iso)pedopeptins that can inhibit drug-resistant gram-negative bacteria including World Health Organization (WHO) top-priority carbapenem-resistant pathogens, and the dipeptide TAN-1057, with activity against methicillin-resistant Staphylococcus aureus (MRSA) strains.
In this review, we comprehensively discuss the bioactivity and biosynthesis of natural products that have already been isolated from Bacteroidetes.
2 Macrolides
Macrolides are hydrophobic polyketides characterized by a large macrocyclic lactone ring decorated with variable side chains and groups. One of the better-known macrolides is erythromycin,15 which has bacteriostatic activity based on the inhibition of bacterial protein biosynthesis.16 Macrolides also show antifungal, antiparasitic, antiviral, cytotoxic and immunosuppressive activities.172.1 Elansolids
The elansolids are unique macrolides produced by Chitinophaga sancti FxGBF13 (DSM 21134)18–20 and Chitinophaga pinensis DSM 2588.21 They share a bicyclo[4.3.0]nonane core originating from an intramolecular Diels–Alder cycloaddition (Fig. 1).22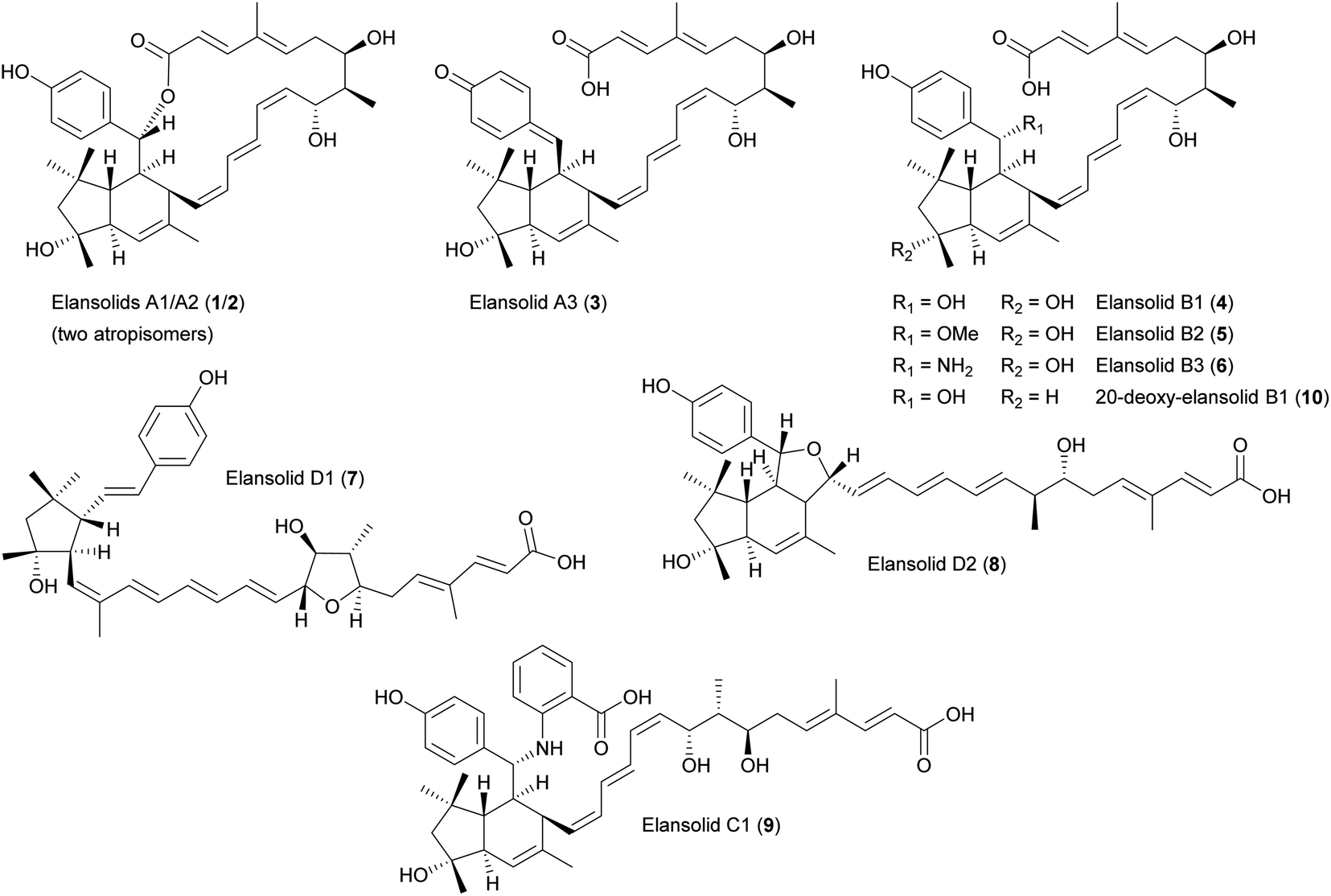 | ||
| Fig. 1 Chemical structures of elansolids (1–10). | ||
Elansolids A1 (1) and A2 (2) are atropisomers featuring a 19-membered macrolactone ring. They are derived from the precursor elansolid A3 (3) by lactonization via a intramolecular Michael addition of the quinone methide group.20,22 The side products elansolids B1–3 (4–6) and D1–2 (7–8) are instead derived from the highly reactive quinone methide moiety in elansolid A3.20,21 The cultivation of C. sancti DSM 21134 in complex media containing different soy peptones, or synthetic medium supplemented with anthranilic acid, yielded elansolid C1 (9).23 This is derived via a nucleophilic addition of the anthranilic acid amino group to the p-quinone methide carbon of elansolid A3.23
The elansolids are produced by trans-acyltransferase (AT) polyketide synthases (PKSs) (Fig. 2A).21,22 The biosynthetic gene cluster (BGC) of C. sancti was identified by scanning a cosmid library.22 In parallel, a closely related set of genes (84.6% nucleotide sequence identity) was identified in C. pinensis DSM 2588, facilitating the isolation of elansolid D2 (8) and the deduction of its biosynthetic pathway.21 Elansolid A3 is probably the mature pathway product (Fig. 2B).21,22 The two trans-AT type I polyketide BGCs consist of either 18 or 19 genes (ela/els PKS) with their assembly line including six multimodular PKS proteins and two trans-ATs.21,22 We also screened 600 publically available Bacteroidetes genomes in silico to examine the sequential and compositional similarity of their BGCs, revealing Chitinophaga sp. YR627 as a third strain carrying a BGC highly similar to the known elansolid BGCs.14 Interestingly, the comparison of all three BGCs suggests the presence of an additional gene encoding a major facilitator superfamily 1 protein, which may transport elansolids across membranes. Major facilitator superfamily proteins are membrane efflux pumps that contribute to the transport of various substances, including macrolides.24
 | ||
| Fig. 2 Elansolid biosynthetic gene clusters (MIBiG BGC0000178) and pathway. (A) Elansolid BCGs found in genomes of three Chitinophaga strains. Identity represents the nucleotide alignment of all five BGCs using MAFFT.156 (B) Model for elansolid biosynthesis.21,22 The genes are indicated by arrows. The genes are denominated differently in the two source publications, so both names are given. The timing of the dehydration centered at C23 and subsequent IMDA reaction cannot be deduced with certainty. ACP, acyl carrier protein; PKS, polyketide synthase; AT, acyltransferase; MFS, major facilitator superfamily; HMGS, hydroxymethylglutaryl-CoA synthase; ECH, enoyl-CoA hydratase/isomerase. | ||
Elansolids A1 (1) and A2 (2) are atropisomers, but only the latter is active against gram-positive bacteria, with minimal inhibitory concentrations (MICs) of 0.2–64 μg mL−1. This product also showed moderate cytotoxicity against L929 mouse fibroblasts, B104 rat cells, SH-SY5Y human neuroblastoma cells, and HeLa cells.18–21 Furthermore, the highly reactive p-quinone methide elansolid A3 (3) can be stabilized as an ammonium salt, which showed antibacterial and cytotoxic activity identical to elansolid A2 (2).20 This reactivity was used to synthesize an elansolid library from crude extracts via Michael-type conjugate addition, using 21 different nucleophiles. None of these precursor-directed derivatives was more potent than elansolids A2 (2) and A3 (3), but all were less cytotoxic than elansolid A2 (2).23
Elansolids A2 (2) and C1 (9) inhibit the secretion-deficient Escherichia coli ΔtolC mutant when combined with the membrane-permeabilizing cationic cyclic peptide polymyxin B nonapeptide (PMBN) at 2 or 8 μg mL−1,25 indicating that a molecular target is also present in gram-negative bacteria but protection is achieved by its outer membrane.
The total chemical synthesis of elansolid B1 (4)26 was followed by the second-generation total synthesis of the same compound and the first synthesis of elansolid B2 (5).27 Further studies to elucidate the biosynthesis of elansolids28,29 revealed a further polyketide, 20-deoxy-elansolid B1 (10), with activity similar to elansolid A2 (2), showing that the hydroxyl group at C20 is not essential for antibacterial activity.28
2.2 YL-02905-A and YL-02905-B
Two Cytophaga strains were found to produce four macrolactones with activity against S. aureus.30–32 First reported in a patent,30 the two 22-membered macrolide ring antibiotics YL-02905-A (also named YM-32890-B, 11) and YL-02905-B (also named YM-32890-A, 12) were isolated from Cytophaga sp. YL-02905S.31 Their natural isomers (13 and 14) complement this group.32 The four macrolactones differ in the aliphatic chain attached to the macrolactone ring at C21 (Fig. 3). Accordingly, molecules 11 and 13 possess a conjugated tetrene system whereas 12 and 14 contain a conjugated triene system, separated by a methylene carbon at C28.31,32 Compound 12 undergoes tautomerization to form 11 (ref. 32) and is generally unstable under light and aerobic conditions.31 Only compounds 12 and 14 displayed activity against S. aureus KCTC 1927 with MICs of ∼10–20 μg mL−1.32 These data reflect the importance of the conjugation pattern of the double bonds along the aliphatic chain.32 The biosynthesis or total synthesis of these compounds has yet to be reported. | ||
| Fig. 3 Chemical structures of YL-02905 A (11) and B (12) as well as their structural isomers 13 and 14. | ||
3 Peptides
Peptide natural products are valuable sources of anti-infective leads and are either synthesized ribosomally or assembled by multi-modular non-ribosomal peptide synthetases (NRPSs), allowing the incorporation of non-proteinogenic amino acids. Optional (multi)cyclization and extensive post-translational modifications result in remarkable structural diversity.333.1 Cyclic depsipeptides
