
Biosynthesis of fungal polyketides by collaborating and trans-acting enzymes
Elizabeth
Skellam
 *
*
Department of Chemistry, BioDiscovery Institute, University of North Texas, 1155 Union Circle, Denton, TX 76203, USA. E-mail: Elizabeth.skellam@unt.edu
First published on 24th November 2021
Abstract
Covering: 1999 up to 2021
Fungal polyketides encompass a range of structurally diverse molecules with a wide variety of biological activities. The giant multifunctional enzymes that synthesize polyketide backbones remain enigmatic, as do many of the tailoring enzymes involved in functional modifications. Recent advances in elucidating biosynthetic gene clusters (BGCs) have revealed numerous examples of fungal polyketide synthases that require the action of collaborating enzymes to synthesize the carbon backbone. This review will discuss collaborating and trans-acting enzymes involved in loading, extending, and releasing polyketide intermediates from fungal polyketide synthases, and additional modifications introduced by trans-acting enzymes demonstrating the complexity encountered when investigating natural product biosynthesis in fungi.
 Elizabeth Skellam | Elizabeth Skellam received an MChem degree from Swansea University, a Masters in Business Administration from the University of North Carolina at Wilmington, and a PhD in Chemistry from the University of Bristol. She has worked as a Technical Writer at ID Business Solutions and as an Akademischer Ratin at Leibniz University Hannover. She is currently an Assistant Professor at the University of North Texas and her research focuses on elucidating and engineering biosynthetic pathways in microorganisms to discover new enzymes and generate novel natural products. |
1. Introduction
Fungal polyketides possess a diverse range of biological activities, some of which are beneficial to human health and have been utilized in medicine and agriculture e.g. lovastatin 1, strobilurin A 2, and fumagillin 3. In contrast, other fungal polyketides are devastating mycotoxins such as aflatoxin 4, patulin 5, zearalenone 6, and fumonisin B1 7, that contaminate foodstuffs (Fig. 1).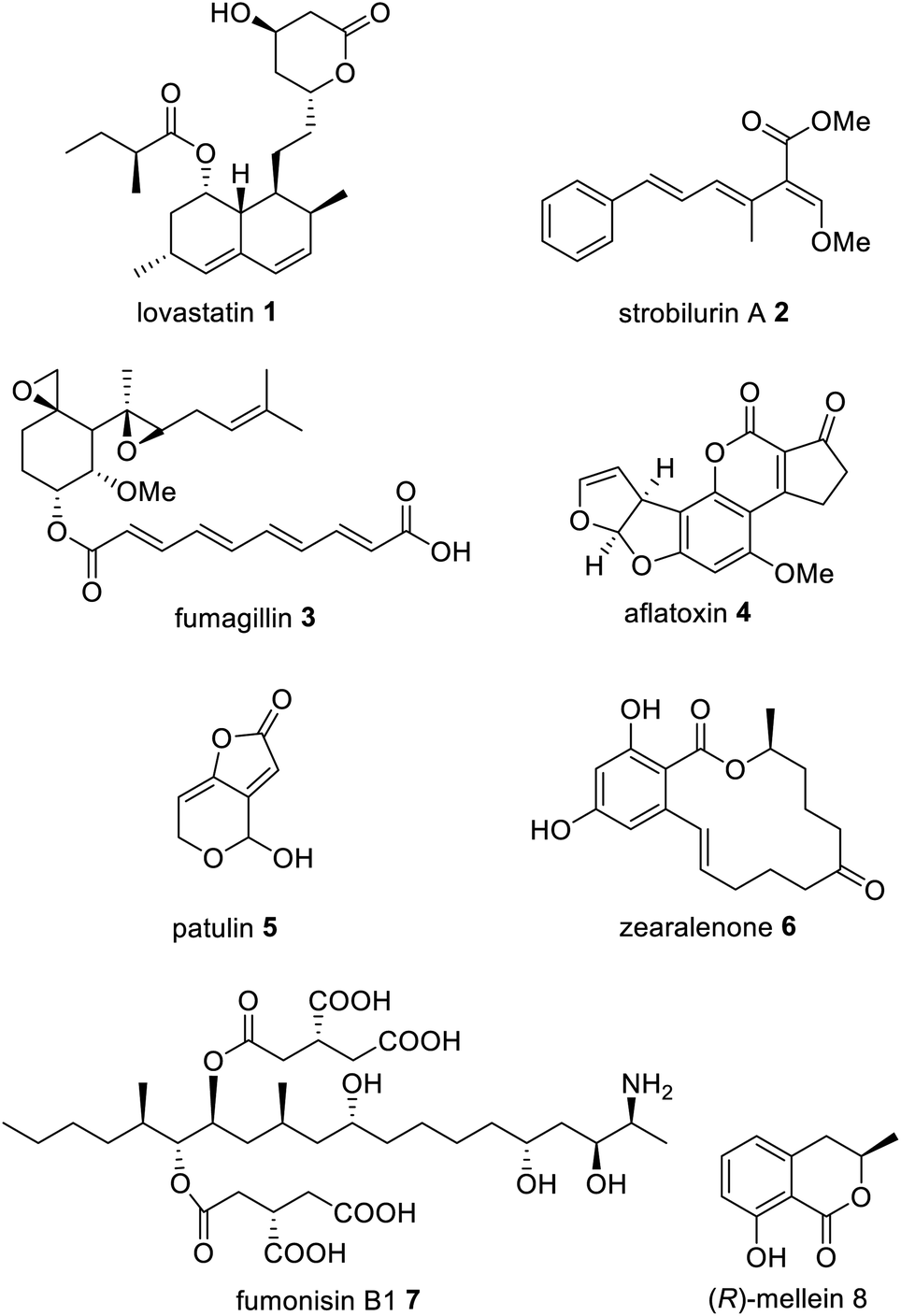 | ||
| Fig. 1 Examples of important fungal polyketide compounds. | ||
In fungi, the majority of polyketides are synthesized by iterative type I multifunctional polyketide synthases (PKS),1–4 although some exceptions exist.5–7 Type I iterative polyketide synthases (PKS) can be subdivided into non-reducing (NR) PKS, partially reducing (PR) PKS and highly reducing (HR) PKS, based on the catalytic domains present and the structure of the resulting polyketide backbone.8,9 Hybrid PKS enzymes are known such as polyketide synthase/non-ribosomal peptide synthases (PKS–NRPS) (Fig. 2).10 Although type III PKS also occur in fungi,11,12 these enzymes are out of scope of this review.
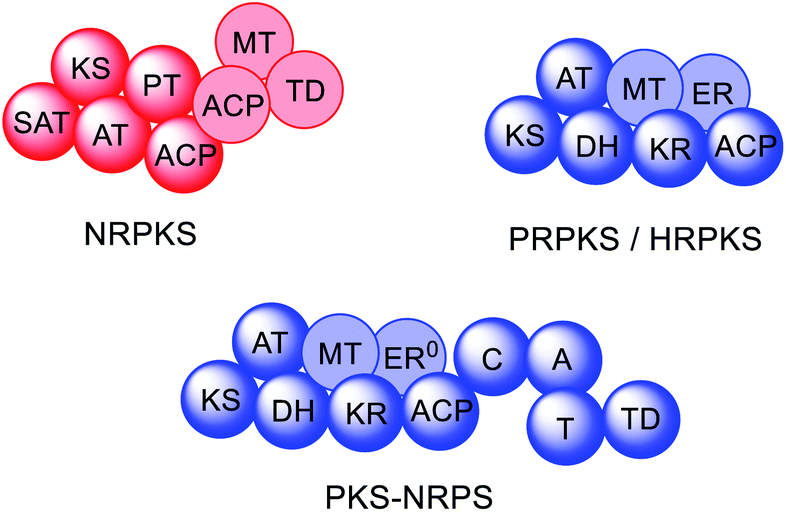 | ||
| Fig. 2 Domain organization of fungal PKS. Faded domains indicate that these domains are not present in every PKS sub-class. ER0 indicates that a domain is present but inactive. Domain abbreviations: SAT = starter unit:ACP-transacylase; KS = ketosynthase; AT = acyltransferase; PT = product template; ACP = acyl carrier protein; MT = C-methyltransferase; TD = terminal domain (involved in release; discussed later); DH = dehydratase (in PRPKS this is a thiolhydrolase (TH) domain13); KR = ketoreductase; ER = enoylreductase; C = condensation; A = adenylation; T = thiolation. | ||
NRPKS contain three catalytic domains that are present in all fungal type I PKS: ketosynthase (KS), acyltransferase (AT) and acyl carrier protein (ACP) domains.14 However, unlike PRPKS and HRPKS they lack the reductive and dehydrative domains active during β-processing, e.g. ketoreductase (KR), enoyl reductase (ER), and dehydratase (DH)/thiolhydrolase (TH)13 domains. Instead, NRPKS contain starter-unit acyl transferase (SAT) and product template (PT) domains.15,16 Additional domains that may be present include: C-methyltransferase (MT) domains, and terminal domains such as thioesterase (TE),17 Claisen cyclase (CLC),18 or reductase (R) domains,19 that are involved in product release rather than β-processing. Aflatoxin 4 and zearalenone 6 are examples of polyketides synthesized by NRPKS.
PRPKS are relatively rare; in addition to KS, AT, and ACP domains they also contain TH and KR domains.13 Only a few PRPKS have been characterized but they typically synthesize 6-methylsalicylic acid,20 the precursor to patulin 5,21 or the common fungal metabolite (R)-mellein 8.22 The products of PRPKS are less reduced than the products of HRPKS due to the presence of only a KR domain, however the enzymes themselves form separate phylogenetic clades from HRPKS and PKS–NRPS.22
HRPKS contain domains required for β-processing e.g. DH, KR, and, usually, ER domains in addition to the standard KS, AT and ACP domains. Often they lack domains involved in product release and consequently are more difficult to study than their NRPKS or PKS–NRPS counterparts. Examples of polyketides synthesized by HRPKS are lovastatin 1, strobilurin A 2, fumagillin 3, and fumonisin B1 7. HRPKS are most similar to fatty acid synthases (FAS) found in mammals in terms of the domains present,14 however the chemistry differs significantly in that the β-processing domains are used in a programmed manner.
Due to the large size of fungal PKS enzymes (>250 kDa),23 and the incredible challenges in obtaining soluble protein for structural studies, there is relatively little structural information available for this class of enzymes. Investigations into the biosynthesis of fungal polyketides, therefore, typically require the use of isotopically labelled precursors and/or intermediates,24 targeted gene inactivation, heterologous expression (omitting one or more genes of interest),25 and enzymology of individual catalytic domains.14–17,26,27 Often, the structures of mFAS28 (Fig. 3) and structurally characterized bacterial PKS domains are used as homologs for fungal PKS enzymes. Only very recently has the structure of a complete fungal PKS been reported.23
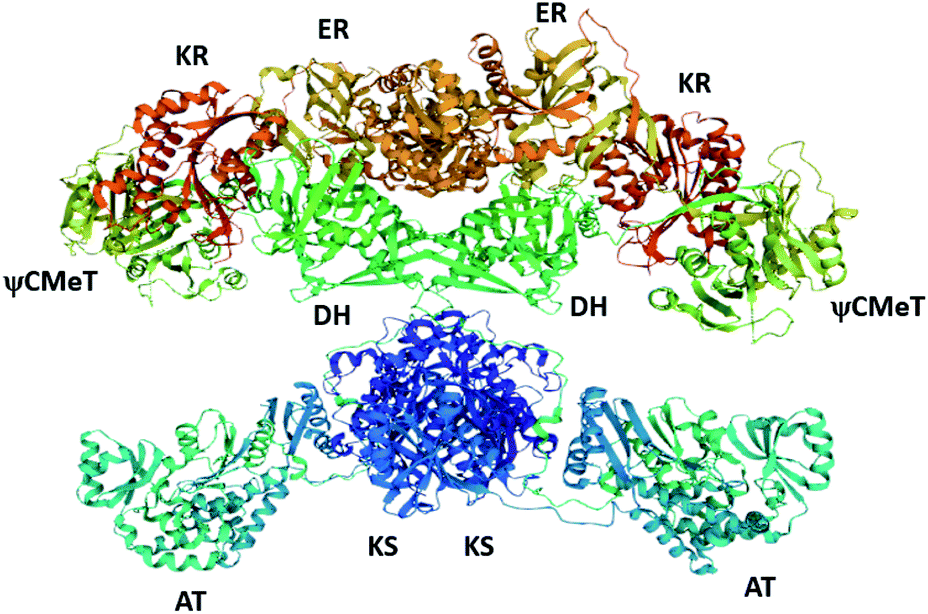
Multifunctional proteins are not unique to secondary metabolism e.g. type I FAS in mammals,29 and the pentafunctional AROM protein required for shikimate production in fungi.30 Multi-domain enzymes are thought to arise from monofunctional enzymes, and be the evolutionary result of smaller ancestral genes encoding the individual enzymes fusing.31,32 The coordinated use of the different catalytic domains by type I PKS is similar to the coordinated use of discrete type II PKS in bacteria, for example, and it is likely that type I PKS arose from fusion of type II PKS genes. An evolutionary analysis of KS domains from PKS and FAS demonstrate that iterative type I PKS are more closely related to modular PKS than fungal FAS.33 Similarly, structural analysis of fungal PT domains, DH domains from FAS, and aromatases/cyclases from bacteria (ARO/CYC) indicate that these enzymes have conserved structural features despite sharing little sequence homology.16,34 These comparisons suggest that the way the individual domains interact with one another may also be conserved.
The recruitment of additional collaborating enzymes is a concept that may be explained by the formation of metabolons,35 where sequential biosynthetic enzymes self-assemble into a multienzyme complex via non-covalent protein–protein interactions.36 These metabolons facilitate efficient substrate transport between the enzymes via channelling and thereby increase the overall flux of the pathway. Metabolons have been reported in primary metabolism, such as the citric acid cycle and fatty acid biosynthesis,37–41 as well as in secondary metabolism in plants.42–45
This review will highlight some of the most well-studied fungal polyketide pathways that require trans-acting and collaborating enzymes e.g. lovastatin 1 and aflatoxin 4 biosynthesis, but also discuss the more unexpected collaborations in fungal polyketide biosynthesis that have been proposed based on isolation of intermediates. From a biosynthetic perspective, the simplest classification system can be defined as collaborations that initiate biosynthesis (PKS loading), collaborations that occur during polyketide chain elongation (PKS extension) or collaborations that terminate biosynthesis (PKS release). However, there are additional examples of tailoring reactions by trans-acting enzymes, and, several increasingly more complex pathways where more than one trans-acting enzyme is required. Ultimately, these observations are intended to be useful when engineering fungal polyketide pathways by factoring in the requirement of collaborating enzymes and considering potential protein–protein interactions.
2. PKS activation by a phosphopantetheinyltransferase (PPT)
Although outside of the scope of this review, it is important to mention that PKS require post-translational modification to activate the ACP domains (Scheme 1). This is achieved by a phosphopanthetheinyltransferase (PPT), a class of enzymes that attach 4′-phosphopantetheine derived from coenzyme A 9 (reviewed elsewhere).46 4′-Phosphopantetheine is considered a “prosthetic arm” that is covalently bound to the ACP and essential for the transfer of all substrates and intermediates. NpgA is a PPT identified in Aspergillus nidulans and shown to be involved in both primary (i.e. lysine biosynthesis)47,48 and secondary metabolism.49,50 NpgA homologs are found in many fungi including the aflatoxin 4 producer Aspergillus parasiticus.51In vitro investigations of NpgA from A. parasiticus demonstrated that this PPT could activate an ACP domain from an iterative PKS (PksA) by transferring 4′-phosphopantetheine, but could not activate an ACP domain from the FAS (HexA).51 Specific ACP/PPT interactions are proposed but not yet elucidated.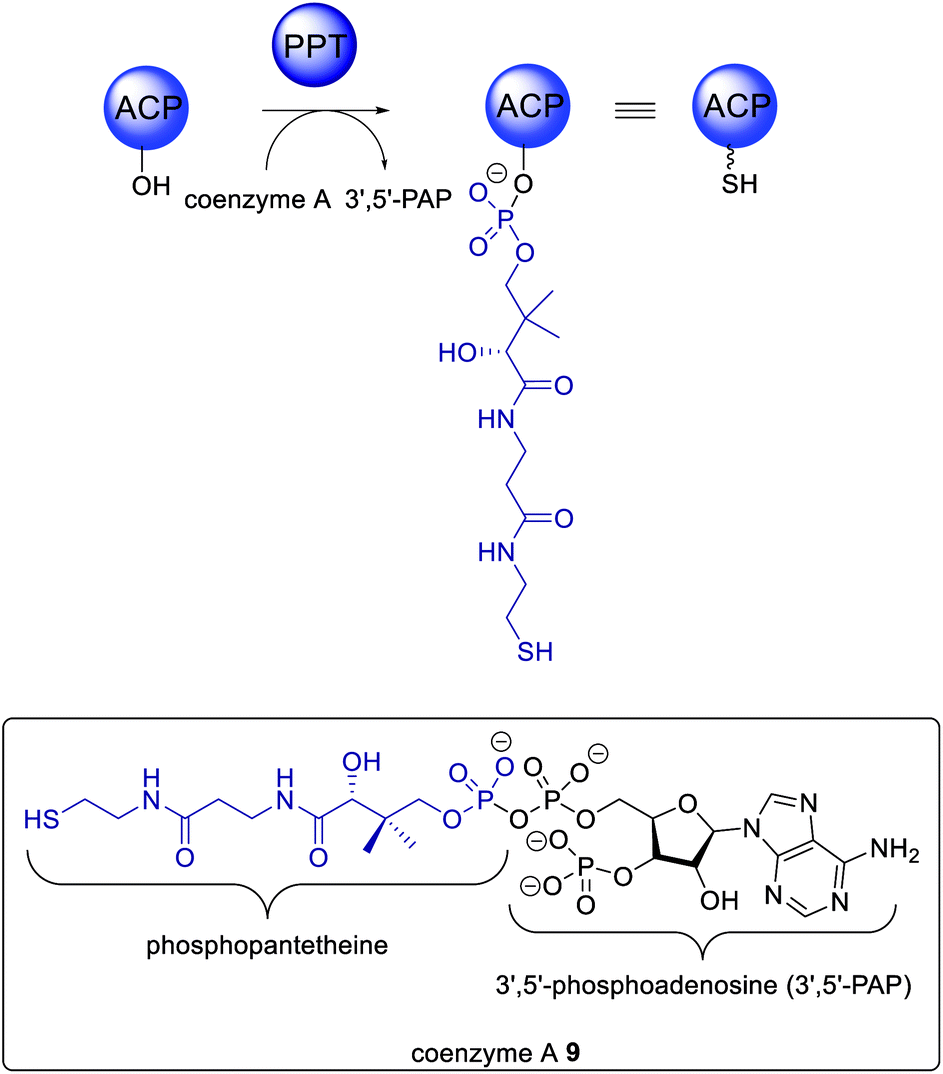 | ||
| Scheme 1 Overview of ACP activation by a trans-acting PPT. | ||
3. PKS loading by collaborating enzymes
Feeding studies with isotopically labelled compounds have demonstrated that the majority of fungal polyketides utilize acetyl-CoA as the starter unit. However, a number of compounds have been identified that are derived from alternative starter units such as: hexanoyl-CoA in norsolorinic acid 10 biosynthesis,52 the precursor of the mycotoxin aflatoxin 4; propionyl-CoA e.g. in pseurotin 11;53 nicotinyl-CoA e.g. in pyripyropene A 12;54 or malonamoyl-CoA in the biosynthesis of viridicatumtoxin 13 (Fig. 4).55 Although there are several examples of HRPKS that utilize non-acetate starter units, such as those involved in the biosynthesis of squalestatin S1 14,56 and strobilurin A 2,57 the rules governing how certain HRPKS select non-acetate starter units remains unknown, however they are not attributed to collaborating enzymes.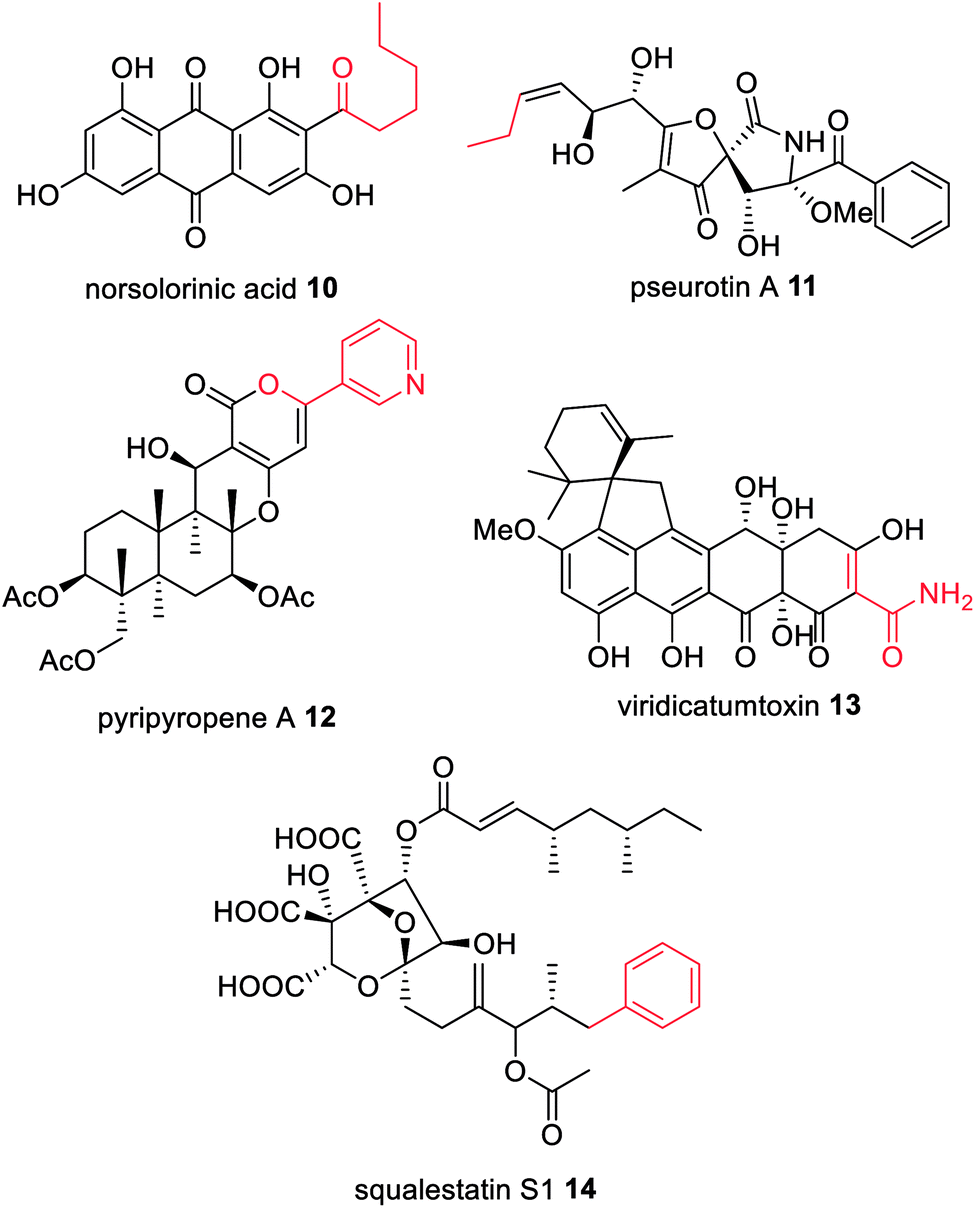 | ||
| Fig. 4 Examples of fungal polyketides derived from non-acetate starter units (highlighted in red). | ||
A breakthrough in the understanding of fungal NRPKS came from the discovery of the starter unit:ACP-transacylase (SAT) domain.15 SAT domains select the starter unit substrate and load the ACP, which then transfers the substrate onto the KS domain. Based on experiments with isolated SAT domains there is some flexibility in which substrates are selected in vitro,58 however SAT domains exhibit a clear selectivity over the chain length and functionality present in the starter unit.
3.1 FAS loading NRPKS enzymes with a starter unit
The biosynthesis of norsolorinic acid 10, the precursor to aflatoxin 4 and other biosynthetically related mycotoxins, results from the collaboration of two FAS subunits, HexA and HexB, with the NRPKS, PksA.59 The two FAS subunits synthesize hexanoyl-CoA that is transferred to the SAT domain of PksA. The N-terminal SAT domain of PksA was cloned, recombinantly expressed, and incubated with holo-ACP and [1-14C]-hexanoyl-CoA, clearly showing that the N-terminal domain covalently binds radiolabelled hexanoyl-CoA and transfers it to the ACP domain.15 In later deconstruction experiments PksA was shown to directly accept the hexanoyl starter unit 15 from the ACP domain of HexA (Scheme 2).60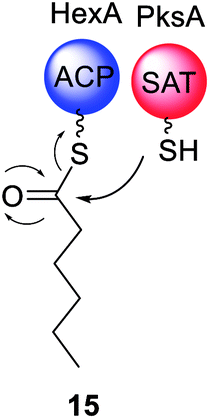 | ||
| Scheme 2 Experimentally confirmed transfer of hexanoyl starter unit 15 from the ACP of the FAS HexA to the SAT domain of the NRPKS PksA. | ||
Besides the fatty acid hexanoyl-CoA, the only other fatty acid to be reported as a starter unit for an SAT domain is octanoyl-CoA in NRPKS from the fungi Coccidioides immitis and C. posadasii.61 These fungi contain biosynthetic gene clusters (BGC) encoding two FAS subunits and an NRPKS, however, the final structure of the metabolite produced by the cryptic cluster remains unknown.