More than 1300 diverse natural cyclic depsipeptides have been described, and are characterized by the presence of ester and amide bonds that contribute to a range of biological properties.34,35 Most cyclic depsipeptides are synthesized by modular NRPSs,36 but some (e.g., microviridins) are synthesized ribosomally.37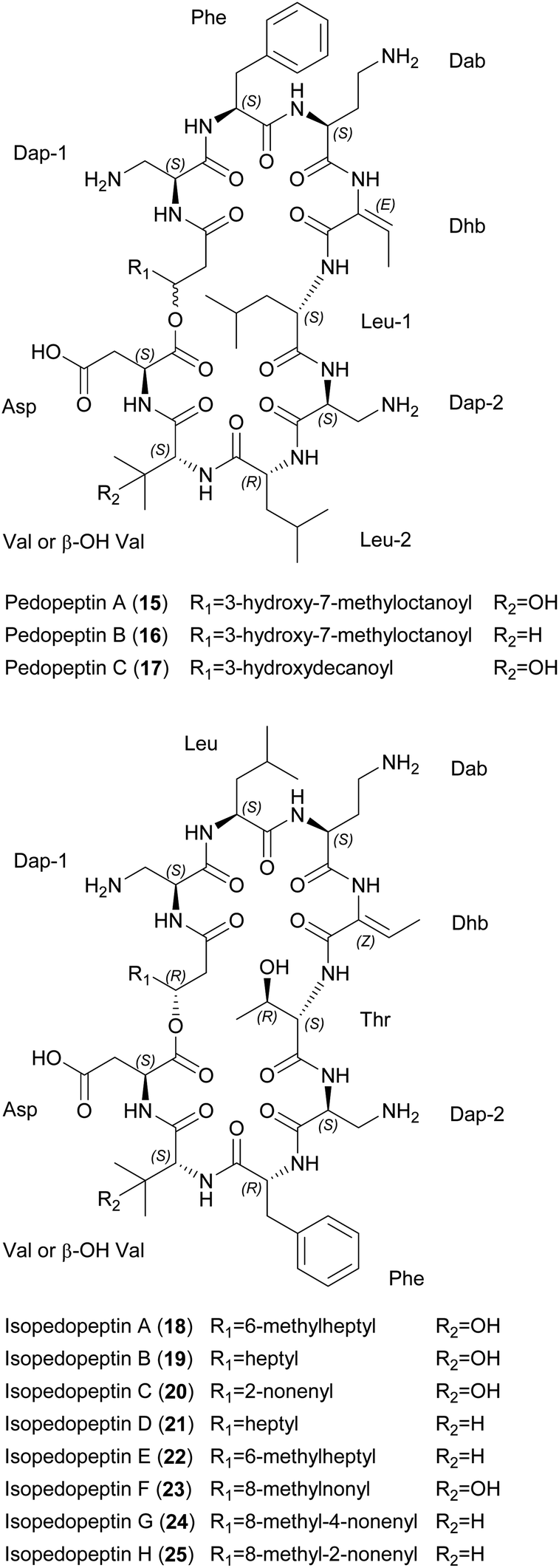 | ||
| Fig. 4 Chemical structures of pedopeptins A–C (15–17) and isopedopeptins A–H (18–25). Dap, diaminopropionic acid; Dab, diaminobutyric acid; Dhb, dehydrobutyrine; β-OH-Val, β-hydroxy valine. | ||
Pedopeptins A (15), B (16) and C (17) inhibit the binding of LPS to CD14 with half-maximal inhibitory concentrations (IC50) of 20, 11 and 47 nM, respectively. LPS-induced cytokine release in cell-based in vitro assays revealed dose-dependent inhibition, with IC50 values of 0.08–0.33 μM (∼1–10 μg mL−1). However, pedopeptins A and B were also cytotoxic at 100 and 30 μg mL−1, respectively. Antimicrobial testing of all three peptides revealed MICs of 2–4 μg mL−1 against two E. coli strains. Interestingly, only pedopeptin B showed strong activity (4 μg mL−1) against two gram-positive bacteria: S. aureus ATCC 6538P and S. epidermis ATCC 14990.39 All isopedopeptins show activity against a broad panel of gram-negative bacteria, including carbapenem-resistant strains on the WHO top-priority list. Isopedopeptin B (19) is the most potent representative, with MICs as low as 1 μg mL−1. In contrast to the pedopeptins, isopedopeptins show minimal activity against gram-positive bacteria (e.g., MIC = 16 μg mL−1 against Bacillus cereus CCUG7414), and none against fungi. Furthermore, isopedopeptins are cytotoxic against HepG2 liver cells (IC50 = 9–50 μM or 10–56 μg mL−1) and are hemolytic (0.8–52.3%). Isopedopeptin B was therefore the most promising candidate due to its potent antibacterial activity and acceptable cytotoxicity.44
The mode of action (MoA) of the cationic pedopeptins is thought to involve interactions with lipid layers due to structural similarities with other cationic cyclic peptides such as polymyxin B and colistin, which are known to interact with anionic LPS.39,45 Interestingly, the polycationic isopedopeptin B (19) showed activity against colistin-resistant strains of Acinetobacter baumannii, E. coli, and Klebsiella pneumoniae with a very low frequency of resistance (<3 × 10−9).44 Using an in vitro bacterial membrane system based on unilamellar liposomes prepared from E. coli phospholipid extracts, isopedopeptin F (23) was shown to cause membrane leakage with a half-maximal effective concentration (EC50) of 2 μM, a similar concentration to other cyclic peptides such as polymyxin B (EC50 = 1 μM) and LL-37 (EC50 = 0.6 μM).44
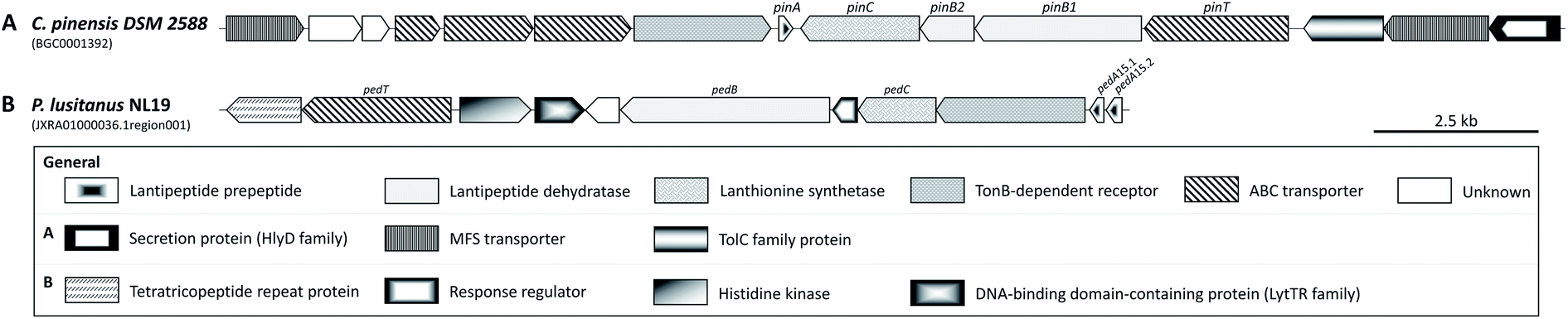
A putative BGC for the pedopeptins has been identified in the pedopeptin producer strain Pedobacter lusitanus NL19 (Fig. 5).46 The NRPS is composed of a large and a small subunit. Genes encoding diaminobutyrate-2-oxoglutarate transaminase and oxygenase (cytochrome P450 family) are found nearby, and may be responsible for the supply of Dab as well as Val hydroxylation. The expression of both NRPS genes correlates with the production of pedopeptins. Furthermore, within this BGC, the C5 domain shows similarity to C-domains that modify incorporated amino acids. In this case, it should catalyze the dehydration of Thr (incorporated by A-domain 4) to Dhb. Genes encoding glutamate synthase and dehydrogenase were also found in the genome, and may be responsible for Dap biosynthesis.46 A BGC for the isopedopeptins has yet to be described. However, the discovery of additional (iso)pedopeptin analogs in several other Pedobacter extracts indicates the existence of related BGCs, suggesting that small changes in A-domain specificity and/or module rearrangement within the pedopeptin BGC may facilitate the biosynthesis of various natural analogs, including the isopedopeptins.42 The total synthesis of these cyclic peptides has not yet been reported.
 | ||
| Fig. 5 Pedopeptin biosynthetic gene cluster. Non-ribosomal peptide synthetase biosynthetic gene cluster and assembly line responsible for the biosynthesis of pedopeptins. In silico predicted amino acids are indicated in the first row and those confirmed by NMR spectroscopy are underlined. Dap, diaminopropionic acid; Dab, diaminobutyric acid; β-OH Val, β-hydroxy valine. | ||
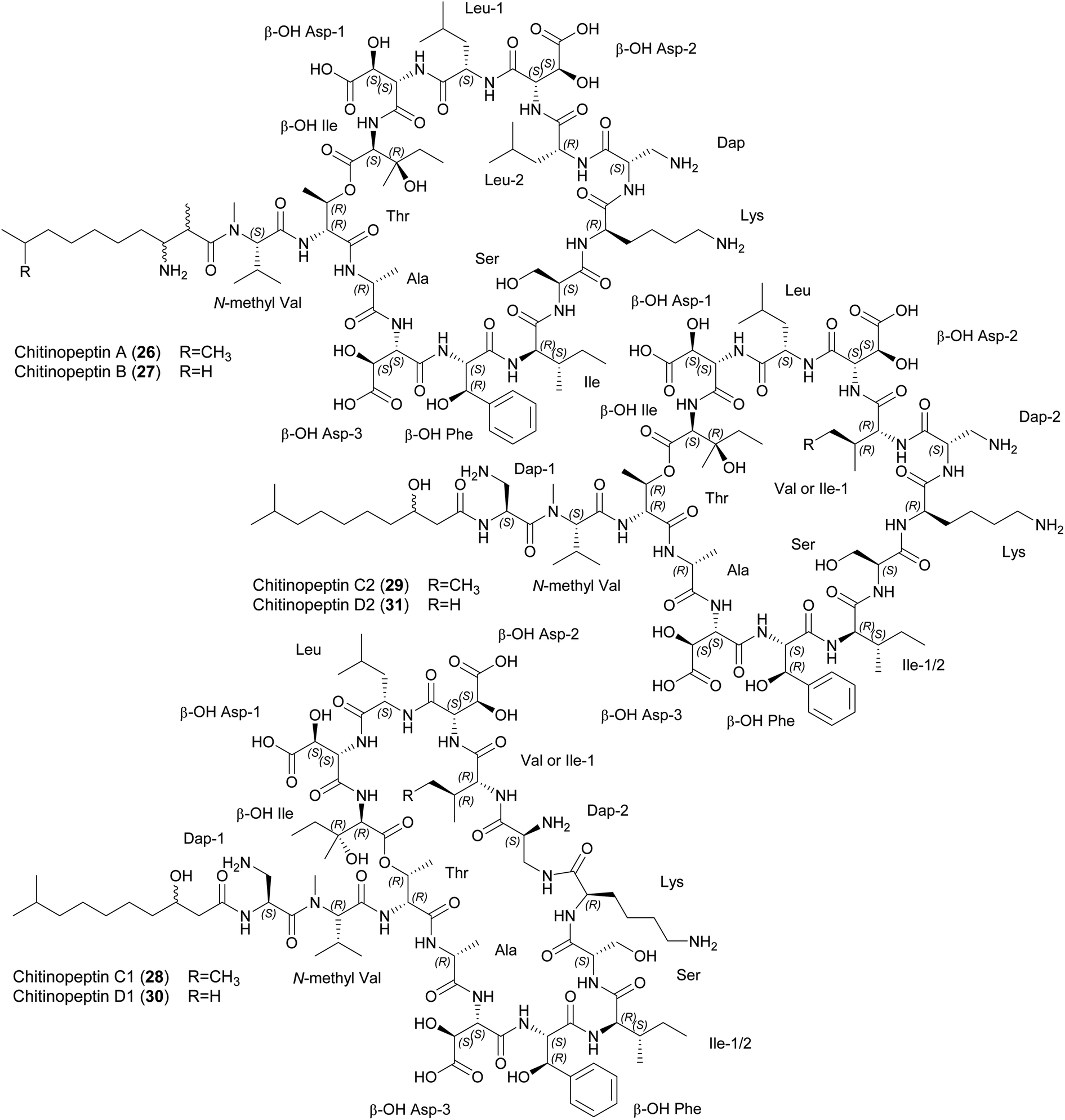 | ||
| Fig. 6 Chemical structures of chitinopeptins A–D (26–31). β-OH Asp, β-hydroxy aspartic acid; β-OH Phe, β-hydroxy phenylalanine; Dap, diaminopropionic acid; β-OH Ile, β-hydroxy isoleucine. | ||
Chitinopeptins inhibit gram-negative and gram-positive bacteria such as Moraxella catarrhalis (MIC = 2 μg mL−1) and Bacillus subtilis (MIC = 4 μg mL−1) as well as the yeast Candida albicans (MICs ≥ 4 μg mL−1). MICs of ≥ 16 μg mL−1 were observed against A. baumannii, Micrococcus luteus and Zymoseptoria tritici. Interestingly, the chitinopeptins bind iron, which in the case of chitinopeptin A (26) reduces but does not abolish its activity.14
Putative NRPS-BGCs have been identified for the synthesis of these CLPs (Fig. 7). The number and predicted substrate specificity of the A-domains, the overall composition of the NRPS assembly line, and genes encoding enzymes responsible for precursor supply and post-assembly modifications matched the structural composition of the chitinopeptins. A BGC-similarity networking approach revealed the presence of homologous BGCs in C. oryziterrae JCM16595 and C. niastensis DSM 24859.14 The total synthesis of chitinopeptins has yet to be disclosed. The synthesis of the β-hydroxylated amino acids Asp, Phe and Ile has been described as an attempt to elucidate the absolute configuration of these amino acids in all chitinopeptin structures.14
 | ||
| Fig. 7 Chitinopeptin biosynthetic gene clusters. Non-ribosomal peptide synthetase biosynthetic gene clusters and assembly lines responsible for the biosynthesis of chitinopeptins. In silico predicted amino acids are indicated and those confirmed by NMR spectroscopy are underlined. β-OH Asp, β-hydroxy aspartic acid; β-OH Phe, β-hydroxy phenylalanine; Dap, diaminopropionic acid; β-OH Ile, β-hydroxy isoleucine. | ||
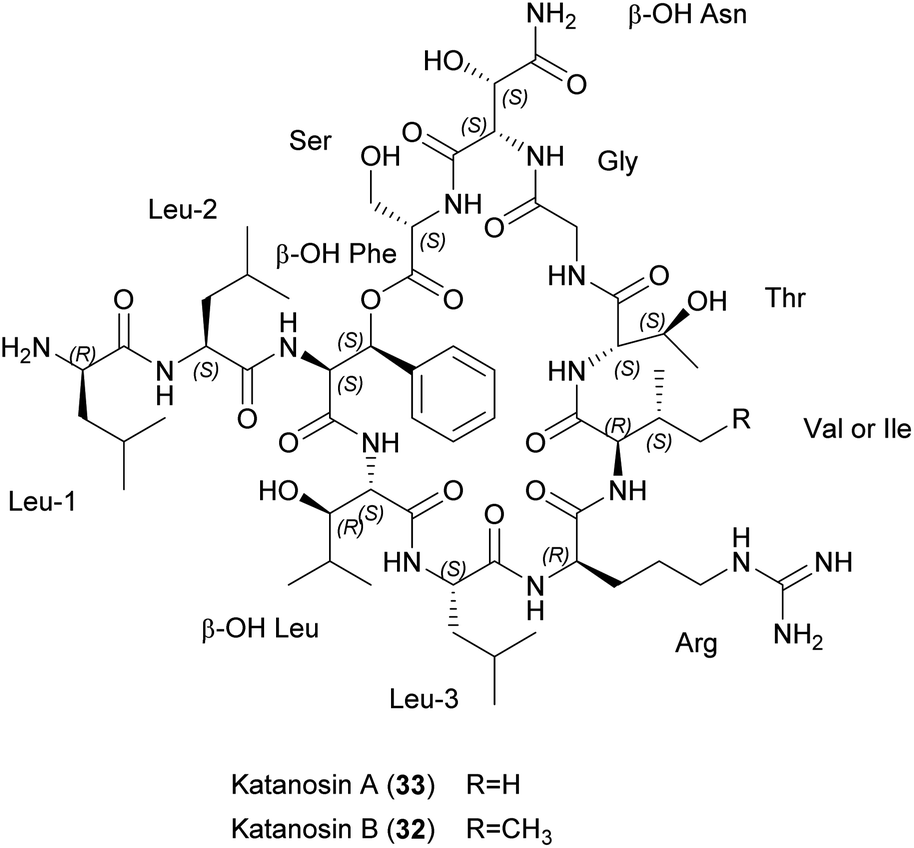 | ||
| Fig. 8 Chemical structures of katanosins A (33) and B (32). β-OH Phe, β-hydroxy phenylalanine; β-OH Asn, β-hydroxy aspargine; β-OH Leu, β-hydroxy leucine. | ||
Both molecules target peptidoglycan synthesis. To gain insight into the MoA, staphylococcal transglycosylation and its preceding reactions were studied by incorporating [14C]GlcNAc into S. aureus peptidoglycan in whole cells, as well as nascent peptidoglycan formation in vitro. Both peptides inhibit nascent peptidoglycan synthesis, but only katanosin B also inhibits lipid intermediate formation.52 Lipid II was identified as the cellular target of katanosin B.53 The MoA of katanosins A and B differs from that of vancomycin and the peptides show a low frequency of resistance, making them suitable for further development. Accordingly, katanosin B and its method of production have been patented.54
A cloning study with a Lysobacter sp. ATCC 53042 genomic library revealed a partial NRPS gene (4.6 kb) as part of the BGC.55 The complete BGC containing two multi-modular NRPSs (lybA and lybB) was discovered by genome sequencing (Fig. 9).56 The termination module showed a tandem thioesterase architecture and both thioesterases were biochemically characterized in vitro. Peptide cyclization and simultaneous release is only mediated by the penultimate thioesterase. The second thioesterase fulfills a proofreading function involving the deacylation of miss-primed peptidyl carrier proteins.56 Synthetic access was established based on modular, solution-phase approaches57 and by solid-phase peptide syntheses.58
 | ||
| Fig. 9 Lysobactin/katanosin biosynthetic gene cluster (MIBiG BGC0000385). Non-ribosomal peptide synthetase biosynthetic gene cluster and assembly line responsible for the biosynthesis of lysobactin/katanosins. In silico predicted amino acids are indicated and those confirmed by NMR spectroscopy are underlined. β-OH Phe, β-hydroxy phenylalanine; β-OH Leu, β-hydroxy leucine; β-OH Asn, β-hydroxy asparagine. | ||
 | ||
| Fig. 10 Chemical structures of YM-47141 (34) and YM-47142 (35). Dah, 2,3-dioxo-4-amino-6-methylheptanoic acid. | ||
3.2 Short linear peptides
Short linear peptides also display remarkably diverse biological activities with biomedical and cosmeceutical applications.63 They tend to have a short in vivo half-life64 and many are immunogenic,65 but generally they are highly selective, can cross biological membranes, and cost-effective synthesis has been reported.63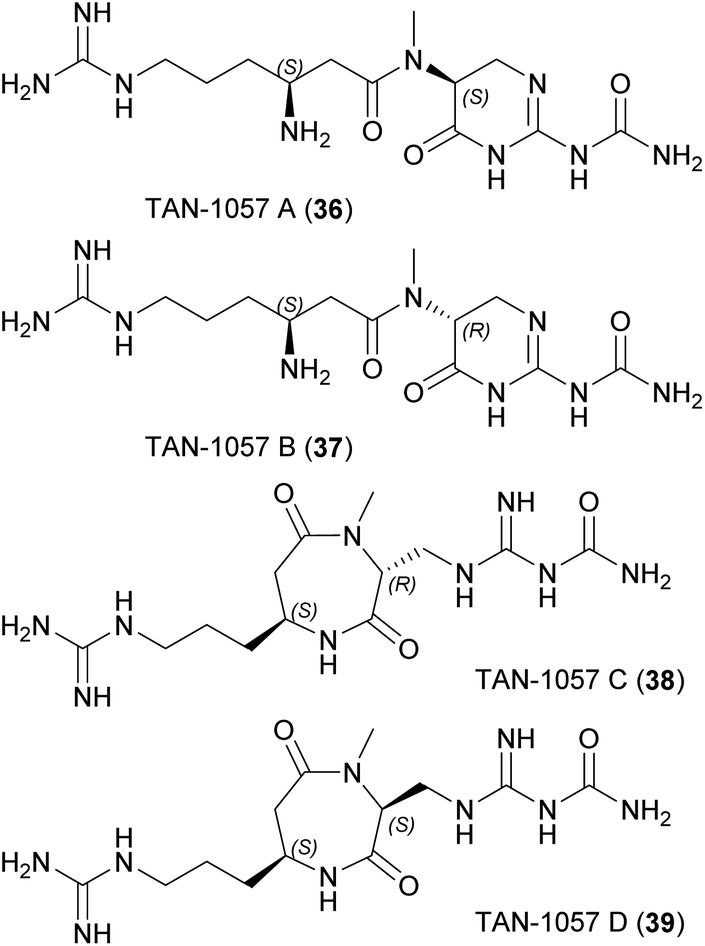 | ||
| Fig. 11 Chemical structures of TAN-1057 A–D (36–39). | ||
MoA studies with the TAN-1057 series showed a negative effect on protein synthesis at the elongation step in whole-cell and cell-free translation assays.67,71 More detailed analysis revealed the target was the peptidyl transferase of the 50S ribosomal subunit, but the binding site appears to differ from that of other inhibitors.67,71,72 Resistance studies indicated a rapid response based on changes in the transport of cellular dipeptides, followed by ribosomal alterations later on.71,73 A BGC congruent with these dipeptide-like metabolites has yet to be identified.
 | ||
| Fig. 12 Chemical structures of falcitidin (40) as well as pentacitidins A (41) and B (42). | ||
Falcitidin displays falcipain-2 enzyme activity in vitro (IC50 = 6 μM)74 but lacks whole-cell activity against Plasmodium falciparum 3D7 (IC50 > 10 μM).75 A liquid-phase synthesis approach produced an N-acyl trifluoromethyl derivative with a hydrophobic N-trityl group on the imidazole residue. In contrast to the isolated natural product, this molecule exhibits whole-cell antimalarial activity (IC50 = 0.14 μM).75 Pentacitidins were produced by solid-phase peptide synthesis.76 Aldehydes are electrophilic warheads that form covalent-reversible hemithioacetal adducts with catalytic Cys residues and covalent-reversible hemiacetal adducts with Ser residues. Testing pentacitidins against human cysteine proteases (cathepsins B and L) and related proteases from parasites (falcipain-2/3 and rhodesain) revealed selective inhibition of the parasitic enzymes with sub-micromolar IC50 values. In addition, the natural products were active against α-chymotrypsin. Docking studies showed favorable distances between the catalytic Cys residue and the pentacitidin aldehyde in its L-configuration.76
A pentacitidin BGC was tentatively identified, consisting of a single NRPS gene (18.5 kb) with five A-domains and a C-starter unit (Fig. 13).76 It is congruent with the pentapeptide structure, strengthening the degradation hypothesis. The BGC contains a terminal reductase domain probably involved in the formation of the C-terminal aldehyde and alcohol analogs via reductive release. These alcohol analogs of pentacitidins were detected by molecular networking analysis and synthesized as pentacitidin intermediates, but showed no bioactivity.76 Similar BGCs were found in the genomes of four other Chitinophaga strains.76
 | ||
| Fig. 13 Pentacitidin biosynthetic gene clusters. Non-ribosomal peptide synthetase biosynthetic gene clusters and assembly lines responsible for the biosynthesis of pentacitidins. In silico predicted amino acids are indicated and those confirmed by NMR spectroscopy are underlined. Identity represents the nucleotide alignment of all five BGCs using MAFFT.156 β-OH Tyr, β-hydroxy tyrosine. | ||
3.3 Ribosomally synthesized and post-translationally modified peptides (RiPPs)
There are more than 20 subclasses of RiPPs, and scientific interest in these peptides has increased in recent decades.77 A few RiPPs have already been isolated from Bacteroidetes, and many more can be expected due to development of new genome mining tools for RiPP discovery.783.3.1.1 Pinensins A and B. Pinensins A (43) and B (44) are produced by C. pinensis DSM 28390. These two cyclic lantipeptides contain 22 and 21 amino acids, respectively, with a rare N-terminal pyruvic acid. Both incorporate two 3-methyl-lanthionine-bridged ring systems with an unusual (2S,3S,6R)-stereochemical configuration (Fig. 14).80
 | ||
| Fig. 14 Chemical structures of pinensins A (43) and B (44). Dhb, dehydrobutyrine; Abu, α-aminobutyric acid. | ||
As a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture, pinensins A and B exhibit broad activity against filamentous fungi and yeast (MIC > 5 μg mL−1) but only weak antibacterial activity. These lantipeptides also inhibit the growth of higher eukaryotic cells (IC50 > 10 μM). The pinensin BGC predicted in silico consists of at least five genes encoding the lantipeptide precursor peptide (PinA), two dehydratases (PinB1 and PinB2), one lanthionine synthetase (PinC) and PinT, required for export and cleavage of the precursor peptide (Fig. 15A). An unusual feature of pinensin biosynthesis is the split dehydratase, with functional domains catalyzing glutamylation and elimination encoded by separate open reading frames.78,80 The precursor peptide PinA has 22 residues like pinensin A, suggesting that pinensin A is converted into pinensin B by the post-translational removal of the terminal Ala residue, but the enzyme responsible remains elusive.80 Pinensin-like BGCs have also been identified in the genomes of other Chitinophaga,14Cryseobacterium, Elizabethkingia, Pedobacter and Sinomicrobium strains.78 Predicted variations in the amino acid sequence of the core peptides indicate a yet undiscovered structural diversity of pinensin-type lantipeptides. A total synthesis approach for these peptides has not been reported.