3.2 HRPKS loading NRPKS enzymes with a starter unit
An emerging class of characterized NRPKS are those that collaborate with a HRPKS lacking a release domain; the HRPKS synthesizes a polyketide starter unit and transfers it directly to the SAT domain of the partnering NRPKS (Scheme 3). To date, di-, tri-, tetra-, penta- and hexaketide starter units have been proposed that are mostly linear and display varying levels of reduction; some of which contain pendant methyl groups (Fig. 5).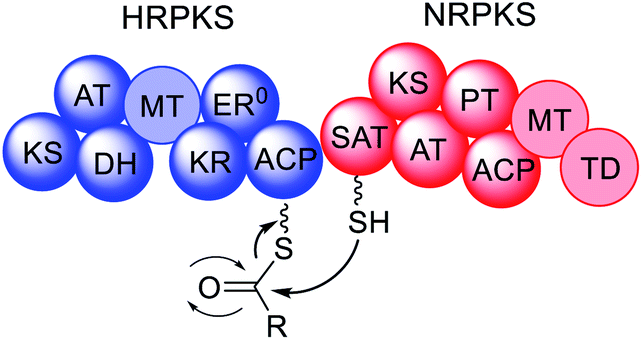 | ||
| Scheme 3 General overview of NRPKS loading by a HRPKS. | ||
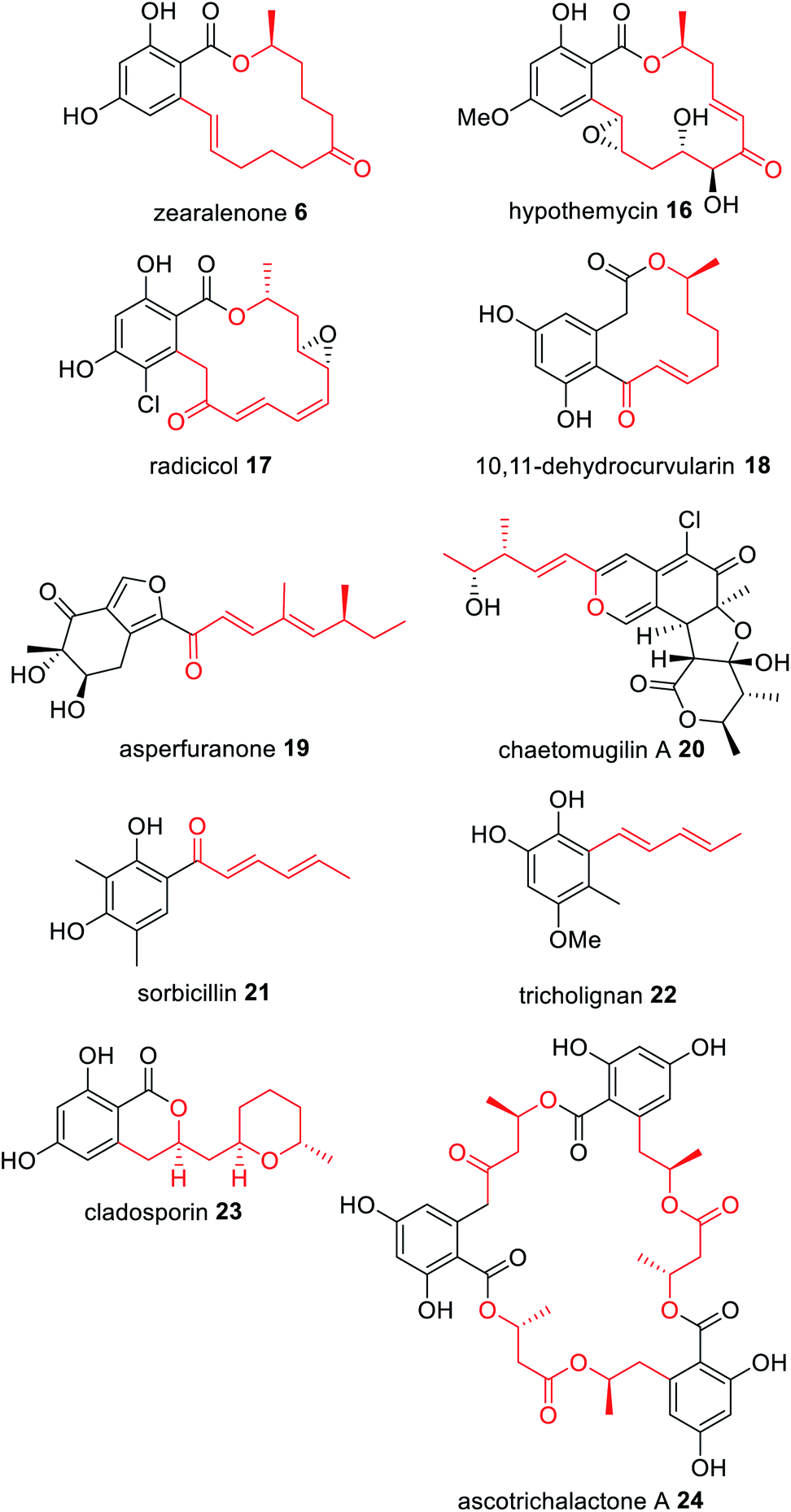 | ||
| Fig. 5 Examples of fungal polyketides derived from collaborating HRPKS and NRPKS. The polyketide transferred by the collaborating HRPKS is highlighted in red. | ||
Zearalenone 6 was the first fungal polyketide recognised as requiring two PKS enzymes for biosynthesis;62,63 the HRPKS PKS4 is proposed to synthesize a very highly reduced hexaketide that is transferred directly to the NRPKS PKS13 (Fig. 5). Similar biosyntheses are proposed for the anti-fungal agents hypothemycin 16,64,65 and radicicol 17,66,67 a HSP90 inhibitor. In contrast, the immune system modulator 10,11-dehydrocurvularin 18 is proposed to arise from an almost fully reduced tetraketide starter unit synthesized by the HRPKS AtCURS1 that primes the NRPKS AtCURS2 (Fig. 5).68
The novel metabolite asperfuranone 19 was discovered by genome mining after activation of the gene cluster encoding a unique HRPKS/NRPKS pair.69 The HRPKS AN1036.3 synthesizes a dimethylated tetraketide that acts as the starter unit for the NRPKS AN1034.3. Since then, a number of fungal polyketide biosynthetic pathways have been shown to involve the collaborative action of HRPKS and NRPKS pairs including: chaetomugilin A 20, derived from a methylated triketide synthesized by the HRPKS CazF;70 the antioxidant sorbicillinoid 21 derived from a triketide diene synthesized by the HRPKS SorA;71 tricholignan 22 that utilizes the triketide starter unit synthesized by the HRPKS TlnA;72 the anti-malarial cladosporin 23, composed of a highly functionalized pentaketide synthesized by the HRPKS Cla2;73 and ascotricholactone A 24, proposed to utilize a reduced oligomeric diketide synthesized by Men1 (Fig. 5).74
The corresponding NRPKS subsequently elongates, cyclizes and releases the polyketide product once it reaches the programmed chain length.75 Some NRPKS contain methyltransferase domains and introduce methyl groups during chain elongation programmatically, and release can be reductive or thioesterase mediated. The polyketide produced through the collaborative action of the HRPKS and NRPKS may be the final product of the biosynthetic pathway e.g.18 and 21, or it may undergo additional modifications by tailoring enzymes encoded by the same gene cluster e.g.19 and 20.
3.3 Structures of SAT domains
X-ray crystal structures of isolated SAT domains indicate an α,β-hydrolase core and a smaller ferrodoxin-like domain. The structure of the SAT domain from the NRPKS CazM (Fig. 6)26 and the SAT–KS–AT tri-domain from NRPKS CTB1 (Fig. 7)76 exhibit hydrophobic residues that line the active site of the SAT domains. These residues are proposed to provide control over the chain length and extent of functional groups of the starter unit entering the cavity.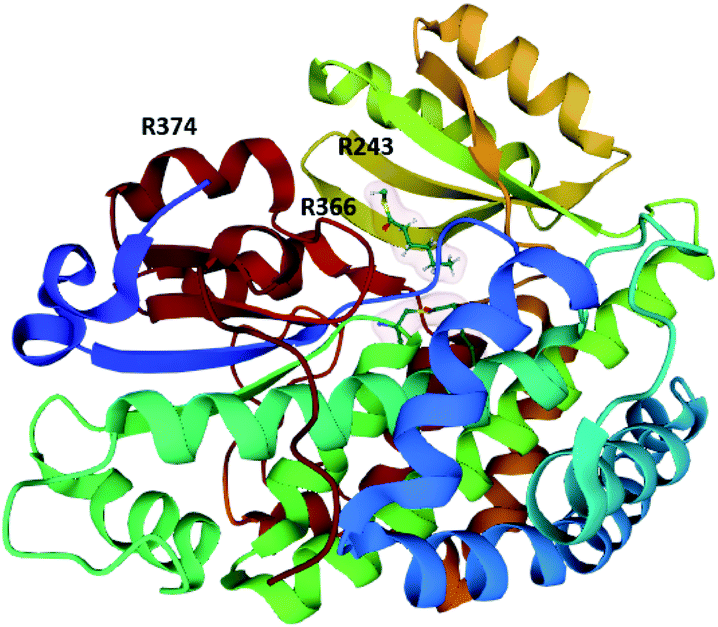 | ||
| Fig. 6 Structure of the CazM SAT domain bound with hexanoyl-CoA (PDB = 4RPM).26 The arginine residues proposed to be involved in inter-/intra-molecular docking with ACP domains are labelled. | ||
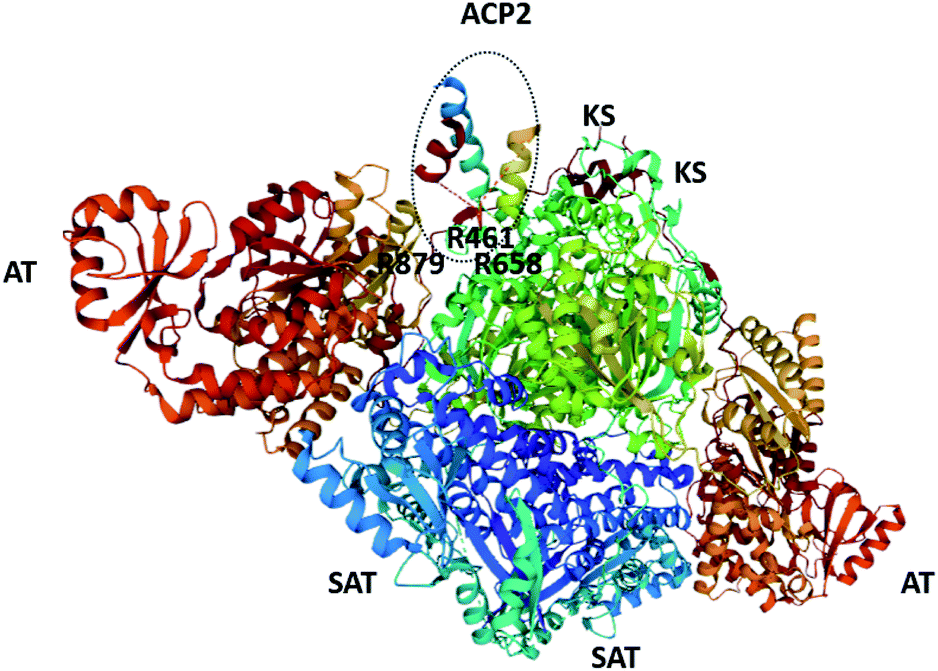 | ||
| Fig. 7 The structure of the SAT–KS–AT tri-domain from CTB1 and ACP2 (also from CTB1; circled. PDB = 6FIK).76 The arginine residues proposed to be involved in inter-/intra-molecular docking with ACP domains are labelled. | ||
Inter- and intramolecular docking of the SAT domain with ACP domains from the collaborating HRPKS (Fig. 6) and the native NRPKS (Fig. 7) are believed to be facilitated by several basic residues lining the entrance of the SAT domain active site.26,76 Therefore, the SAT domain has at least three fundamental roles: docking with the downstream ACP domain for starter unit transfer, selecting the preferred starter unit, and docking with the ACP domain from the collaborating enzyme.
To date, SAT domains have been reported to interact with collaborating FAS enzymes or HRPKS enzymes that provide a non-acetate starter unit. It is not yet possible to predict the starter unit selected by the SAT domain; this has to be confirmed experimentally using isotope labelled precursors, isolated domain assays, or via heterologous expression of the NRPKS with its collaborating enzyme partner.