:1 mixture, pinensins A and B exhibit broad activity against filamentous fungi and yeast (MIC > 5 μg mL−1) but only weak antibacterial activity. These lantipeptides also inhibit the growth of higher eukaryotic cells (IC50 > 10 μM). The pinensin BGC predicted in silico consists of at least five genes encoding the lantipeptide precursor peptide (PinA), two dehydratases (PinB1 and PinB2), one lanthionine synthetase (PinC) and PinT, required for export and cleavage of the precursor peptide (Fig. 15A). An unusual feature of pinensin biosynthesis is the split dehydratase, with functional domains catalyzing glutamylation and elimination encoded by separate open reading frames.78,80 The precursor peptide PinA has 22 residues like pinensin A, suggesting that pinensin A is converted into pinensin B by the post-translational removal of the terminal Ala residue, but the enzyme responsible remains elusive.80 Pinensin-like BGCs have also been identified in the genomes of other Chitinophaga,14Cryseobacterium, Elizabethkingia, Pedobacter and Sinomicrobium strains.78 Predicted variations in the amino acid sequence of the core peptides indicate a yet undiscovered structural diversity of pinensin-type lantipeptides. A total synthesis approach for these peptides has not been reported.
 | ||
| Fig. 15 Biosynthetic gene clusters of the ribosomally synthesized and post-translationally modified peptides pinensin (MIBiG BGC0001392) (A) and pedA15.1 (B). | ||
3.3.1.2 PedA peptides. The computational prediction of precursor peptide sequences revealed several BGCs that may provide novel lathipeptides.78 A BGC from Pedobacter lusitanus NL19 encoding two precursor peptides (Fig. 15B) could not be elicited in the native producer, so a heterologous expression strategy was implemented to gain access to these compounds. Co-expression of precursor peptide genes pedA15.1 and pedA15.2 in E. coli, together with their modifying enzymes, led to the discovery of the PedA15 peptides.81 Only the structure of PedA15.1 (45) was elucidated in detail by NMR analysis (Fig. 16). This contains a novel 14-membered bis-lanthionine ring system in the LL-lanthionine stereochemical configuration. The 4-fold or 5-fold dehydrated peptide carries three lanthionine bridges between Dha7 and Cys16 nested with the Cys8–Dha15 ring, and a final ring between Dha18 and Cys21. This lantipeptide ring pattern has not been described in other lantipeptides. PedA15.1 carries further dehydrated but non-cyclized amino acids as well as a Ser that escaped dehydration in the heterologous host. The biosynthetic machinery of E. coli thus allows the production of modified peptides, but the heterologous products may not be the same as those synthesized in the native host, potentially explaining why PedA15.1 and PedA15.2 lack antibacterial and antifungal activity.81 As for the pinensins, the total synthesis of PedA15.1 has not been described.
 | ||
| Fig. 16 Chemical structure of PedA15.1 (45). Dha, dehydroalanine. | ||
 | ||
| Fig. 17 Chemical structure of FR901451 (46). | ||
4 Polyketide – non-ribosomal peptide hybrids
Modular PKS and NRPS systems present analogous assembly lines with similar mechanisms used to incorporate building blocks in the nascent chain. Genes encoding enzymes involved in either polyketide or non-ribosomal peptide synthesis can be combined, resulting in hybrid natural products that greatly expand the potential chemical diversity.86 One example is the immunosuppressant rapamycin, with a pipecolate group in its polyketide skeleton.87 Several of these complex polyketide–amino acid/peptide hybrids, featuring both acyl and aminoacyl building blocks, have been described in Bacteroidetes.4.1 Ariakemicins A and B
Ariakemicins A (47) and B (48) were isolated from a marine gliding bacterium of the genus Rapidithrix and were described as an inseparable mixture.88 The linear structures carry on one side an aromatic moiety substituted with one hydroxyl and one methoxy group, and on the other a methylated oxazol moiety decorated with a primary amide. The two compounds differ in the location of the double bond, which is found in position Δ12 and Δ11 in ariakemicins A and B, respectively (Fig. 18). The ariakemicin mixture was shown to inhibit gram-positive bacteria, with MICs ranging from 0.46 μg mL−1 against S. aureus IFO12732 to 83 μg mL−1 against B. subtilis IFO3134, but was inactive against gram-negative bacteria and C. albicans. Cytotoxicity was observed against human lung cancer cell line A549 and baby hamster kidney (BHK) cells with IC50 values of 25 and 15 μg mL−1, respectively.88 We were able to dereplicate ariakemicin A in extracts from Rapidithrix thailandica s80 (ref. 89) and subsequent isolation yielded pure ariakemicin A (47). This was active against E. coli ATCC25922 ΔtolC (our unpublished results), indicating that a molecular target for this compound is also present in gram-negative bacteria. One drawback of the ariakemicins is their rapid decomposition and loss of activity.88 This may also explain why these compounds have not yet been synthesized.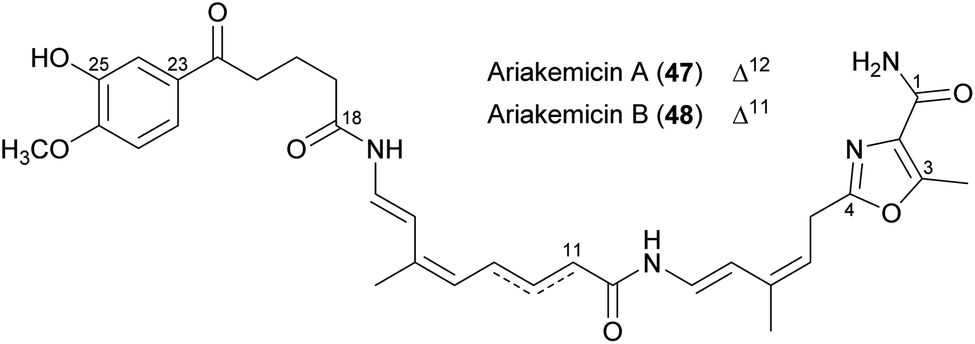 | ||
| Fig. 18 Chemical structures of ariakemicins A (47) and B (48). | ||
5 Quinolines
The heterocyclic aromatic compound quinoline was first extracted from coal tar in the 18th century, and its structural motif can be found in many plant alkaloids, including quinine. However, there are also prominent bacterial examples, such as the antifungal compound pyrrolnitrin in Pseudomonas spp.90 This aminophenylpyrrole alkaloid became a lead structure for the development of foliar fungicides. Additional structural variants have been isolated mainly from marine bacteria of the order Cytophagales.5.1 G1499-2
The first quinoline isolated from the genus Cytophaga was 3-methyl-2-(2-pentylidenecyclopropyl)-4-quinolinone, also known as G1499-2 (49) (Fig. 19).91 It has weak activity against a few bacteria, such as Flavobacterium sp. 1980, a marine species, and no toxicity in mice. The BGC is unknown and total synthesis has not been reported for this compound. | ||
| Fig. 19 Chemical structure of G1499-2 (49). | ||
5.2 Marinoquinolines and marinoazepinones
Several natural products featuring a rarely described 3H-pyrollo[2,3-c]quinoline ring system have also been identified (Fig. 20), beginning with marinoquinoline A (50), which was isolated from Rapidithrix thailandica.92 This product is an acetylcholinesterase inhibitor with an IC50 value of 4.9 μM.93 Five derivatives known as marinoquinolines B–F (51–55) are produced by Ohtaekwangia kribbensis.94 All five showed toxicity against three cancer cell lines in the low micromolar range (0.3–8.0 μg mL−1) and two (51 and 55) showed potent activity against P. falciparum K1 (IC50 = 1.8 and 1.7 μM, respectively). | ||
| Fig. 20 Chemical structures of marinoquinolines A–K (50–60) as well as marinoazepinones A (61) and B (62). | ||
Several more marine Bacteroidetes were isolated by cultivation in seawater-based media, leading to the discovery of marinoquinolines G–K (56–60).95 Furthermore, the prolific producer strain Mooreia alkaloidigena CNX-216T synthesizes the structurally-related marinoazepinones A (61) and B (62), as well as other alkaloids.
To investigate quinoline biosynthesis in Bacteroidetes, the BGCs predicted in silico were expressed in E. coli along with other key enzymes for in vitro assays.96 This revealed that only a single enzyme-catalyzed reaction is essential, in which tryptophan (63) is converted to 3-(2′-aminophenyl)-pyrrole (64), which is another potent acetylcholinesterase inhibitor already isolated from a Rapidithrix strain.97 This active intermediate can be coupled to a variety of aldehydes via a spontaneous Pictet–Spengler-like reaction under physiological conditions, producing diverse products using a minimal complement of enzymes (Fig. 21).