4. PKS requiring collaborating enzymes for polyketide extension
4.1 Trans-acting ER enzymes
The first example of a fungal PKS being reliant on a trans-acting accessory protein for correct polyketide extension was in the biosynthesis of the cholesterol-lowering drug lovastatin 1.77 The biosynthetic gene cluster contains two genes encoding PKS enzymes; lovB encodes lovastatin nonaketide synthase (LNKS) and lovF encodes lovastatin diketide synthase (LDKS). LNKS, like many HRPKS, has an inactive ER domains (ER0) based on the absence of key active site residues. Also contained in the cluster is lovC, encoding a stand-alone protein with sequency similarity to PKS ER domains.77 In heterologous expression experiments in A. nidulans, when LNKS is expressed alone, instead of producing the expected dihydromonacolin L 25, shunt polyene compounds 26 and 27 were detected (Scheme 4A). Furthermore, the shunt products had shorter carbon chain lengths than expected, indicating a loss of LNKS programming fidelity. Co-expression of LNKS and LovC led to synthesis of the expected polyketide 25 (Scheme 4A) demonstrating that the associated trans-acting ER protein, LovC, not only reduces the LNKS olefins selectively but also contributes to overall PKS programming control.77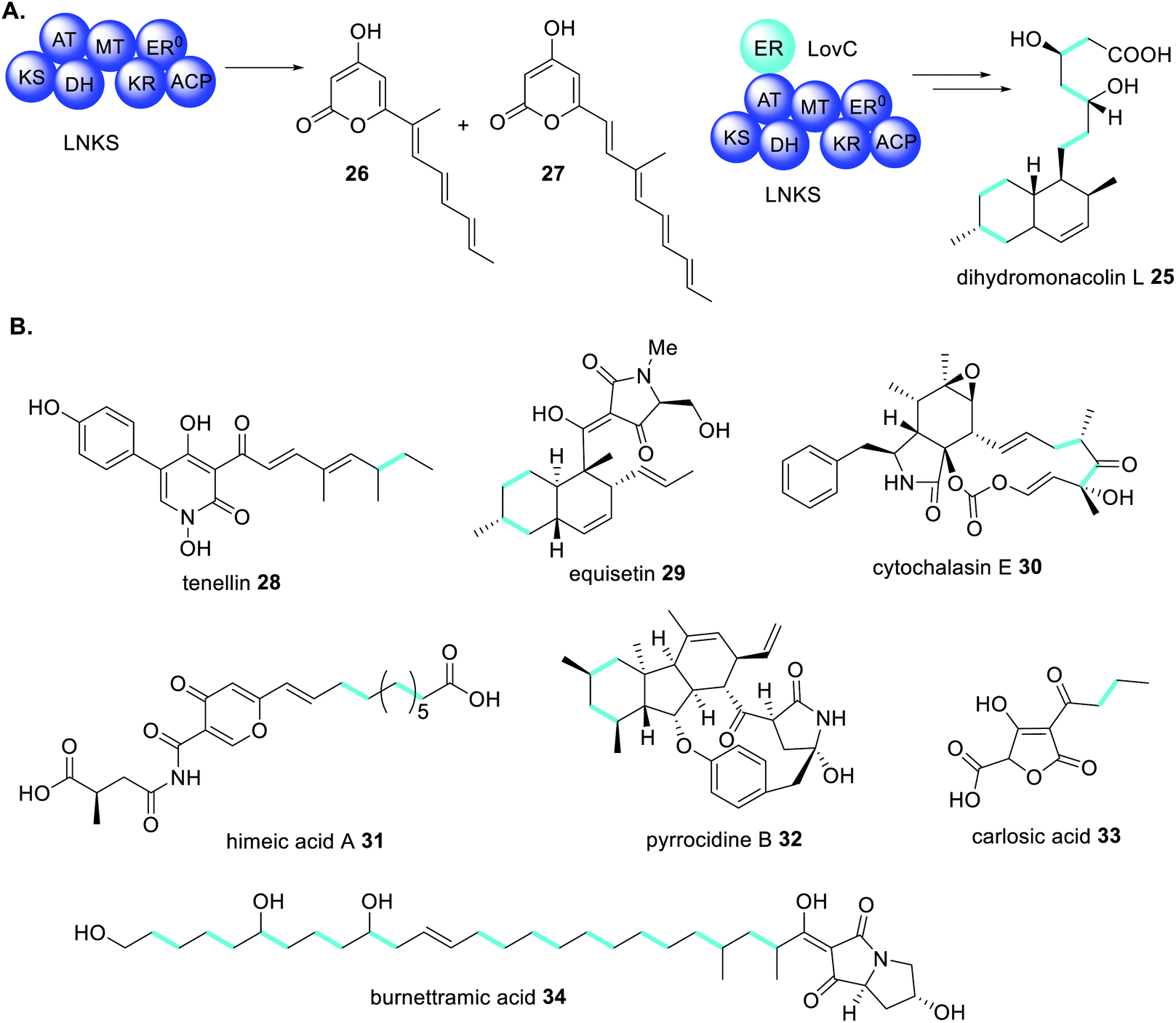 | ||
| Scheme 4 (A) The trans-acting ER enzyme LovC contributes to the correct programming of LNKS in dihydromonacolin L 25 biosynthesis.77 (B) Examples of fungal polyketides biosynthesized through the actions of trans-acting ER enzymes. Chemical modifications introduced by trans-acting ER enzymes are highlighted in light blue. | ||
Trans-acting ER enzymes have been confirmed in a number of other fungal polyketide biosynthetic pathways including tenellin 28 and other pyridones,78–84 equisetin 29 and other decalins,85–91 cytochalasin E 30 and other cytochalasans,92–98 himeic acid 31,99 pyrrocidines 32,100 carlosic acid 33,101 and other acyl tetronic acids,102 burnettramic acid A 34,103 and other lipid-like polyketides104 (Scheme 4B). It is now widely accepted, even expected, that a gene encoding a HRPKS with an ER0 domain, in close proximity to a trans-ER gene in a BGC, must be co-expressed for correct polyketide synthesis.
To date, only a single crystal structure of a fungal trans-acting ER enzyme, LovC, has been reported.105 LovC contains a medium chain dehydrogenase/reductase (MDR) fold and exists as a monomer. Between the catalytic domains and the NADPH co-factor binding domain are two loops that are highly conserved amongst fungal trans-acting ERs. These extended loops are proposed to prevent dimerization of LovC, since they induce steric clash in computational models.105 The substrate binding pocket is adjacent to the bound NADP+ and contains an electropositive bridge including the residue K54, proposed to dock the phosphopanthetheine arm of the LNKS ACP domain (Fig. 8). The monomeric form of LovC was proposed as being necessary for protein–protein interactions with LNKS and it was hypothesized that LovC forms a heterodimer with LNKS–ER0. However, no complex was detected when LovC was incubated with LNKS ER0 indicating a different mode of protein–protein interaction.105
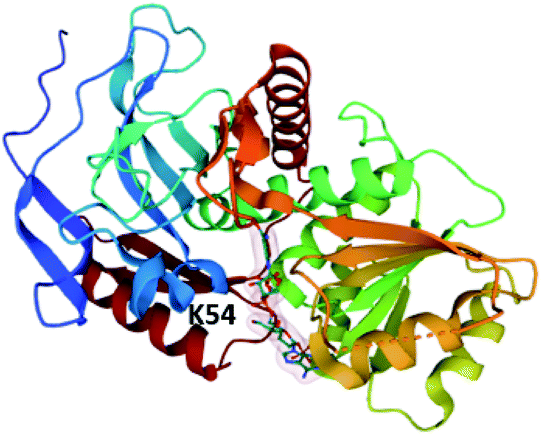 | ||
| Fig. 8 The structure of LovC in complex with NADP+ (PDB ID = 3B70).105 The putative PPT docking site K54 is labelled. | ||
The structure of LovC reveals important information about substrate specificity and the final structure of the nonaketide synthesized by LNKS. Due to hydrophobic binding, the substrate pocket of LovC accommodates tetra- and pentaketide intermediates more efficiently than the di- and triketide intermediates, and so they undergo enoyl reduction.105 However, the hexaketide intermediate forms a decalin due to Diels–Alder cyclization being favoured over enoyl reduction.105
Recently, the cryo-EM structure of the LovC-LNKS complex was reported.23 LNKS is considered as a HRPKS even though it contains a terminal condensation (C) domain, similar to those observed in non-ribosomal peptide synthetase (NRPS) enzymes. Although the ACP and C domains could not be solved, this is the most complete fungal PKS enzyme structure containing eight connected catalytic domains. The LovC-LNKS complex is an X-shaped dimer and LovC interacts with the AT domain, forming two L-shaped chimeric catalytic chambers (Fig. 9),23 reminiscent of the structure of mFAS (Fig. 3).
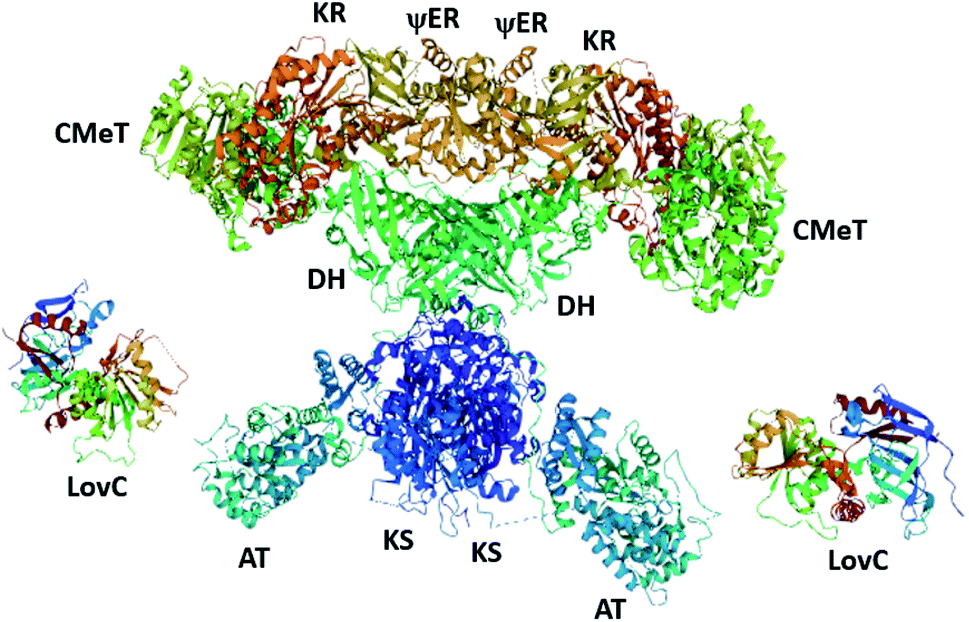
Unfortunately, the resolution of the LovC-LNKS interface was not high enough to model protein–protein interactions, but computational docking identified the docking region between residues 695–757 of the LNKS AT domain and the loop region of LovC. Mutating the loop region of LovC and co-expressing with LNKS–AT resulted in the two proteins eluting separately. Furthermore, mutating residues 747–750 within LNKS–AT also causes the two proteins to elute separately, thereby establishing the trans-acting ER interaction between the loop region and a helix of the AT domain.23
4.2 Trans-acting MT enzymes
Pseurotin A 11 is a PKS–NRPS derived polyketide displaying neuritogenic activities amongst other bioactivities.106,107 A gene cluster for biosynthesis of 11 was identified in Aspergillus fumigatus as it was the only cluster encoding a PKS–NRPS enzyme.108 The function of the PKS–NRPS encoding gene, psoA, was confirmed as being essential for biosynthesis of 11 by both knockout and overexpression.108 The PKS–NRPS product of this gene, PsoA, had all the domains expected for the biosynthesis of 11, including a MT domain, albeit with low homology to other fungal MT domains (Scheme 5).108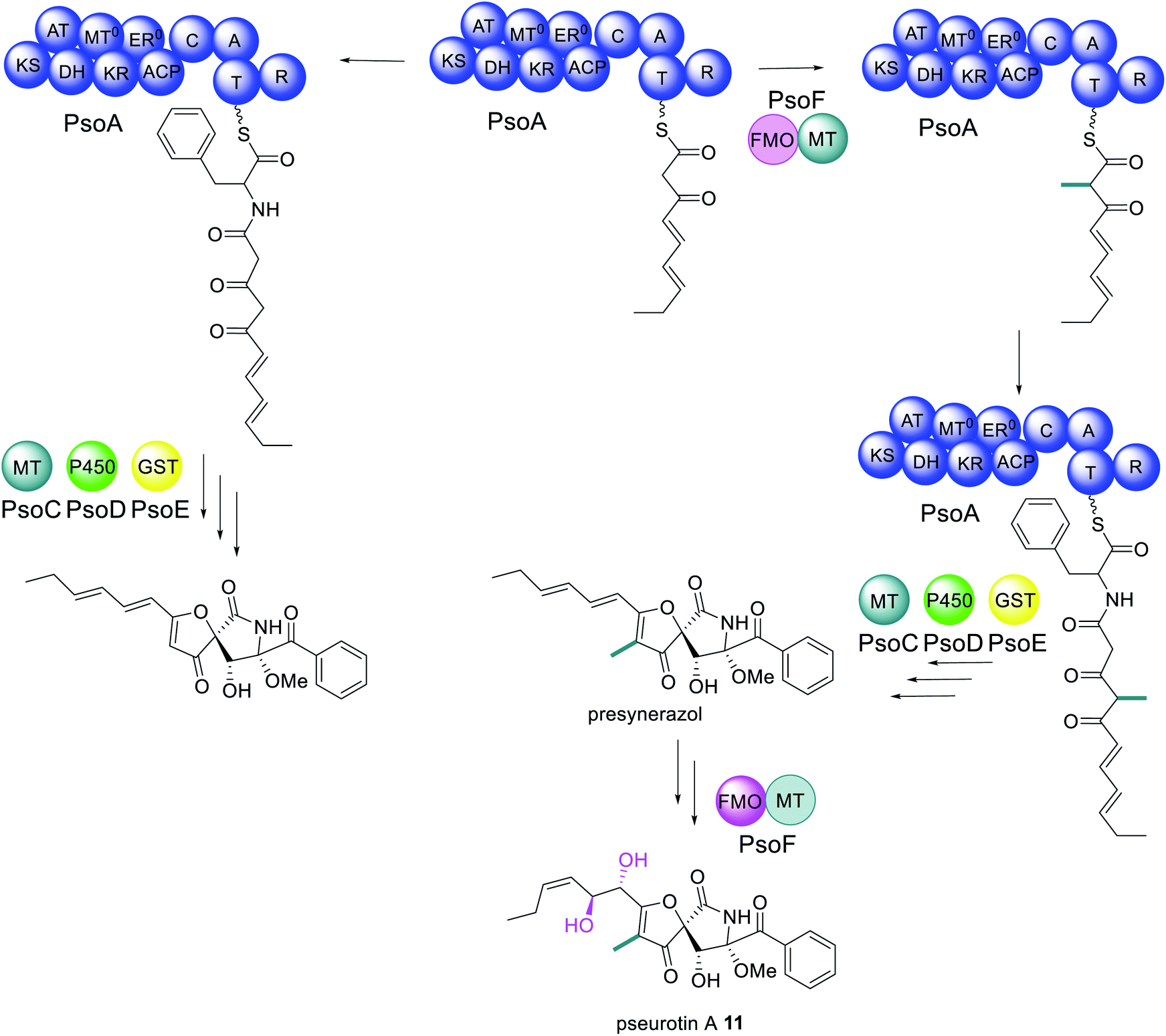 | ||
| Scheme 5 Biosynthetic pathway of pseurotin A 11.110,111 Abbreviations: as in Fig. 2; GST = glutathione S-transferase. Only chemical modifications introduced by the bifunctional PsoF are highlighted in colour. | ||
Unexpectedly, the pseurotin gene cluster was later found to be interlinked with the genes required for the biosynthesis of fumagillin 3, within a supercluster.109 An additional two genes were subsequently identified as being essential for biosynthesis of 11; psoF, encoding a dual function monooxygenase/methyltransferase, and psoG, encoding a hypothetical protein.110 Characterization of the modification enzymes: PsoC, an O-methyltransferase; PsoD, a P450; PsoE, a glutathione S-transferase; and the bifunctional PsoF, established precise functions for PsoC–E and indicated that PsoF participated in trans-C-methylation of the growing polyketide (Scheme 5), despite the MT domain of the PKS–NRPS retaining some methylation activity in in vitro studies.111 The role of PsoG remains unknown as it has no homology to any known protein.