 | ||
| Fig. 21 Tryptophan (63) is enzymatically converted to 3-(2′-aminophenyl)-pyrrole (64). This intermediate reacts spontaneously with a variety of aldehydes, resulting in marinoquinolines such as marinoquinoline A (50). | ||
The synthesis of a pyrroloquinoline system has been achieved using several approaches,98–102 including the synthesis of four different marinoquinolines via Heck–Matsuda arylation and Pictet–Spengler cyclisation.100 Brønsted acid-promoted arene-ynamide cyclization was applied to produce high yields of marinoquinolines A (50) and C (52).101 More recently, the total synthesis of marinoquinoline A was achieved in four steps starting with the palladium-catalyzed Ullmann coupling of o-bromonitrobenzene to iodinated pyrrole.102 Also, an intramolecular Diels–Alder oxazole cycloaddition reaction was used to produce marinoquinoline A from oxazole with an overall yield of 12%.99
6 Lactams
The clinical introduction of penicillin in the 1940s is a milestone in medical history. Even today, β-lactam antibiotics are ranked among the most important antimicrobials. Following the penicillins, further β-lactam antibiotic classes (e.g., cephalosporins, carbapenems and clavams) were discovered in fungi and actinomycetes. By expanding the search to include Proteobacteria and Bacteroidetes, the global β-lactam discovery effort culminated in the description of the monobactams in the early 1980s.103 All reports on lactams from Bacteroidetes date back to a short period between 1982 and 1987, so there is not yet any information about the genetic or metabolic basis of such compounds and their total synthesis has not been reported.6.1 Monobactams
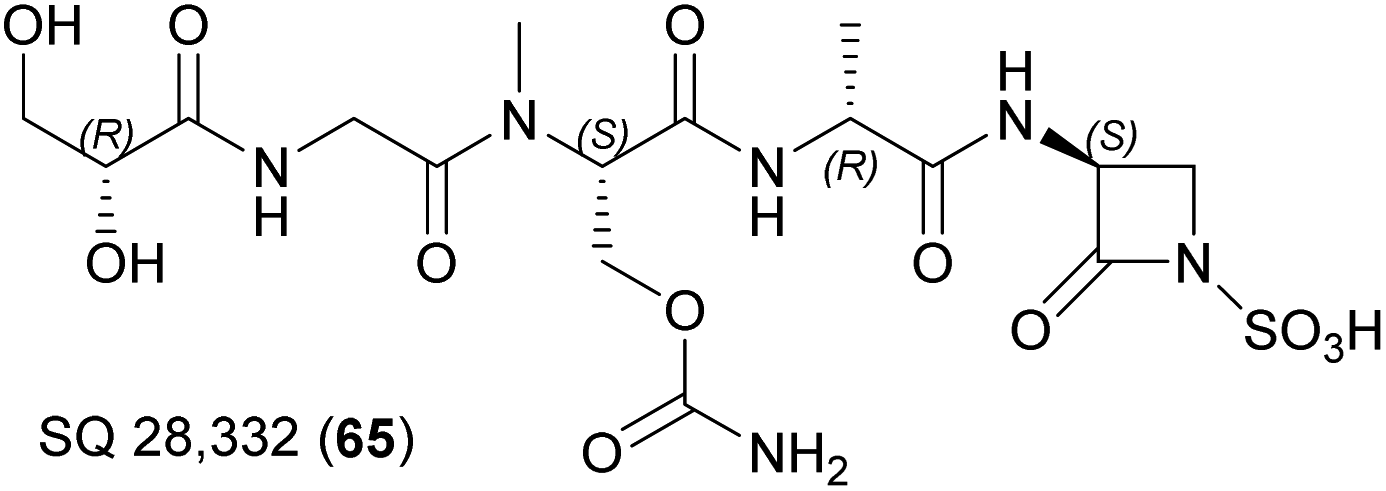 | ||
| Fig. 22 Chemical structure of SQ 28332 (63). | ||
 | ||
| Fig. 23 Chemical structures of PB-5266 A–C (64–66). | ||
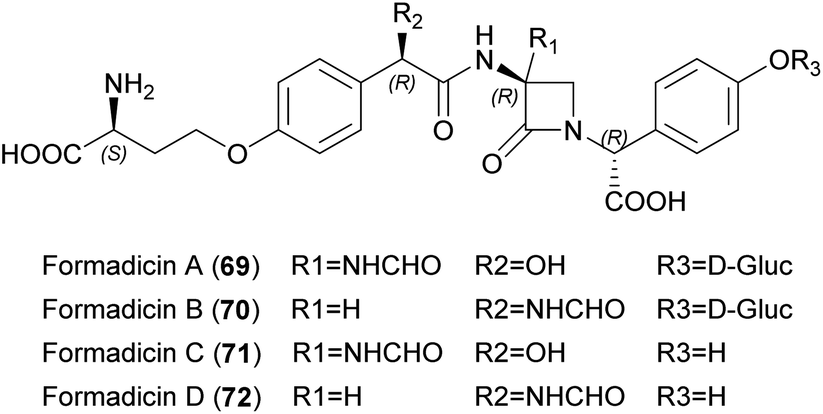 | ||
| Fig. 24 Chemical structures of formadicins A–D (67–70). | ||
6.2 Cephems
 | ||
| Fig. 25 Chemical structure of desacetoxycephalosporin C (71). | ||
 | ||
| Fig. 26 Chemical structures of SQ 28516 (72) and SQ 28517 (73). | ||
 | ||
| Fig. 27 Chemical structures of chitinovorins A–D (74–77). | ||
7 Additional Bacteroidetes natural products
7.1 Pigments
The best-known Bacteroidetes pigments are flexirubin-like compounds,115 which are phylogenetically conserved and were used as chemotaxonomic markers for the phylum (formerly known as the Cytophaga–Flavobacterium–Bacteroides group) upon their discovery in Chitinophaga filiformis (formerly Flexibacter elegans Fx e1).116 The total synthesis117 and biosynthesis118 of these pigments has been reviewed.115 Flexirubin analogs have been evaluated for their antioxidant activity.119 Other pigments, found in many Bacteroidetes, include carotenoids such as saproxanthin, myxol, zeaxanthin, flexixanthin and deoxyflexixanthin, some of which also possess antioxidant activity.120 The flexixanthin BGC has been identified.1217.2 Siderophores
Siderophores are iron-chelating compounds. Hydroxamate siderophores (tenacibactins A–D and bisucaberin B) have been isolated from Tenacibaculum spp.,122,123 and fulvivirgamides from Fulvivirga sp. W222.124 The bisucaberin B and fulvivirgamid BGCs have been experimentally validated.123–1257.3 Lipids
Chryseobacterium spp. produce sulfonolipids (sulfobacin A and B) that are von Willebrand factor (vWF) receptor antagonists126 and potential anti-inflammatory127 and anticarcinogenic128 leads. Flavocristamides A and B (whereby flavocristamide B is structurally identical to the before mentioned sulfobacin A) were isolated from a marine Flavobacterium species and were shown to inhibit DNA polymerase α.129 Other sulfonolipids (the capnoids capnine and N-acylcapnine)130 were shown to be required for gliding motility in Capnocytophaga spp.131 The N-acetylated sulfonolipids RIF-1 and RIF-2 (rosette inducing factors) and IOR-1 (inhibitor of rosettes) isolated from Algoriphagus machipongonensis were shown to influence morphogenesis in the choanoflagellate Salpingoeca rosetta.132 The total synthesis133 and biosynthesis134 of most of these sulfonolipids has been investigated.Other than sulfonolipids, the major lipids isolated from Bacteroidetes include lipoamino acids (LAAs), phospholipids and N-acyl amino acids (NAAAs). LAAs consist of a single de-esterified fatty acid or two fatty acids linked by esterification at C3 with different degrees of unsaturation. The hydrophobic fatty acid part is linked via an amide to Gly, Ser and ornithine, or a combination of two or three of these amino acids. The physicochemical properties and bioactivities differ significantly between stereoisomers in many cases, including WB-3559 A–D;135 the topostins B553, B567 (also known as cytolipin), D640 and D654 (also known as WB-3559D);136–138 and flavolipin139,140 (also known as lipid 654 (ref. 141 and 142).143 In terms of biological properties, these LAAs act as mammalian DNA topoisomerase I inhibitors,136,137 macrophage activators,144 promoters of hemagglutination,140 bacterial virulence factors,145,146 N-type calcium channel blockers,147 and ligands for Toll-like receptor 2.141,142,148 Accordingly, they are linked to the development of two chronic inflammatory diseases (periodontitis and atherosclerosis146), multiple sclerosis,149 as well as showing antimicrobial activities.150
7.4 Others
Other diverse compounds produced by Bacteroidetes include indole analogs. For example, Cytophaga strain AM13.1 produces 2,5-bis(3-indolylmethyl)pyrazine and pharacine, a natural p-cyclophane.151Cytophaga johnsonae AJ 12589 produces resorcinin, which stimulates the growth of NIH 3T3 mouse fibroblasts.152 Furthermore, four neoverrucosane diterpenoids were identified as products of Saprospira grandis ATCC 23116,153 whereas the algal morphogen thallusin is produced by a Zobellia strain and has been synthesized.1548 Concluding remarks
The phylum Bacteroidetes produces a wide range of metabolites that highlight its status as a valuable resource for natural products research. Although the isolation of interesting compounds from Bacteroidetes traces back to the golden era of natural product discovery (1950–1960), the total number of chemical entities is still remarkably low.The recent identification of novel natural products is exemplified by the isopedopeptins and chitinopeptins, supporting the hypothesis that the natural product diversity in this phylum has yet to be exploited comprehensively. Of outstanding significance are the isopedopeptins, which show potent activity against gram-negative bacteria on the WHO's top-priority list, a low frequency of resistance, and acceptable levels of cytotoxicity, making them promising new candidate antibiotics.38,44 The β-lactams are a more historical (but still extremely relevant) class of natural products that were isolated from Bacteroidetes in the 1980s. Their clinical use as antibiotics that inhibit bacterial penicillin-binding proteins and also act as β-lactamase inhibitors with the potential to break AMR is of great importance to international healthcare. Resistance to β-lactam antibiotics and fluoroquinolones is a major societal burden that accounts for more than 70% of all deaths attributed to AMR.1 The β-lactams are still an essential drug class and a cornerstone of the current antibiotics market. Therefore, we must develop new solutions to counteract the steady loss of existing products. The naturally evolved strategy of combining β-lactam antibiotics with a β-lactamase inhibitor sharing the same warhead is therefore crucial to address the AMR crisis. There is a constant need for the development new β-lactamase inhibitors that maintain or even expand the antibacterial range of our clinical β-lactam/β-lactamase inhibitor combinations.155 Although not directly pursued as a therapeutic strategy, we also consider β-lactams from the Bacteroidetes as very much part of this success story in learning from naturally evolved concepts and translating microbial diversity (and the corresponding structural and functional diversity of natural products) into life-saving drugs. Accordingly, the Bacteroidetes join other β-lactam producers such as the Actinobacteria, Proteobacteria and fungi.
Even compounds not followed up in the past such, as the highly active but unstable ariakemicin, may be suitable as candidates for repurposing strategies that aim to generate more applicable structures based on a natural scaffold. Following this approach, our recent omics studies revealed the immense genomic potential of this phylum that corresponds to uncharted chemical space, which differs significantly from the more widely investigated producer taxa.14 In our opinion, this pronounced and unique biosynthetic capacity offers the potential for groundbreaking new compounds in the near future. Considering their historically proven role in the discovery of structurally diverse natural products, the Bacteroidetes have not received the attention they deserve during the latest renaissance in natural products research. It is therefore time to reconsider the phylum Bacteroidetes as a source for the discovery of interesting and valuable natural products to fill the future drug development pipeline.
9 Conflicts of interest
There are no conflicts to declare.10 Acknowledgements
The authors would like to acknowledge the financial support of their original work by the Hessiann State Ministry of Higher Education, Research and the Arts (HMWK) via the state initiative for the development of scientific and economic excellence for the LOEWE Center for Insect Biotechnology and Bioresources, and the German Centre for Infection Research (DZIF).11 References
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. de Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef.
- A. Talebi Bezmin Abadi, A. A. Rizvanov, T. Haertlé and N. L. Blatt, J. Bionanosci., 2019, 9, 778–788 CrossRef.
- T. F. Schäberle and I. M. Hack, Trends Microbiol., 2014, 22, 165–167 CrossRef PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- N. Ziemert, M. Alanjary and T. Weber, Nat. Prod. Rep., 2016, 33, 988–1005 RSC.
- F. Thomas, J.-H. Hehemann, E. Rebuffet, M. Czjzek and G. Michel, Front. Microbiol., 2011, 2, 93 Search PubMed.
- R. L. Hahnke, J. P. Meier-Kolthoff, M. García-López, S. Mukherjee, M. Huntemann, N. N. Ivanova, T. Woyke, N. C. Kyrpides, H.-P. Klenk and M. Göker, Front. Microbiol., 2016, 7, 2003 CrossRef PubMed.
- M. J. McBride and Y. Zhu, J. Bacteriol., 2013, 195, 270–278 CrossRef CAS PubMed.
- M. J. McBride, in The Prokaryotes. Other Major Lineages of Bacteria and The Archaea, ed. E. F. DeLong, S. Lory, E. Stackebrandt, F. Thompson and E. Rosenberg, Springer, Berlin, Heidelberg, 2014, pp. 643–676 Search PubMed.
- J. W. Clark and S. Kambhampati, Mol. Phylogenet. Evol., 2003, 26, 82–88 CrossRef CAS PubMed.
- D. P. Bayley, E. R. Rocha and C. J. Smith, FEMS Microbiol. Lett., 2000, 193, 149–154 CrossRef CAS PubMed.
- (a) D. Vingadassalom, A. Kolb, C. Mayer, T. Rybkine, E. Collatz and I. Podglajen, Mol. Microbiol., 2005, 56, 888–902 CrossRef CAS PubMed; (b) S. Chen, M. Bagdasarian, M. G. Kaufman, A. K. Bates and E. D. Walker, J. Bacteriol., 2007, 189, 5108–5118 CrossRef CAS PubMed.
- C. Borsetto, G. C. A. Amos, U. N. da Rocha, A. L. Mitchell, R. D. Finn, R. F. Laidi, C. Vallin, D. A. Pearce, K. K. Newsham and E. M. H. Wellington, Microbiome, 2019, 7, 78 CrossRef PubMed.
- S. Brinkmann, M. Kurz, M. A. Patras, C. Hartwig, M. Marner, B. Leis, A. Billion, Y. Kleiner, A. Bauer, L. Toti, C. Pöverlein, P. E. Hammann, A. Vilcinskas, J. Glaeser, M. S. Spohn and T. F. Schäberle, bioRxiv, 2021 DOI:10.1101/2021.07.30.454449.
- (a) J. M. McGUIRE, R. L. BUNCH, R. C. ANDERSON, H. E. BOAZ, E. H. FLYNN, H. M. POWELL and J. W. SMITH, Antibiot. Chemother., 1952, 2, 281–283 CAS; (b) C. W. Pettinga, W. M. Stark and F. R. van Abeele, J. Am. Chem. Soc., 1954, 76, 569–571 CrossRef CAS.
- D. Jelić and R. Antolović, Antibiotics, 2016, 5, 29 CrossRef PubMed.
- K. D. Lenz, K. E. Klosterman, H. Mukundan and J. Z. Kubicek-Sutherland, Toxins, 2021, 13 Search PubMed.
- G. Klaus, H. Steinmetz and G. Höfle, European patent., EP2093212A1, 2008 Search PubMed.
- H. Steinmetz, K. Gerth, R. Jansen, N. Schläger, R. Dehn, S. Reinecke, A. Kirschning and R. Müller, Angewandte Chemie (International ed. in English), 2011, 50, 532–536 CrossRef CAS PubMed.
- R. Jansen, K. Gerth, H. Steinmetz, S. Reinecke, W. Kessler, A. Kirschning and R. Müller, Chem. - Eur. J., 2011, 17, 7739–7744 CrossRef CAS PubMed.
- R. Teta, M. Gurgui, E. J. N. Helfrich, S. Künne, A. Schneider, G. van Echten-Deckert, A. Mangoni and J. Piel, Chembiochem, 2010, 11, 2506–2512 CrossRef CAS PubMed.
- R. Dehn, Y. Katsuyama, A. Weber, K. Gerth, R. Jansen, H. Steinmetz, G. Höfle, R. Müller and A. Kirschning, Angewandte Chemie (International ed. in English), 2011, 50, 3882–3887 CrossRef CAS PubMed.
- H. Steinmetz, W. Zander, M. A. M. Shushni, R. Jansen, K. Gerth, R. Dehn, G. Dräger, A. Kirschning and R. Müller, Chembiochem, 2012, 13, 1813–1817 CrossRef CAS PubMed.
- S. Kumar, G. He, P. Kakarla, U. Shrestha, K. C. Ranjana, I. Ranaweera, T. M. Willmon, S. R. Barr, A. J. Hernandez and M. F. Varela, Infect. Disord. - Drug Targets, 2016, 16, 28–43 CrossRef CAS PubMed.
- A. Beckmann, S. Hüttel, V. Schmitt, R. Müller and M. Stadler, Microb. Cell Factories, 2017, 16, 143 CrossRef PubMed.
- A. Weber, R. Dehn, N. Schläger, B. Dieter and A. Kirschning, Org. Lett., 2014, 16, 568–571 CrossRef CAS PubMed.
- L.-L. Wang and A. Kirschning, Beilstein J. Org. Chem., 2017, 13, 1280–1287 CrossRef CAS PubMed.
- L. L. Wang, D. Candito, G. Dräger, J. Herrmann, R. Müller and A. Kirschning, Chem. - Eur. J., 2017, 23, 5291–5298 CrossRef CAS PubMed.
- L.-L. Wang, D. Candito, G. Dräger and A. Kirschning, Eur. J. Org. Chem., 2017, 2017, 5582–5591 CrossRef CAS.
- Yamanouchi Pharmaceutical Co. Ltd, JP06340651, 1994.
- K. Kamigiri, T. Tokunaga, T. Sugawara, K. Nagai, M. Shibazaki, B. Setiawan, R. M. Rantiatmodjo, M. Morioka and K. Suzuki, J. Antibiot., 1997, 50, 556–561 CrossRef CAS PubMed.
- N.-G. Jung and B.-T. Kim, J. Korean Chem. Soc., 2013, 57, 416–419 CrossRef CAS.
- T. Dang and R. D. Süssmuth, Acc. Chem. Res., 2017, 50, 1566–1576 CrossRef CAS PubMed.
- L. Taevernier, E. Wynendaele, B. Gevaert and B. de Spiegeleer, Curr. Protein Pept. Sci., 2017, 18, 425–452 CrossRef CAS PubMed.
- S. Sivanathan and J. Scherkenbeck, Molecules, 2014, 19, 12368–12420 CrossRef PubMed.
- D. A. Alonzo and T. M. Schmeing, Protein Sci., 2020, 29, 2316–2347 CrossRef CAS PubMed.
- N. Ziemert, K. Ishida, A. Liaimer, C. Hertweck and E. Dittmann, Angewandte Chemie (International ed. in English), 2008, 47, 7756–7759 CrossRef CAS PubMed.
- N. Mutsuo, H. Yuki, A. Osamu, K. Nahojiyu, K. Shiho and F. Daisuke, JP2005200324(A), 2005.
- S. Kozuma, Y. Hirota-Takahata, D. Fukuda, N. Kuraya, M. Nakajima and O. Ando, J. Antibiot., 2014, 67, 237–242 CrossRef CAS PubMed.
- Y. Hirota-Takahata, S. Kozuma, N. Kuraya, D. Fukuda, M. Nakajima and O. Ando, J. Antibiot., 2014, 67, 243–251 CrossRef CAS PubMed.
- A. Broberg, C. Nord, J. J. Levenfors, J. Bjerketorp, B. Guss and B. Öberg, Amino Acids, 2021, 53, 323–331 CrossRef CAS PubMed.
- J. Bjerketorp, J. J. Levenfors, C. Nord, B. Guss, B. Öberg and A. Broberg, Front. Microbiol., 2021, 12, 642829 CrossRef PubMed.
- B. Öberg, A. Broberg, B. Guss, J. Levenfors, J. Bjerketorp and C. Nord, WO2020/046190A1, 2020.
- C. Nord, J. Bjerketorp, J. J. Levenfors, S. Cao, A. A. Strömstedt, B. Guss, R. Larsson, D. Hughes, B. Öberg and A. Broberg, ACS Chem. Biol., 2020, 15, 2937–2944 CrossRef CAS PubMed.
- M. J. Trimble, P. Mlynárčik, M. Kolář and R. E. W. Hancock, Cold Spring Harbor Perspect. Med., 2016, 6, a025288 CrossRef PubMed.
- C. Covas, B. Almeida, A. C. Esteves, J. Lourenço, P. Domingues, T. Caetano and S. Mendo, New Biotechnol., 2021, 60, 62–71 CrossRef CAS PubMed.
- J. Shoji, H. Hinoo, K. Matsumoto, T. Hattori, T. Yoshida, S. Matsuura and E. Kondo, J. Antibiot., 1988, 41, 713–718 CrossRef CAS PubMed.
- T. Kato, H. Hinoo, Y. Terui, J. Kikuchi and J. Shoji, J. Antibiot., 1988, 41, 719–725 CrossRef CAS PubMed.
- J. O'Sullivan, J. E. McCullough, A. A. Tymiak, D. R. Kirsch, W. H. Trejo and P. A. Principe, J. Antibiot., 1988, 41, 1740–1744 CrossRef PubMed.
- D. P. Bonner, J. O'Sullivan, S. K. Tanaka, J. M. Clark and R. R. Whitney, J. Antibiot., 1988, 41, 1745–1751 CrossRef CAS.
- A. A. Tymiak, T. J. McCormick and S. E. Unger, J. Org. Chem., 1989, 54, 1149–1157 CrossRef CAS.
- H. Maki, K. Miura and Y. Yamano, Antimicrob. Agents Chemother., 2001, 45, 1823–1827 CrossRef CAS PubMed.
- W. Lee, K. Schaefer, Y. Qiao, V. Srisuknimit, H. Steinmetz, R. Müller, D. Kahne and S. Walker, J. Am. Chem. Soc., 2016, 138, 100–103 CrossRef CAS PubMed.
- S. Anlauf, M.-A. Bruening, N. A. Brunner, R. Endermann, C. Fuerstner, E. Hartmann, J. Koebberling, J. Ragot, G. Schiffer, J. Schuhmacher, N. Svenstrup, J. Telser and F. von Nussbaum, WO2004099239A1, 2004.
- F. Bernhard, G. Demel, K. Soltani, H. V. Döhren and V. Blinov, DNA Sequence, 1996, 6, 319–330 CrossRef CAS PubMed.
- J. Hou, L. Robbel and M. A. Marahiel, Chem. Biol., 2011, 18, 655–664 CrossRef CAS PubMed.
- (a) F. von Nussbaum, S. Anlauf, J. Benet-Buchholz, D. Häbich, J. Köbberling, L. Musza, J. Telser, H. Rübsamen-Waigmann and N. A. Brunner, Angewandte Chemie (International ed. in English), 2007, 46, 2039–2042 CrossRef CAS PubMed; (b) A. Guzman-Martinez, R. Lamer and M. S. VanNieuwenhze, J. Am. Chem. Soc., 2007, 129, 6017–6021 CrossRef CAS PubMed.
- (a) B. J. Egner and M. Bradley, Tetrahedron, 1997, 53, 14021–14030 CrossRef CAS; (b) E. A. Hall, E. Kuru and M. S. VanNieuwenhze, Org. Lett., 2012, 14, 2730–2733 CrossRef CAS PubMed.
- K. Yasumuro, Y. Suzuki, M. Shibazaki, K. Teramura, K. Abe and M. Orita, J. Antibiot., 1995, 48, 1425–1429 CrossRef CAS PubMed.
- M. Orita, K. Yasumuro, K. Kokubo, M. Shimizu, K. Abe, T. Tokunaga and H. Kaniwa, J. Antibiot., 1995, 48, 1430–1434 CrossRef CAS PubMed.
- H. H. Wasserman, J.-H. Chen and M. Xia, J. Am. Chem. Soc., 1999, 121, 1401–1402 CrossRef CAS.
- K. Teramura, K. Yasumuro and K. Abe, J. Enzym. Inhib., 1996, 11, 33–38 CrossRef CAS PubMed.
- V. Apostolopoulos, J. Bojarska, T.-T. Chai, S. Elnagdy, K. Kaczmarek, J. Matsoukas, R. New, K. Parang, O. P. Lopez, H. Parhiz, C. O. Perera, M. Pickholz, M. Remko, M. Saviano, M. Skwarczynski, Y. Tang, W. M. Wolf, T. Yoshiya, J. Zabrocki, P. Zielenkiewicz, M. AlKhazindar, V. Barriga, K. Kelaidonis, E. M. Sarasia and I. Toth, Molecules, 2021, 26, 430 CrossRef CAS PubMed.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed.
- Y. A. Haggag, BJSTR, 2018, 8, 6659–6662 Search PubMed.
- H. Ono, Y. Funabashi, S. Harada, European Pat., EP0339596, 1989 Search PubMed.
- N. Katayama, S. Fukusumi, Y. Funabashi, T. Iwahi and H. Ono, J. Antibiot., 1993, 46, 606–613 CrossRef CAS PubMed.
- Y. Funabashi, S. Tsubotani, K. Koyama, N. Katayama and S. Harada, Tetrahedron, 1993, 49, 13–28 CrossRef CAS.
- (a) C. Yuan and R. M. Williams, J. Am. Chem. Soc., 1997, 119, 11777–11784 CrossRef CAS; (b) R. M. Williams, C. Yuan, V. J. Lee and S. Chamberland, J. Antibiot., 1998, 51, 189–201 CrossRef CAS PubMed; (c) V. V. Sokolov, S. I. Kozhushkov, S. Nikolskaya, V. N. Belov, M. Es-Sayed and A. de Meijere, Eur. J. Org. Chem., 1998, 1998, 777–783 CrossRef; (d) P. Lin and A. Ganesan, Synthesis, 2000, 2000, 2127–2130 CrossRef; (e) N. Aguilar and J. Krüger, Molecules, 2002, 7, 469–474 CrossRef CAS; (f) M. Brands, R. Endermann, R. Gahlmann, J. Krüger and S. Raddatz, Bioorg. Med. Chem. Lett., 2003, 13, 241–245 CrossRef CAS PubMed; (g) M. Brands, Y. C. Grande, R. Endermann, R. Gahlmann, J. Krüger and S. Raddatz, Bioorg. Med. Chem. Lett., 2003, 13, 2641–2645 CrossRef CAS PubMed; (h) V. N. Belov, V. V. Sokolov, B. D. Zlatopolskiy and A. de Meijere, Eur. J. Org. Chem., 2011, 2011, 4093–4097 CrossRef CAS.
- M. Brands, R. Endermann, R. Gahlmann, J. Krüger, S. Raddatz, J. Stoltefuss, V. N. Belov, S. Nizamov, V. V. Sokolov and A. de Meijere, J. Med. Chem., 2002, 45, 4246–4253 CrossRef CAS PubMed.
- N. Böddeker, G. Bahador, C. Gibbs, E. Mabery, J. Wolf, L. Xu and J. Watson, RNA, 2002, 8, 1120–1128 CrossRef.
- W. S. Champney, J. Pelt and C. L. Tober, Curr. Microbiol., 2001, 43, 340–345 CrossRef CAS PubMed.
- E. Limburg, R. Gahlmann, H.-P. Kroll and D. Beyer, Antimicrob. Agents Chemother., 2004, 48, 619–622 CrossRef CAS PubMed.