The MT domain of PsoF was cloned and expressed in E. coli for structural and mechanistic studies (Fig. 10).112 The structure of PsoF identified a structural fold characteristic of class I S-adenosyl-methionine (SAM) dependent methyltransferases that act on small molecules. Computational docking studies suggested that there is a hydrophobic tunnel, formed by the SAM-binding domain and substrate binding domain, that can accommodate a tetraketide intermediate.112 Mutagenesis of any of the active site residues Tyr687, His799 or Glu825 significantly reduced enzyme activity. Tyr687 is proposed to be essential for substrate binding and catalysis and Tyr833 is proposed to be involved in controlling the chain length of the intermediate that is accepted into the active site. Currently, it is still unknown how exactly the trans-acting PsoF interacts with the PKS–NRPS PsoA.
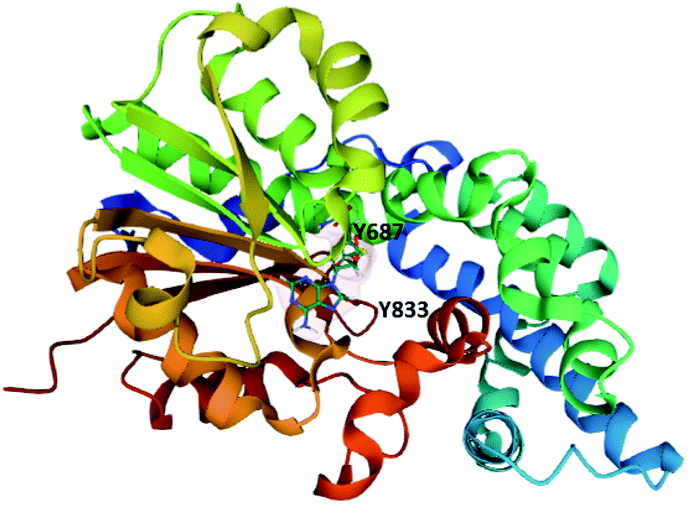 | ||
| Fig. 10 The structure of PsoF MT domain in complex with SAH (PDB ID = 6KJI).112 The tyrosine residues proposed to be involved in substrate binding and recognition are labelled. | ||
Compared to trans-acting ER enzymes, reports of trans-acting MT enzymes are relatively rare. A trans-acting MT domain has also been identified in the biosynthesis of the fungal metabolites trichnolignan 22 and calbistrin A 35 (Fig. 11).72,113 In calbistrin A 35 biosynthesis the function of the trans-acting MT CalH′ was first established through heterologous expression with the HRPKS CalA′ in Aspergillus oryzae.113 A mixture of compounds 36–39 was detected including 36 that has a carbon backbone identical to the polyene portion of 35 (Scheme 6). Compounds 36 and 38 were also observed in in vitro enzymatic reactions, and assays determined that CalH′ acts on the growing polyketide chain while it is still attached to the PKS.113
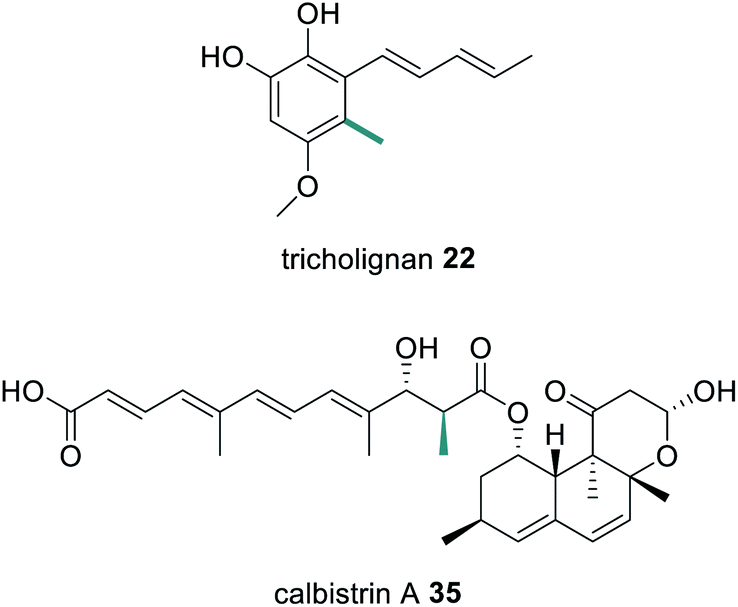 | ||
| Fig. 11 Structures of tricholignan 22 and calbistrin A 35 indicating the position of a methyl group introduced by a trans-acting MT enzyme. Chemical modifications introduced by the trans-acting MT enzymes are highlighted in green. | ||
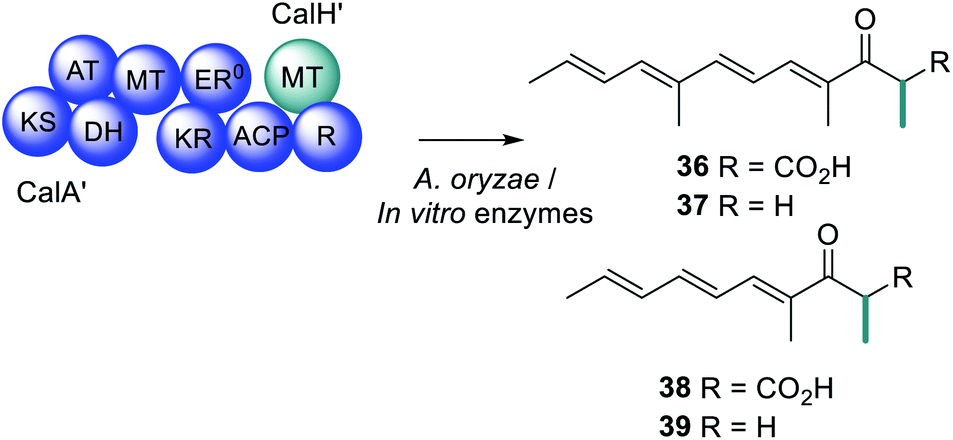 | ||
| Scheme 6 Investigations into the biosynthetic products of CalA′ and the trans-acting MT CalH′. | ||
Similar to the structure of PsoF-MT, CalH′ contains catalytic residues Tyr157, His266 and Glu292 that are well conserved, however the hydrophobic substrate tunnel of CalH′ is larger, to accommodate a longer polyene substrate (Fig. 12).113 Furthermore, the surface of CalH′, near to the substrate tunnel, also consists of positively charged residues. A computational docking study suggests that the negatively charged region of the CalA′ ACP fully covers the entrance of the active site due to the interaction between the oppositely charged protein surfaces, orchestrating the methylation reaction.113
The biosynthesis of both tricholignan 22 and calbistrin A 35 contain a number of additional unusual features and so will be discussed further in Section 7.
4.3 Trans-acting KR enzymes
The immunosuppressant agent dalmanol 40 contains a rare 6/6/6/6/6 ring system proposed to derive from 1,3,6,8-tetrahydroxynaphthalene (4HN) 41 and 1-(2,6-dihydroxyphenyl)but-2-en-1-one (PBEO) 42 (Scheme 7).114 The genes required for the biosynthesis of 40 are located in two different gene clusters, physically separated on the genome of Daldinia eschscholzii. One gene cluster encodes a PRPKS, ChrA, adjacent to a ketoreductase enzyme, ChrB, while the second gene cluster encodes an NRPKS, pksTL. Both gene clusters were confirmed as being required for biosynthesis of 40 by gene inactivation and heterologous expression in Aspergillus oryzae.112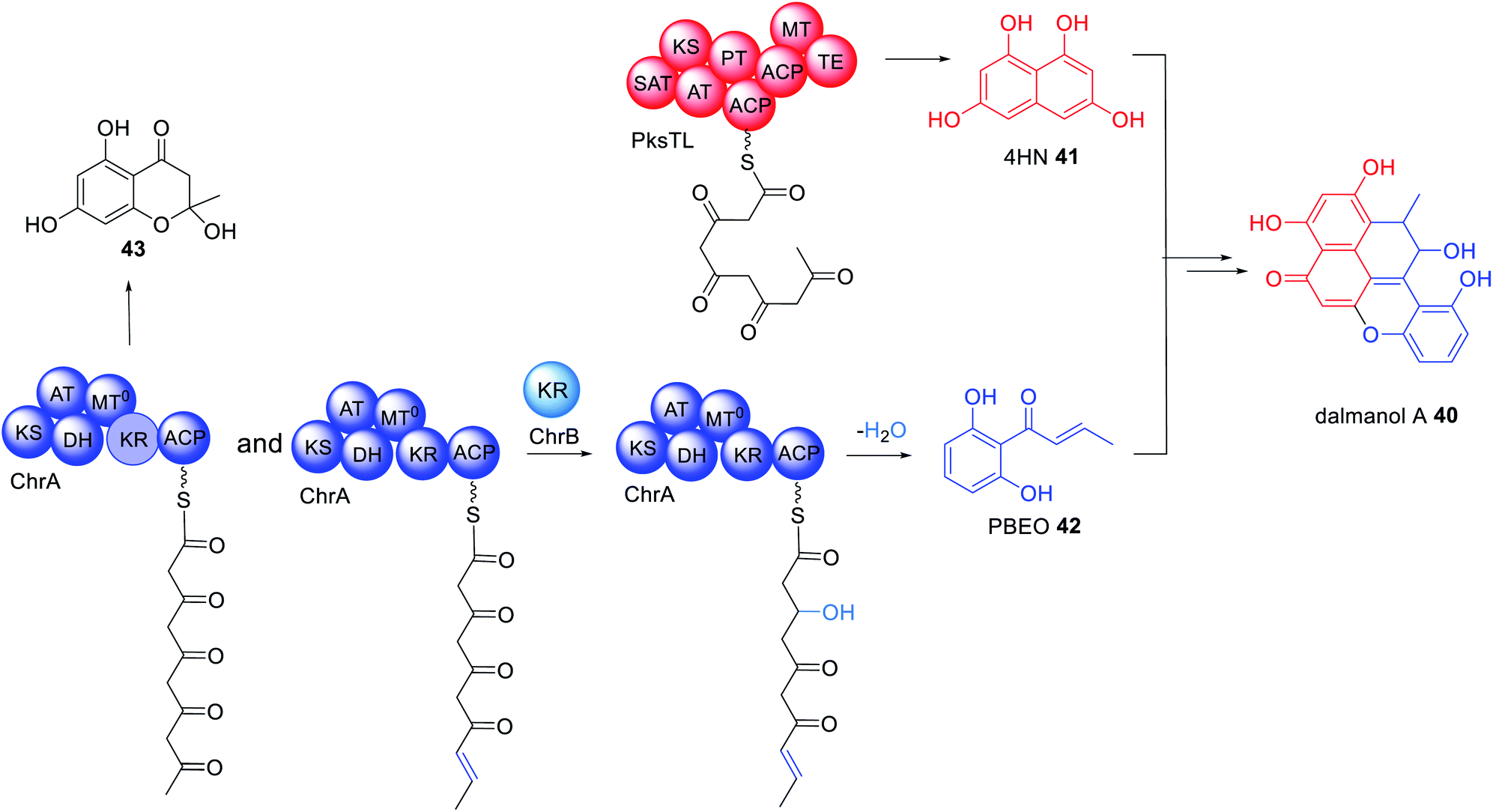 | ||
| Scheme 7 Biosynthetic pathway of dalmanol A 40. The HRPKS ChrA contains an active KR domain however, the trans-acting KR enzyme ChrB appears to be utilized to diversify the range of polyketide products observed. The chemical modification introduced by the trans-acting KR enzyme ChrB is highlighted in light blue. | ||
ChrA is an unusual PRPKS with an inactive MT domain. Unexpectedly, heterologous expression experiments of ChrA in A. oryzae determined that the KR domain is active during the first round of chain extension due to isolation of 43. However, when the KR enzyme, ChrB, was co-expressed with ChrA, 42 was observed, indicating that ChrB is active in the fourth round of chain extension, specifically reducing the C-3 carbonyl to a hydroxyl group (Scheme 7).114
A trans-acting PKS-like multidomain enzyme, TerB, has been identified as essential for 6-hydroxymellein (6-HM, 44) biosynthesis,115 a precursor to the phytotoxin terrein.116 6-HM is synthesized through the collaboration of TerA, an NRPKS, and TerB, an unusual enzyme containing an active KR domain and inactive DH and MT domains, in an organization similar to a truncated HRPKS. Heterologous expression of TerA alone in either Aspergillus niger or A. oryzae led to production of triketide 45, tetraketide 46, and pentaketide 47 products indicating an unusual relaxation of the TE domain with respect to both the polyketide length and mode of release i.e. lactonization vs. hydrolysis (Scheme 8).115,116 Co-expression of TerA and TerB led to a production of 6-HM 44 in a substantially higher titer than 45–47, pointing to a collaborative mode of action between the two enzymes.115
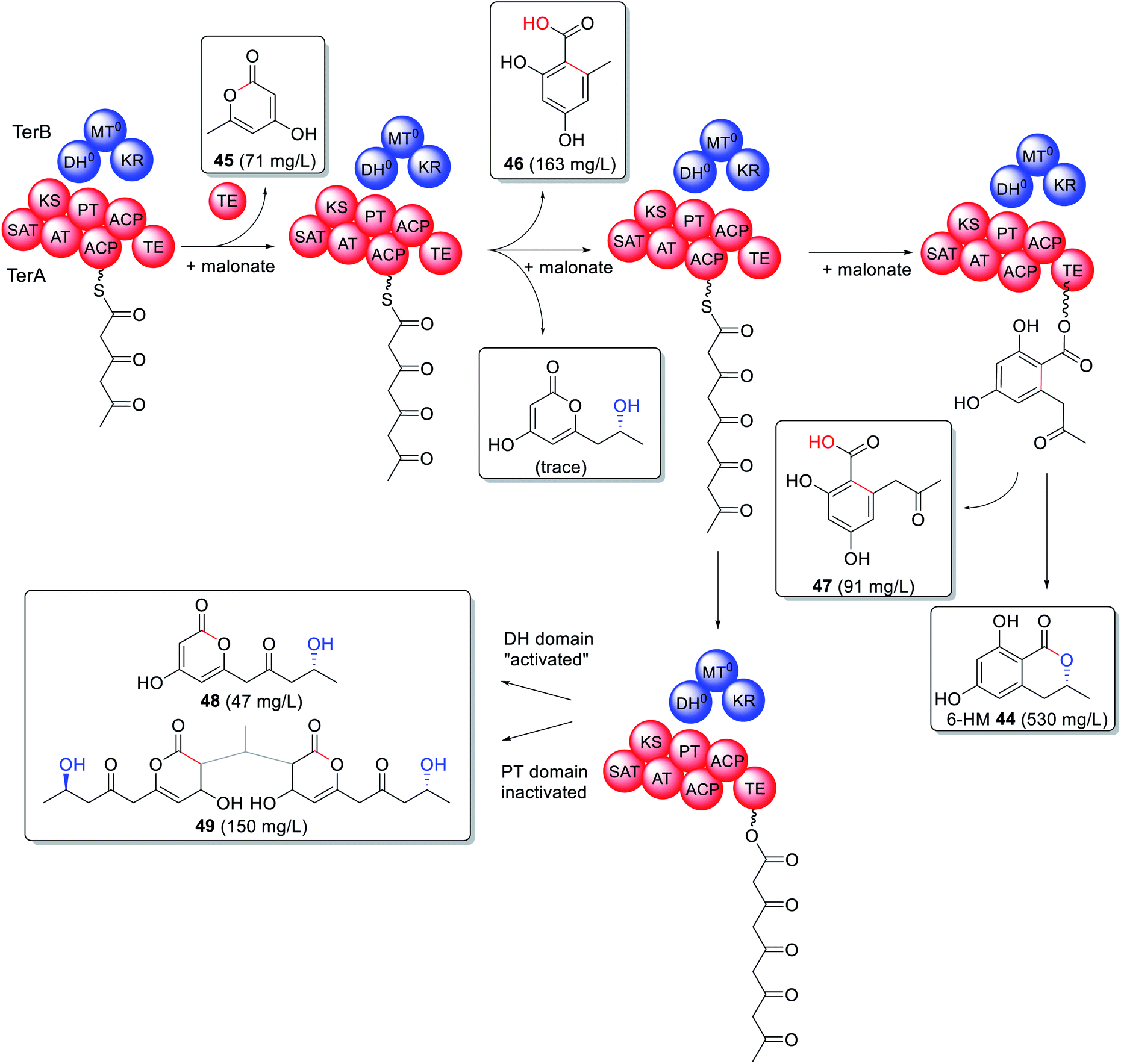 | ||
| Scheme 8 Biosynthesis of 6-HM 44 requires collaborating NRPKS TerA and the PKS-like enzyme TerB. Site-directed mutations determined that the interaction site is between the PT and DH domains. The chemical modifications introduced by the trans-acting PKS-like enzyme TerB is highlighted in blue. | ||
The interaction between TerA and TerB was investigated via site directed mutations of specific catalytic sites including both ACPs, the TE, the DH and the KR domains. Surprisingly inactivating the catalytic site of the PT domain (H1320A) had the same effect as “activating” the catalytic site of the DH domain (Q46H) during co-expression experiments (Scheme 8). The change in product profile to 48 and 49 indicated that mutating the DH domain within TerB affected the PT domain within TerA, preventing it from functioning as expected and providing insight into where the two enzymes interact.115 6-HM 44 has also been established as being required for the biosynthesis of cyclohelminthols 50, palmaenones 51, roussoellatide 52 (Fig. 13),117,118 and is a shunt product in the biosynthesis of 24.74
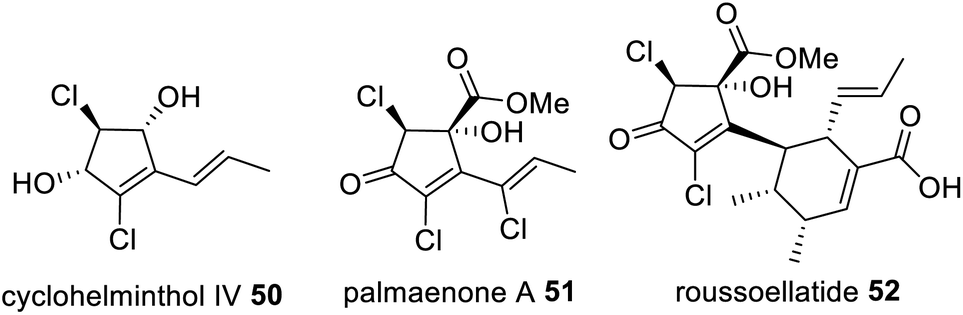 | ||
| Fig. 13 Structures of 6-HM derived metabolites that require the collaboration of a NRPKS and trans-acting HRPKS-like enzyme. | ||
5. PKS requiring collaborating enzymes for polyketide release
There are relatively few HRPKS that contain a terminal domain for product release,104,118–120 instead they are often found fused to an NRPS module generating a PKS–NRPS, or collaborating with NRPKS enzymes, as observed in the biosynthesis of 16–24. HRPKS enzymes lacking a terminal off-loading domain appear to collaborate with trans-acting enzymes for polyketide release and there are various classes of enzymes known to facilitate this polyketide off-loading.Similarly, NRPKS that belong to group V have no domain for polyketide release,121 instead a trans-acting TE, or more specifically a metallo-β-lactamase protein, is necessary for product release. Finally, there are trans-acting enzymes with proof-reading roles. Although mechanistically they are not required for polyketide release they greatly facilitate the efficiency of the biosynthesis leading to higher titers of natural product obtained.
5.1 Trans-acting α,β-hydrolases
α,β-Hydrolases are a large class of enzymes with both catalytic and non-catalytic functions that include thioesterases. These enzymes contain a catalytic triad comprised of a nucleophilic residue, an acidic residue (e.g. Glu or Asp) and a catalytic His.122 These collaborating α,β-hydrolases will therefore be categorized according to their role in biosynthesis.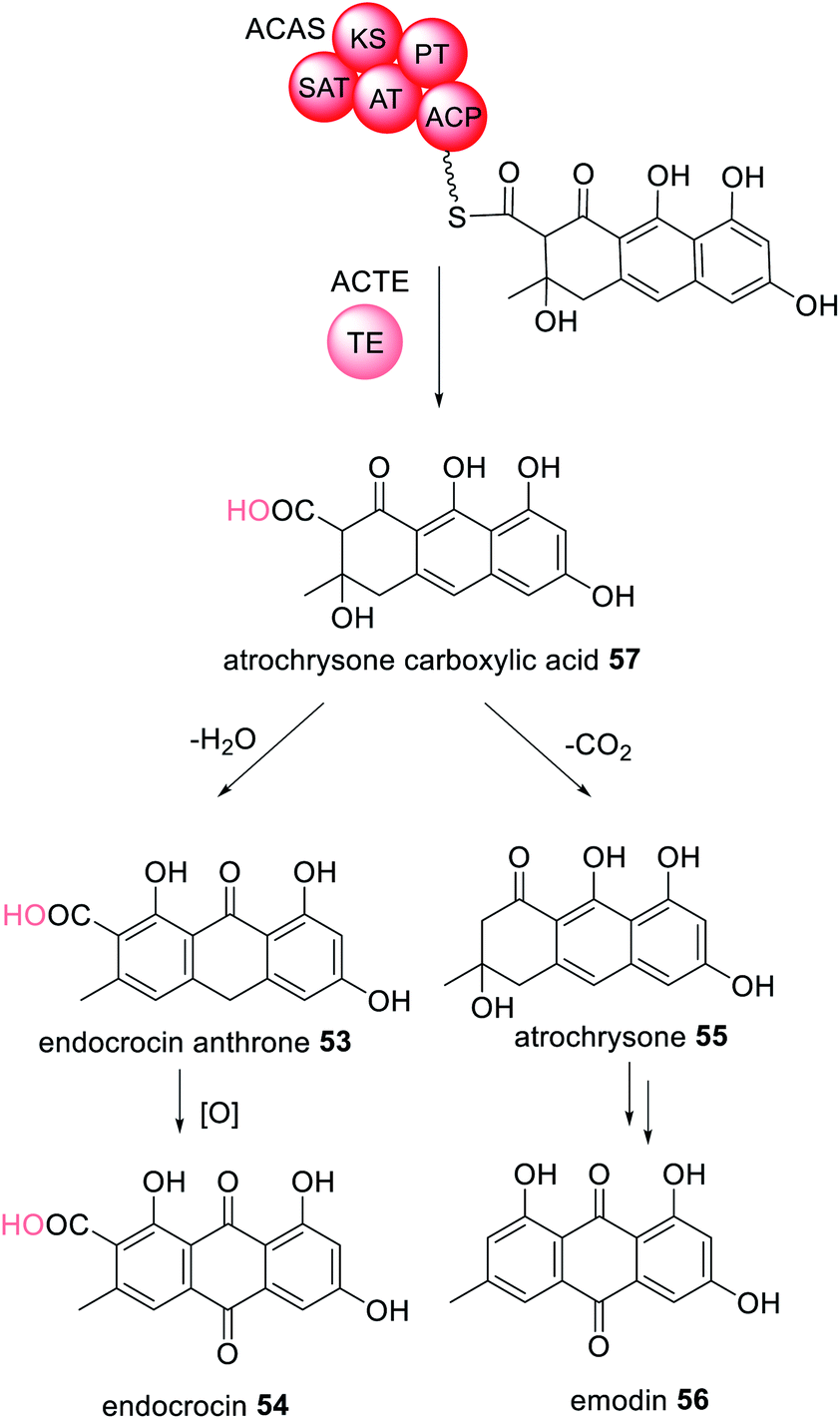 | ||
| Scheme 9 The TE enzyme ACTE releases the octaketide intermediate attached to the NRPKS ACAS by hydrolysis to generate 57. The chemical modification introduced by the trans-acting TE enzyme is highlighted in light pink. | ||
TE-less NRPKS with trans-acting MβL-TE have also been identified in the biosynthesis of viridicatumtoxin 13,55 neosartoricin 58,124 monodictyphenone 59,125 asperthecin 60,126 trypacidin 61,127 geodin 62,128 (Fig. 14) and many other anthraquinone and xanthone products derived from emodin 56.129–132 A common feature of this class of metabolites is spontaneous decarboxylation of the carboxylic acid group generated by the associated trans-acting MβL-TE.