- B. Somanadhan, S. R. Kotturi, C. Yan Leong, R. P. Glover, Y. Huang, H. Flotow, A. D. Buss, M. J. Lear and M. S. Butler, J. Antibiot., 2013, 66, 259–264 CrossRef CAS PubMed.
- S. R. Kotturi, B. Somanadhan, J.-H. Chng, K. S.-W. Tan, M. S. Butler and M. J. Lear, Tetrahedron Lett., 2014, 55, 1949–1951 CrossRef CAS.
- S. Brinkmann, S. Semmler, C. Kersten, M. A. Patras, M. Kurz, N. Fuchs, S. J. Hammerschmidt, J. Legac, P. E. Hammann, A. Vilcinskas, P. J. Rosenthal, T. Schirmeister, A. Bauer and T. F. Schäberle, ACS Chem. Biol., 2022 DOI:10.1021/acschembio.1c00861.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- T. Caetano, W. van der Donk and S. Mendo, Microbiol. Res., 2020, 235, 126441 CrossRef CAS PubMed.
- J. Delves-Broughton, P. Blackburn, R. J. Evans and J. Hugenholtz, Antonie Leeuwenhoek, 1996, 69, 193–202 CrossRef CAS PubMed.
- K. I. Mohr, C. Volz, R. Jansen, V. Wray, J. Hoffmann, S. Bernecker, J. Wink, K. Gerth, M. Stadler and R. Müller, Angewandte Chemie (International ed. in English), 2015, 54, 11254–11258 CrossRef CAS PubMed.
- I. R. Bothwell, T. Caetano, R. Sarksian, S. Mendo and W. A. van der Donk, ACS Chem. Biol., 2021, 16, 1019–1029 CrossRef CAS PubMed.
- T. Fujita, H. Hatanaka, K. Hayashi, N. Shigematsu, S. Takase, M. Okamoto, M. Okuhara, K. Shimatani and A. Satoh, J. Antibiot., 1994, 47, 1359–1364 CrossRef CAS PubMed.
- H. Hatanaka, S. Takase, T. Fujita, M. Okamoto and M. Okuhara, WO9302203A1, 1992.
- T. Kinoshita, T. Kitatani, M. Warizaya and T. Tada, Acta Crystallogr. F, 2005, 61, 808–811 CrossRef CAS PubMed.
- T. Fujita, Y. Shinguh, A. Yamazaki, K. Nakahara, M. Okamoto and M. Okuhara, J. Antibiot., 1994, 47, 1365–1368 CrossRef CAS PubMed.
- A. Miyanaga, F. Kudo and T. Eguchi, Nat. Prod. Rep., 2018, 35, 1185–1209 RSC.
- Y. J. Yoo, H. Kim, S. R. Park and Y. J. Yoon, J. Ind. Microbiol. Biotechnol., 2017, 44, 537–553 CrossRef CAS PubMed.
- N. Oku, K. Adachi, S. Matsuda, H. Kasai, A. Takatsuki and Y. Shizuri, Org. Lett., 2008, 10, 2481–2484 CrossRef CAS PubMed.
- L. Linares-Otoya, V. Linares-Otoya, L. Armas-Mantilla, C. Blanco-Olano, M. Crüsemann, M. L. Ganoza-Yupanqui, J. Campos-Florian, G. M. König and T. F. Schäberle, Mar. Drugs, 2017, 15, 308 CrossRef PubMed.
- K. H. van Pée and J. M. Ligon, Nat. Prod. Rep., 2000, 17, 157–164 RSC.
- J. R. Evans, E. J. Napier and R. A. Fletton, J. Antibiot., 1978, 31, 952–958 CrossRef CAS PubMed.
- (a) P. Srisukchayakul, C. Suwanachart, Y. Sangnoi, A. Kanjana-opas, S. Hosoya, A. Yokota and V. Arunpairojana, Int. J. Syst. Evol. Microbiol., 2007, 57, 2275–2279 CrossRef CAS PubMed; (b) A. Kanjana-opas, S. Panphon, H.-K. Fun and S. Chantrapromma, Acta Crystallogr. E, 2006, 62, o2728–o2730 CrossRef CAS.
- Y. Sangnoi, O. Sakulkeo, S. Yuenyongsawad, A. Kanjana-opas, K. Ingkaninan, A. Plubrukarn and K. Suwanborirux, Mar. Drugs, 2008, 6, 578–586 CrossRef CAS PubMed.
- P. W. Okanya, K. I. Mohr, K. Gerth, R. Jansen and R. Müller, J. Nat. Prod., 2011, 74, 603–608 CrossRef CAS PubMed.
- E. J. Choi, S.-J. Nam, L. Paul, D. Beatty, C. A. Kauffman, P. R. Jensen and W. Fenical, Chem. Biol., 2015, 22, 1270–1279 CrossRef CAS PubMed.
- L. Linares-Otoya, Y. Liu, V. Linares-Otoya, L. Armas-Mantilla, M. Crüsemann, M. L. Ganoza-Yupanqui, J. Campos-Florian, G. M. König and T. F. Schäberle, ACS Chem. Biol., 2019, 14, 176–181 CrossRef CAS PubMed.
- Y. Sangnoi, O. Sakulkeo, S. Yuenyongsawad, A. Kanjana-opas, K. Ingkaninan, A. Plubrukarn and K. Suwanborirux, Mar. Drugs, 2008, 6, 578–586 CrossRef CAS PubMed.
- (a) X. Ma, Y. Vo, M. G. Banwell and A. C. Willis, Asian J. Org. Chem., 2012, 1, 160–165 CrossRef CAS; (b) J. P. Mahajan, Y. R. Suryawanshi and S. B. Mhaske, Org. Lett., 2012, 14, 5804–5807 CrossRef CAS PubMed; (c) L. Ni, Z. Li, F. Wu, J. Xu, X. Wu, L. Kong and H. Yao, Tetrahedron Lett., 2012, 53, 1271–1274 CrossRef CAS; (d) B. Patel and S. Hilton, Synlett, 2014, 26, 79–83 CrossRef; (e) B. Bolte, C. S. Bryan, P. P. Sharp, S. Sayyahi, C. Rihouey, A. Kendrick, P. Lan, M. G. Banwell, C. J. Jackson, N. J. Fraser, A. C. Willis and J. S. Ward, J. Org. Chem., 2020, 85, 650–663 CrossRef CAS PubMed.
- M. Osano, D. P. Jhaveri and P. Wipf, Org. Lett., 2020, 22, 2215–2219 CrossRef CAS PubMed.
- C. S. Schwalm and C. R. D. Correia, Tetrahedron Lett., 2012, 53, 4836–4840 CrossRef CAS.
- Y. Yamaoka, T. Yoshida, M. Shinozaki, K.-i. Yamada and K. Takasu, J. Org. Chem., 2015, 80, 957–964 CrossRef CAS PubMed.
- F. Khan, M. Dlugosch, X. Liu and M. G. Banwell, Acc. Chem. Res., 2018, 51, 1784–1795 CrossRef CAS PubMed.
- W. L. Parker, J. O'Sullivan and R. B. Sykes, in Advances in Applied Microbiology, Elsevier, 1986, vol. 31, pp. 181–205 Search PubMed.
- P. D. Singh, J. H. Johnson, P. C. Ward, J. S. Wells, W. H. Trejo and R. B. Sykes, J. Antibiot., 1983, 36, 1245–1251 CrossRef CAS PubMed.
- R. Cooper, K. Bush, P. A. Principe, W. H. Trejo, J. S. Wells and R. B. Sykes, J. Antibiot., 1983, 36, 1252–1257 CrossRef CAS PubMed.
- T. Kato, H. Hinoo, J. Shoji, K. Matsumoto, T. Tanimoto, T. Hattori, K. Hirooka and E. Kondo, J. Antibiot., 1987, 40, 135–138 CrossRef CAS PubMed.
- T. Kato, H. Hinoo, Y. Terui, J. Nishikawa, Y. Nakagawa, Y. Ikenishi and J. Shoji, J. Antibiot., 1987, 40, 139–144 CrossRef CAS PubMed.
- N. Katayama, Y. Nozaki, K. Okonogi, H. Ono, S. Harada and H. Okazaki, J. Antibiot., 1985, 38, 1117–1127 CrossRef CAS PubMed.
- T. Hida, S. Tsubotani, N. Katayama, H. Okazaki and S. Harada, J. Antibiot., 1985, 38, 1128–1140 CrossRef CAS PubMed.
- C. E. Higgens, R. L. Hamill, T. H. Sands, M. M. Hoehn and N. E. Davis, J. Antibiot., 1974, 27, 298–300 CrossRef CAS PubMed.
- P. D. Singh, P. C. Ward, J. S. Wells, C. M. Ricca, W. H. Trejo, P. A. Principe and R. B. Sykes, J. Antibiot., 1982, 35, 1397–1399 CrossRef CAS PubMed.
- P. D. Singh, M. G. Young, J. H. Johnson, C. M. Cimarusti and R. B. Sykes, J. Antibiot., 1984, 37, 773–780 CrossRef CAS PubMed.
- J. Shoji, T. Kato, R. Sakazaki, W. Nagata, Y. Terui, Y. Nakagawa, M. Shiro, K. Matsumoto, T. Hattori and T. Yoshida, J. Antibiot., 1984, 37, 1486–1490 CrossRef CAS PubMed.
- J. Shoji, R. Sakazaki, T. Kato, Y. Terui, K. Matsumoto, T. Tanimoto, T. Hattori, K. Hirooka and E. Kondo, J. Antibiot., 1985, 38, 538–540 CrossRef CAS PubMed.
- C. A. Aruldass, L. Dufossé and W. A. Ahmad, J. Clean. Prod., 2018, 180, 168–182 CrossRef CAS.
- (a) H. Achenbach, Arch. Microbiol., 1974, 101, 131–144 CrossRef CAS PubMed; (b) H. Achenbach, W. Kohl, H. Reichenbach and H. Kleinig, Tetrahedron Lett., 1974, 15, 2555–2556 CrossRef.
- H. Achenbach and J. Witzke, Angew. Chem., Int. Ed. Engl., 1977, 16, 191–192 CrossRef.
- (a) H. Achenbach, A. Böttger, W. Kohl, E. Fautz and H. Reichenbach, Phytochemistry, 1979, 18, 961–963 CrossRef CAS; (b) E. Fautz and H. Reichenbach, Phytochemistry, 1979, 18, 957–959 CrossRef CAS; (c) H. Achenbach, A. Bttger-Vetter, E. Fautz and H. Reichenbach, Arch. Microbiol., 1982, 132, 241–244 CrossRef CAS; (d) M. J. McBride, G. Xie, E. C. Martens, A. Lapidus, B. Henrissat, R. G. Rhodes, E. Goltsman, W. Wang, J. Xu, D. W. Hunnicutt, A. M. Staroscik, T. R. Hoover, Y.-Q. Cheng and J. L. Stein, Appl. Environ. Microbiol., 2009, 75, 6864–6875 CrossRef CAS PubMed; (e) S. W. Fuchs, K. A. J. Bozhüyük, D. Kresovic, F. Grundmann, V. Dill, A. O. Brachmann, N. R. Waterfield and H. B. Bode, Angew. Chem., Int. Ed. Engl., 2013, 52, 4108–4112 CrossRef CAS PubMed; (f) T. A. Schöner, S. W. Fuchs, C. Schönau and H. B. Bode, J. Microb. Biotechnol., 2014, 7, 232–241 CrossRef PubMed.
- (a) M. E. P. Jiménez, C. M. B. Pinilla, E. Rodrigues and A. Brandelli, Nat. Prod. Res., 2019, 33, 1541–1549 CrossRef PubMed; (b) A. Mogadem, M. A. Almamary, N. A. Mahat, K. Jemon, W. A. Ahmad and I. Ali, Molecules, 2021, 26 Search PubMed.