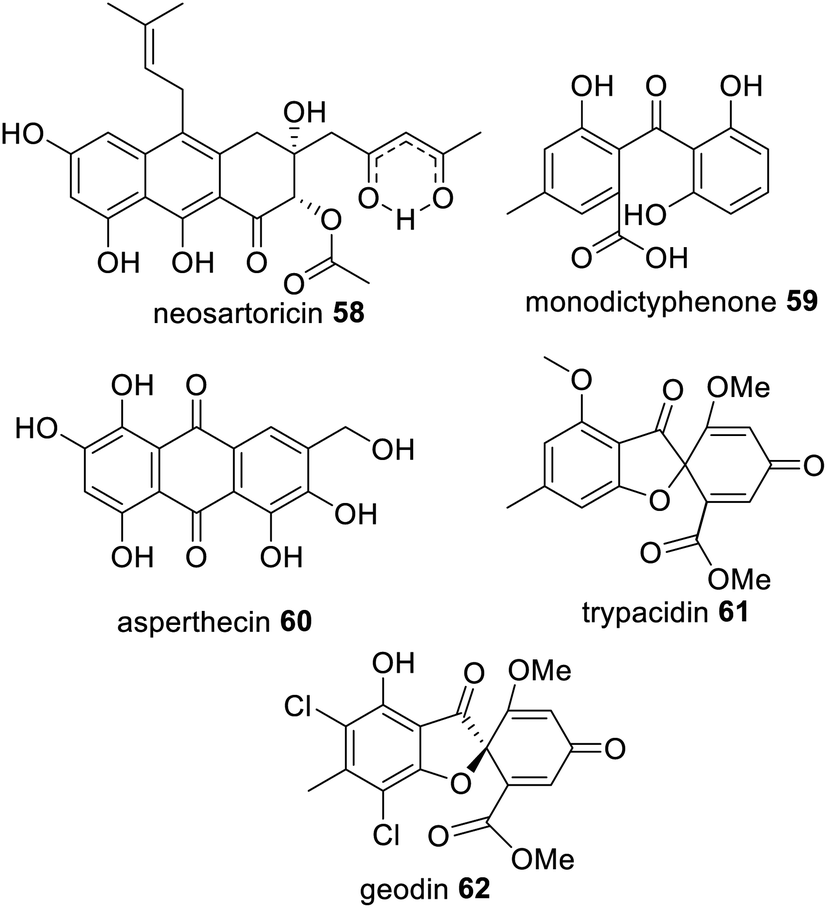 | ||
| Fig. 14 Examples of fungal polyketides that utilize a trans-acting TE enzyme for hydrolytic polyketide release from an NRPKS. | ||
Trans-acting hydrolases required for hydrolytic release are also found collaborating with HRPKS, e.g. in lovastatin 1,133 squalestatin S1 14,56 byssochalamic acid 63,134 fusarielin H 64,135 and fusaric acid 65 biosynthesis136 (Fig. 15). In lovastatin biosynthesis, LovG, a serine hydrolase, was investigated through gene knock-out experiments where disruption caused a drop in production of 1 by over 95%.133In vitro studies confirmed the role of LovG in releasing polyketide products from LNKS.
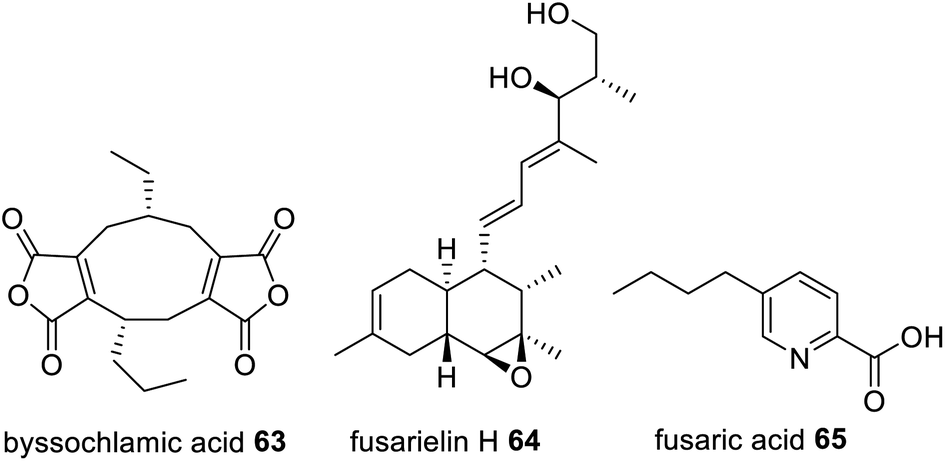 | ||
| Fig. 15 Examples of fungal polyketides that utilize a trans-acting hydrolase enzyme for hydrolytic polyketide release from a HRPKS. | ||
In the biosynthesis of the hexaketide portion of 14, a citrate synthase is proposed to act on the polyketide chain tethered to the HRPKS SQHKS (Clz14) and then the hydrolase (Clz11) releases the modified polyketide product 66 (Scheme 10).56 The functions of these enzymes were elucidated in the fungal host A. nidulans; if Clz11 was omitted or replaced with β-lactamase Clz13, there was more than a 99% reduction in titers of 14.56 The biosynthesis of byssochalamic acid 63 and other maleic anhydrides have also been shown to require the co-ordination of both a citrate synthase and hydrolase,134,137 indicating this is another common general release mechanism requiring two collaborating and trans-acting enzymes.138
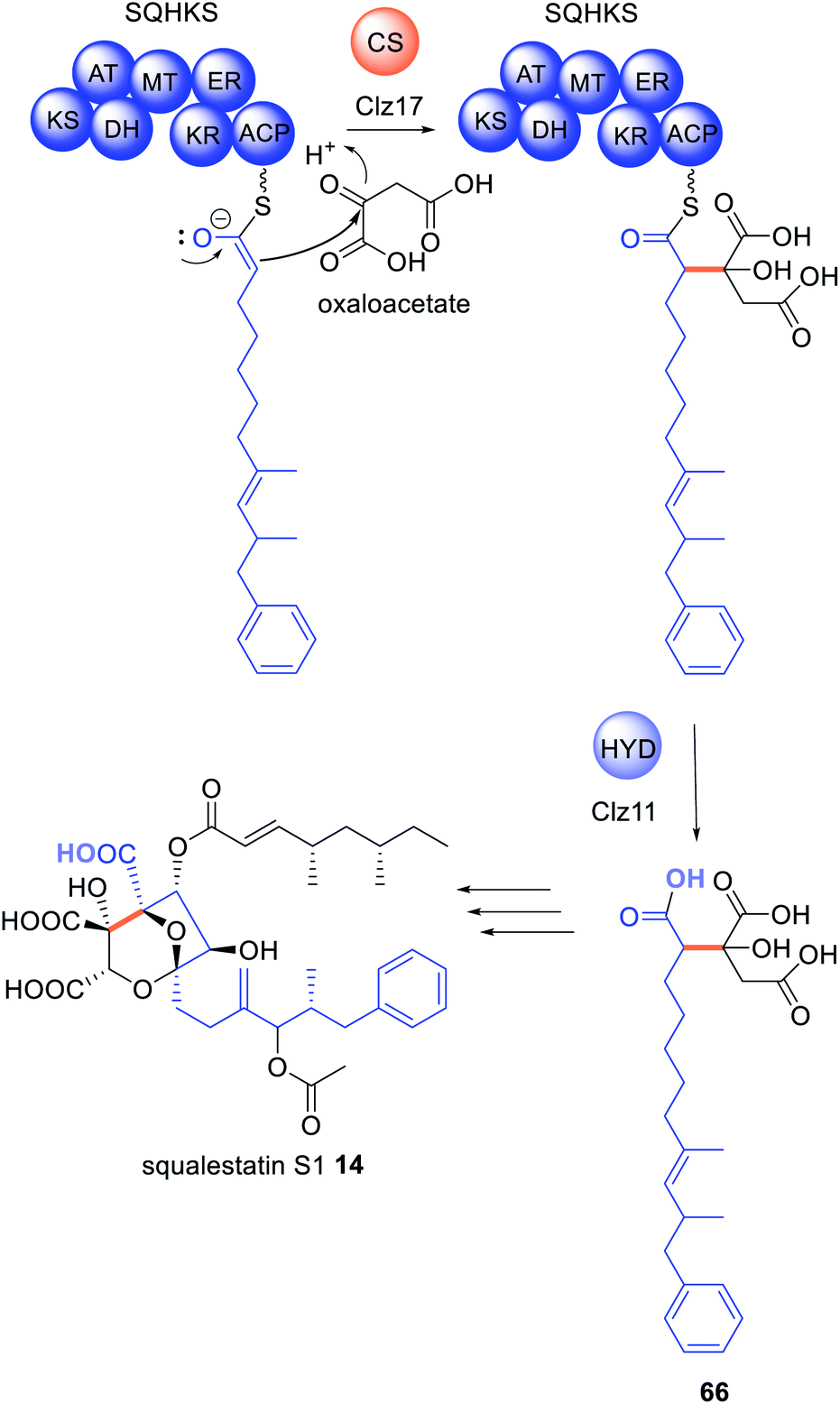 | ||
| Scheme 10 Biosynthesis of squalestatin hexaketide 66 requires both a citrate synthase and hydrolase. The chemical modifications introduced by the trans-acting enzymes are highlighted in colour. | ||
Often, when HRPKS collaborate with trans-acting hydrolases, lactones or macrolides are formed e.g.67–74 (Fig. 16). The biosynthesis of brefeldin A 67, for example, requires a HRPKS (Bref-PKS) and a thiohydrolase (Bref-TH).139In vitro investigation of Bref-PKS led to production of acylic nonaketide products e.g.75 (Scheme 11) which was surprising since 67 is a cyclic octaketide. Including the Bref-TH produced acyclic octaketides e.g.76, revealing a dual role for Bref-TH in hydrolysing the polyketide product as well as controlling the polyketide chain length (Scheme 11).139
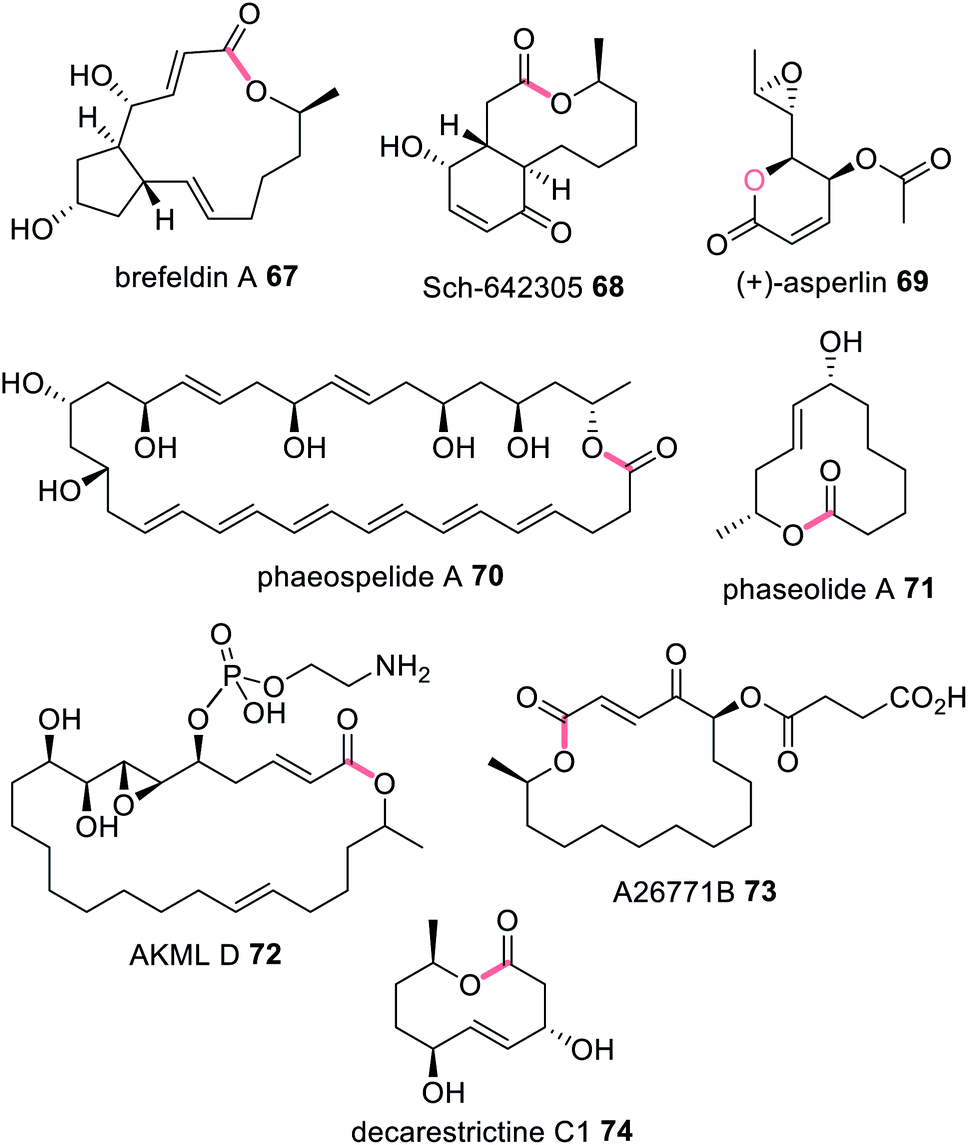 | ||
| Fig. 16 Examples of fungal polyketides that utilize a trans-acting TE enzyme for polyketide release from a HRPKS. Chemical modifications introduced by the trans-acting TH/TE enzymes are highlighted in light pink. | ||
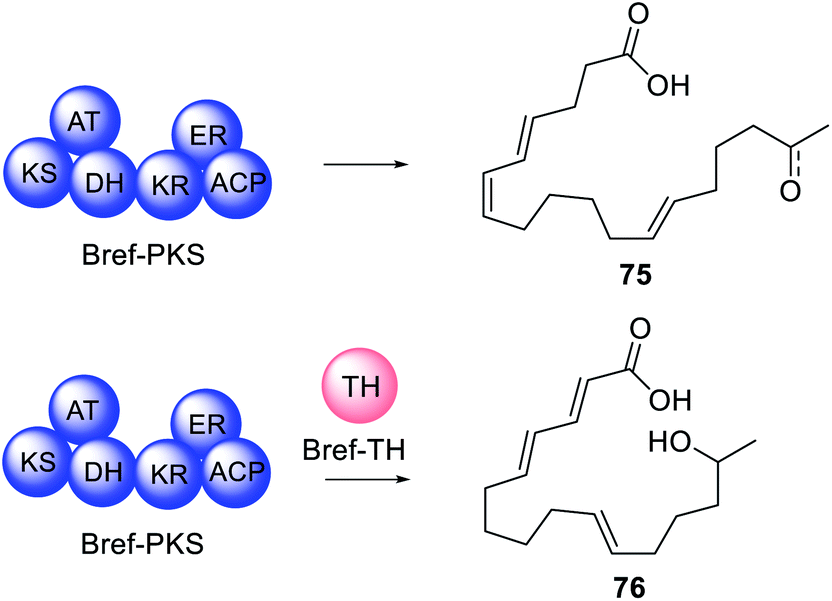 | ||
| Scheme 11 Investigations into the role of Bref-TH. | ||
A remaining question is why macrolactonization of 76 was not observed in the assays and whether Bref-TH is required. It was proposed that the cyclopentane moiety observed in 67 may need to be formed first, suggesting that additional trans-acting enzymes remain to be discovered.139 Similarly, the biosynthesis of Sch-642305 68, a bicyclic macrolide similar to 67, also suggests a P450 enzyme acting early in the biosynthesis. Heterologous expression of the PKS (SchPKS) and the corresponding hydrolase (SchR1) did not reveal any linear or cyclic polyketide products, however when the P450 SchR2 was co-expressed with SchPKS and SchR1 both linear polyketides and macrolides were observed for the first time.140 Knock-out of SchR2 in Phomopsis sp. CMU-LMA, the producer of 68, also abolished biosynthesis and no linear or cyclic polyketides were observed.
Trans-acting hydrolases were revealed in the biosynthesis of 69 by transcriptional factor engineering and by heterologous expression for 70–73 (Fig. 16).141–146 Recently the thioesterase, DcsB, was shown to collaborate with the HRPKS DcsA during the biosynthesis of the 10-membered lactone decarestrictine C1 74.145 DcsB was shown to be able to hydrolyse a number of polyketide mimics with varying chain lengths demonstrating its use as a biocatalyst. The crystal structure was obtained showing that DcsB is a homodimer containing an α,β-hydrolase fold (Fig. 17).145 DcsB was co-crystallized with a HRPKS thioester mimic enabling visualization of the active site, however protein–protein interactions with DcsA were not reported.
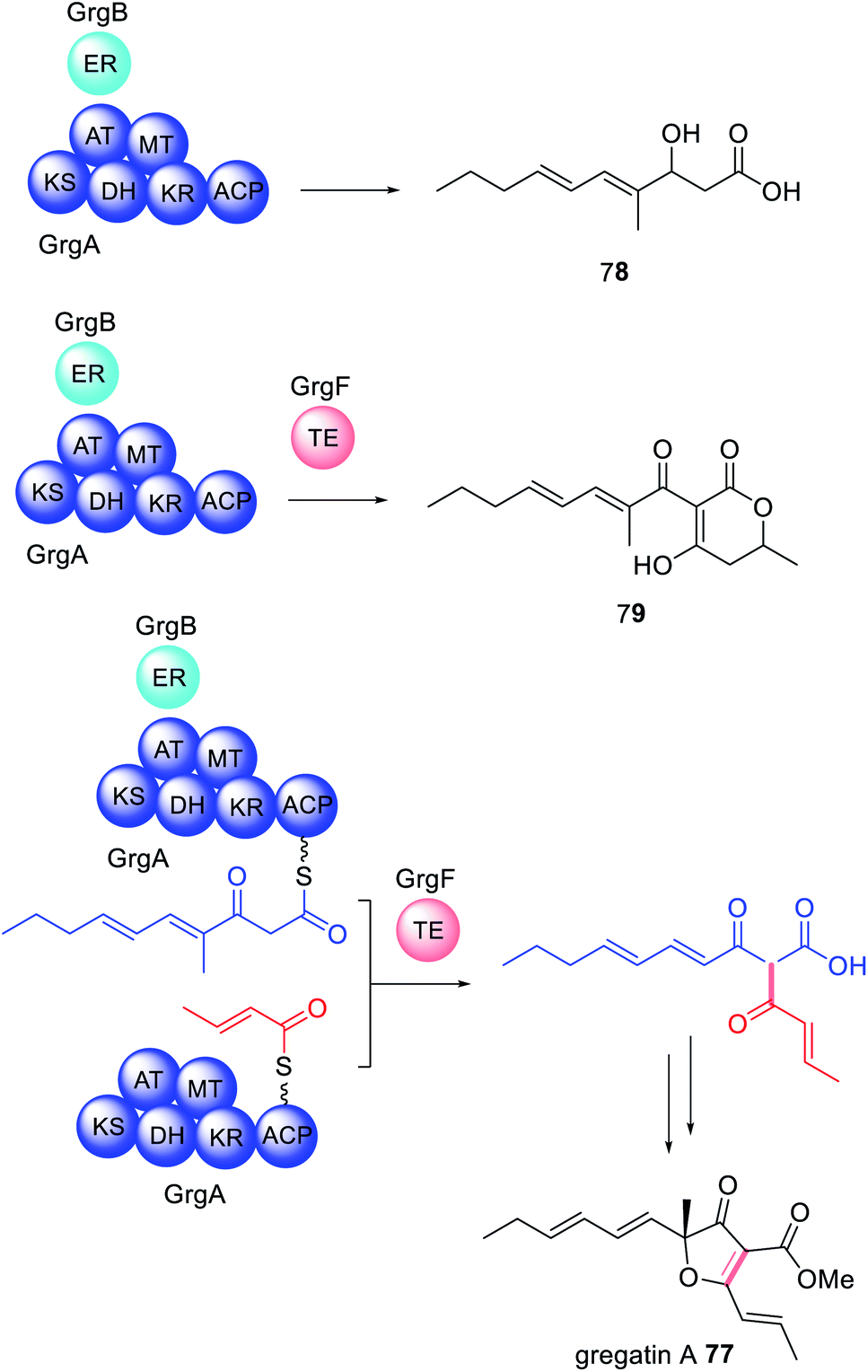 | ||
| Scheme 12 The biosynthesis of gregatin A 77 revealed that GrgF is a trans-acting TE that fuses two different length polyketides synthesized by the HRPKS GrgA with and without collaboration of trans-acting ER GrgB. | ||
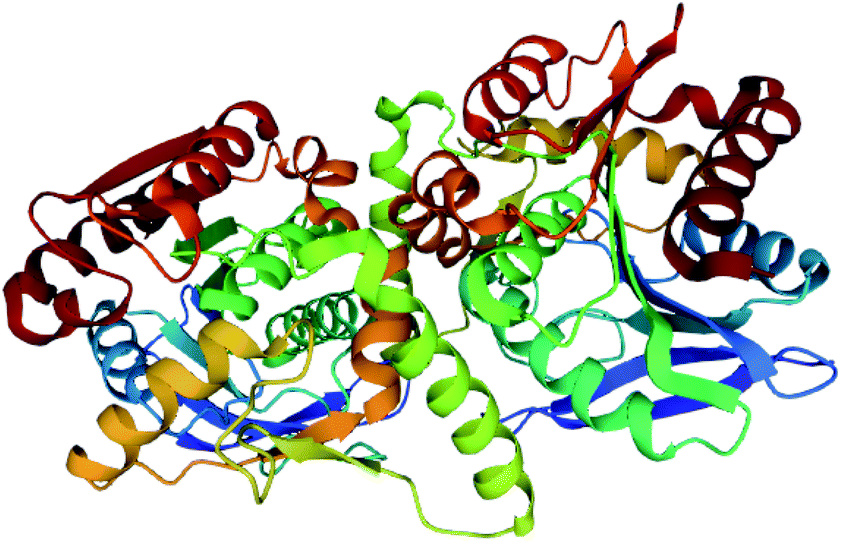
The crystal structure of GrgF demonstrates an α,β-hydrolase fold and contains a typical Cys–His–Asp catalytic triad (Fig. 18).147 A highly hydrophobic chamber was identified as the substrate binding site and confirmed via docking studies and molecular dynamic simulations. At this time how GrgF interacts with the ACP of GrgA remains unknown but future structural biology studies are proposed.
α,β-Hydrolases are commonly found in PKS–NRPS gene clusters.85,86,88,92–98,110,148,149 The α,β-hydrolase PyiE, involved in the biosynthesis of the cytochalasan pyrichalasin H, was recently proposed to control the formation a specific pyrrolinone tautomer after synthetic and in vivo investigations.150 Protein–protein interactions are proposed between the PKS–NRPS, PyiS, and hydrolase, PyiE, to enable precise control over the rapid tautomerization, but remain to be confirmed. Similarly, the thioesterase PynI is identified in the biosynthesis of pyranonigrin as being essential for PKS–NRPS product release and tetramic acid formation.151
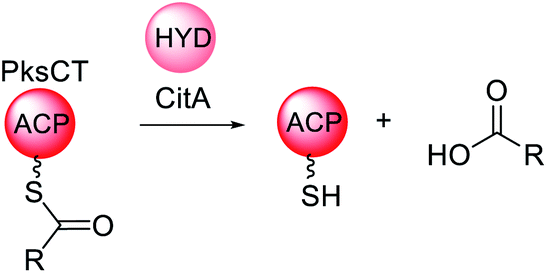 | ||
| Scheme 13 Overview of the function of the trans-acting hydrolase CitA in the biosynthesis of citrinin. | ||
Trans-acting hydrolases with similar proof-reading activities have been identified in the biosynthesis of lovastatin 1,133 asperfuranone 19,69 azanigerone 80,154 and various Monascus azaphilone-derived pigments.155 Similar roles for cis-acting TE domains have been characterized in NRPKS such as PksA involved in the biosynthesis of 10.156,157
5.2 Trans-acting acyl transferases
As mentioned, a distinctive feature of HRPKS is the frequent lack of a domain for polyketide release. The biosynthesis of lovastatin 1 requires two HRPKS, LNKS and LDKS, and a trans-acting hydrolase is required for release of the nonaketide from LNKS. In contrast, the stand alone acyltransferase LovD is required for release of the diketide from LDKS.5 The mode of release was studied in vitro by recombinant expression of LDKS in yeast. After incubating with malonyl-CoA, SAM and NADPH; no diketide product was detected, indicating that LDKS is unable to release the polyketide. When LovD and monacolin J acid 81 were included in the reaction mixture 82 could be detected. If LovD was not included, or the LovD S27A active site mutant was used as a replacement, 82 was not detected, indicating that LovD transfers the diketide from LDKS to 81 (Scheme 14A).5