- (a) D. L. Fox and R. A. Lewin, Can. J. Microbiol., 1963, 9, 753–768 CrossRef CAS; (b) A. J. Aasen and S. L. Jensen, Acta Chem. Scand., 1966, 20, 811–819 CrossRef CAS PubMed; (c) A. J. Aasen and S. L. Jensen, Acta Chem. Scand., 1966, 20, 1970–1988 CrossRef CAS PubMed; (d) A. J. Aasen and S. L. Jensen, Acta Chem. Scand., 1966, 20, 2322–2324 CrossRef CAS PubMed; (e) A. J. Aasen, S. Liaaen-Jensen, G. Borch, A. F. Andresen, W. B. Pearson and V. Meisalo, Acta Chem. Scand., 1972, 26, 404–405 CrossRef CAS; (f) K. Shindo, K. Kikuta, A. Suzuki, A. Katsuta, H. Kasai, M. Yasumoto-Hirose, Y. Matsuo, N. Misawa and S. Takaichi, Appl. Microbiol. Biotechnol., 2007, 74, 1350–1357 CrossRef CAS PubMed; (g) K. Shindo and N. Misawa, Mar. Drugs, 2014, 12, 1690–1698 CrossRef PubMed.
- L. Tao, H. Yao, H. Kasai, N. Misawa and Q. Cheng, Mol. Genet. Genomics, 2006, 276, 79–86 CrossRef CAS PubMed.
- J.-H. Jang, K. Kanoh, K. Adachi, S. Matsuda and Y. Shizuri, J. Nat. Prod., 2007, 70, 563–566 CrossRef CAS PubMed.
- M. J. Fujita, K. Nakano and R. Sakai, Molecules, 2013, 18, 3917–3926 CrossRef CAS PubMed.
- Z.-J. Wang, H. Zhou, G. Zhong, L. Huo, Y.-J. Tang, Y. Zhang and X. Bian, Org. Lett., 2020, 22, 939–943 CrossRef CAS PubMed.
- M. J. Fujita, Y. Goto and R. Sakai, Mar. Drugs, 2018, 16, 342 CrossRef PubMed.
- (a) T. Kamiyama, T. Umino, Y. Itezono, Y. Nakamura, T. Satoh and K. Yokose, J. Antibiot., 1995, 48, 929–936 CrossRef CAS PubMed; (b) T. Kamiyama, T. Umino, T. Satoh, S. Sawairi, M. Shirane, S. Ohshima and K. Yokose, J. Antibiot., 1995, 48, 924–928 CrossRef CAS PubMed.
- J. Maeda, M. Nishida, H. Takikawa, H. Yoshida, T. Azuma, M. Yoshida and Y. Mizushina, Int. J. Mol. Med., 2010, 26, 751–758 CAS.
- P. N. Chaudhari, K. S. Wani, B. L. Chaudhari and S. B. Chincholkar, Appl. Biochem. Biotechnol., 2009, 158, 231–241 CrossRef CAS PubMed.
- J. 'i. Kohayashi, S. Mikami, H. Shigemori, T. Takao, Y. Shimonishi, S. Izuta and S. Yoshida, Tetrahedron, 1995, 51, 10487–10490 CrossRef.
- (a) W. Godchaux and E. R. Leadbetter, J. Bacteriol., 1980, 144, 592–602 CrossRef CAS PubMed; (b) D. R. Abbanat, W. Godchaux and E. R. Leadbetter, Arch. Microbiol., 1988, 149, 358–364 CrossRef CAS.
- (a) D. R. Abbanat, E. R. Leadbetter, W. Godchaux and A. Escher, Nature, 1986, 324, 367–369 CrossRef CAS; (b) W. Godchaux and E. R. Leadbetter, J. Bacteriol., 1983, 153, 1238–1246 CrossRef CAS PubMed; (c) W. Godchaux and E. R. Leadbetter, J. Biol. Chem., 1984, 259, 2982–2990 CrossRef CAS PubMed.
- (a) R. A. Alegado, L. W. Brown, S. Cao, R. K. Dermenjian, R. Zuzow, S. R. Fairclough, J. Clardy and N. King, Elife, 2012, 1, e00013 CrossRef PubMed; (b) A. M. Cantley, A. Woznica, C. Beemelmanns, N. King and J. Clardy, J. Am. Chem. Soc., 2016, 138, 4326–4329 CrossRef CAS PubMed; (c) A. Woznica, A. M. Cantley, C. Beemelmanns, E. Freinkman, J. Clardy and N. King, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7894–7899 CrossRef CAS PubMed; (d) C. Beemelmanns, A. Woznica, R. A. Alegado, A. M. Cantley, N. King and J. Clardy, J. Am. Chem. Soc., 2014, 136, 10210–10213 CrossRef CAS PubMed.
- (a) N. Irako and T. Shioiri, Tetrahedron Lett., 1998, 39, 5793–5796 CrossRef CAS; (b) N. Irako and T. Shioiri, Tetrahedron Lett., 1998, 39, 5797–5798 CrossRef CAS; (c) H. Takikawa, S.-e. Muto, D. Nozawa, A. Kayo and K. Mori, Tetrahedron Lett., 1998, 39, 6931–6934 CrossRef CAS; (d) H. Takikawa, D. Nozawa, A. Kayo, S.-e. Muto and K. Mori, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 2467–2477 RSC; (e) T. Shioiri and N. Irako, Tetrahedron, 2000, 56, 9129–9142 CrossRef CAS; (f) O. Labeeuw, P. Phansavath and J.-P. Genêt, Tetrahedron Lett., 2003, 44, 6383–6386 CrossRef CAS; (g) O. Labeeuw, P. Phansavath and J.-P. Genêt, Tetrahedron: Asymmetry, 2004, 15, 1899–1908 CrossRef CAS; (h) P. Gupta, S. V. Naidu and P. Kumar, Tetrahedron Lett., 2004, 45, 9641–9643 CrossRef CAS; (i) A. Sharma, S. Gamre and S. Chattopadhyay, Tetrahedron Lett., 2007, 48, 3705–3707 CrossRef CAS.
- (a) D. R. Abbanat, W. Godchaux, G. Polychroniou and E. R. Leadbetter, Biochem. Biophys. Res. Commun., 1985, 130, 873–878 CrossRef CAS PubMed; (b) R. H. White, J. Bacteriol., 1984, 159, 42–46 CrossRef CAS PubMed; (c) M. Á. Vences-Guzmán, R. Peña-Miller, N. A. Hidalgo-Aguilar, M. L. Vences-Guzmán, Z. Guan and C. Sohlenkamp, Environ. Microbiol., 2021, 23, 2448–2460 CrossRef PubMed.
- (a) I. Uchida, K. Yoshida, Y. Kawai, S. Takase, Y. Itoh, H. Tanaka, M. Kohsaka and H. Imanaka, J. Antibiot., 1985, 38, 1476–1486 CrossRef CAS PubMed; (b) K. Yoshida, M. Iwami, Y. Umehara, M. Nishikawa, I. Uchida, M. Kohsaka, H. Aoki and H. Imanaka, J. Antibiot., 1985, 38, 1469–1475 CrossRef CAS PubMed.
- K. Suzuki, H. Yamaguchi, S. Miyazaki, K. Nagai, S. Watanabe, T. Saito, K. Ishii, M. Hanada, T. Sekine and Y. Ikegami, J. Antibiot., 1990, 43, 154–157 CrossRef CAS PubMed.
- Y. Ikegami, N. Takeuchi, M. Hanada, Y. Hasegawa, K. Ishii, T. Andoh, T. Sato, K. Suzuki, H. Yamaguchi and S. Miyazaki, J. Antibiot., 1990, 43, 158–162 CrossRef CAS PubMed.
- (a) T. Nemoto, M. Ojika, Y. Takahata, T. Andoh and Y. Sakagami, Tetrahedron, 1998, 54, 2683–2690 CrossRef CAS; (b) T. Shioiri, Y. Terao, N. Irako and T. Aoyama, Tetrahedron, 1998, 54, 15701–15710 CrossRef CAS.
- M. Shiozaki, N. Deguchi, T. Mochizuki, T. Wakabayashi, T. Ishikawa, H. Haruyama, Y. Kawai and M. Nishijima, Tetrahedron, 1998, 54, 11861–11876 CrossRef CAS.
- Y. Kawai, I. Yano and K. Kaneda, Eur. J. Biochem., 1988, 171, 73–80 CrossRef CAS PubMed.
- R. B. Clark, J. L. Cervantes, M. W. Maciejewski, V. Farrokhi, R. Nemati, X. Yao, E. Anstadt, M. Fujiwara, K. T. Wright, C. Riddle, C. J. La Vake, J. C. Salazar, S. Finegold and F. C. Nichols, Infect. Immun., 2013, 81, 3479–3489 CrossRef CAS PubMed.
- Y.-H. Wang, R. Nemati, E. Anstadt, Y. Liu, Y. Son, Q. Zhu, X. Yao, R. B. Clark, D. W. Rowe and F. C. Nichols, Bone, 2015, 81, 654–661 CrossRef CAS PubMed.
- R. Nemati, C. Dietz, E. J. Anstadt, J. Cervantes, Y. Liu, F. E. Dewhirst, R. B. Clark, S. Finegold, J. J. Gallagher, M. B. Smith, X. Yao and F. C. Nichols, J. Lipid Res., 2017, 58, 1999–2007 CrossRef CAS PubMed.
- Y. Kawai and K. Akagawa, Infect. Immun., 1989, 57, 2086–2091 CrossRef CAS PubMed.
- O. Teng, C. K. E. Ang and X. L. Guan, Front. Immunol., 2017, 8, 1836 CrossRef PubMed.
- I. Olsen and F. C. Nichols, Infect. Immun., 2018, 86, e00035-18 CrossRef PubMed.
- T. Morishita, A. Sato, M. Hisamoto, T. Oda, K. Matsuda, A. Ishii and K. Kodama, J. Antibiot., 1997, 50, 457–468 CrossRef CAS PubMed.
- (a) C. S. Mirucki, M. Abedi, J. Jiang, Q. Zhu, Y.-H. Wang, K. E. Safavi, R. B. Clark and F. C. Nichols, J. Endod., 2014, 40, 1342–1348 CrossRef PubMed; (b) F. C. Nichols, W. J. Housley, C. A. O'Conor, T. Manning, S. Wu and R. B. Clark, Am. J. Surg. Pathol., 2009, 175, 2430–2438 CrossRef CAS PubMed; (c) K. Gomi, K. Kawasaki, Y. Kawai, M. Shiozaki and M. Nishijima, J. Immunol., 2002, 168, 2939–2943 CrossRef CAS PubMed.
- V. Farrokhi, R. Nemati, F. C. Nichols, X. Yao, E. Anstadt, M. Fujiwara, J. Grady, D. Wakefield, W. Castro, J. Donaldson and R. B. Clark, Clin. Transl. Immunol., 2013, 2, e8 CrossRef CAS PubMed.
- (a) Y. K.-H. Schneider, K. Ø Hansen, J. Isaksson, S. Ullsten, E. H Hansen and J. Hammer Andersen, Molecules, 2019, 24, 3991 CrossRef CAS PubMed; (b) M.-K. Bill, S. Brinkmann, M. Oberpaul, M. A. Patras, B. Leis, M. Marner, M.-P. Maitre, P. E. Hammann, A. Vilcinskas, S. M. M. Schuler and T. F. Schäberle, Molecules, 2021, 26, 5195 CrossRef CAS PubMed.
- M. Shaaban, R. P. Maskey, I. Wagner-Döbler and H. Laatsch, J. Nat. Prod., 2002, 65, 1660–1663 CrossRef CAS PubMed.
- S. Imai, K. Fujioka, K. Furihata, R. Fudo, S. Yamanaka and H. Seto, J. Antibiot., 1993, 46, 1319–1322 CrossRef CAS PubMed.
- A. Spyere, D. C. Rowley, P. R. Jensen and W. Fenical, J. Nat. Prod., 2003, 66, 818–822 CrossRef CAS PubMed.
- (a) Y. Matsuo, H. Imagawa, M. Nishizawa and Y. Shizuri, Science, 2005, 307, 1598 CrossRef CAS PubMed; (b) H. Yamamoto, Y. Takagi, N. Yamasaki, T. Mitsuyama, Y. Kasai, H. Imagawa, Y. Kinoshita, N. Oka and M. Hiraoka, Tetrahedron, 2018, 74, 7173–7178 CrossRef CAS.
- (a) K. M. Papp-Wallace and R. A. Bonomo, Infect. Dis. Clin., 2016, 30, 441–464 CrossRef PubMed; (b) S. Andrei, G. Droc and G. Stefan, Discoveries, 2019, 7, e102 CrossRef PubMed.
- K. Katoh and D. M. Standley, Mol. Biol. Evol., 2013, 30, 772–780 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |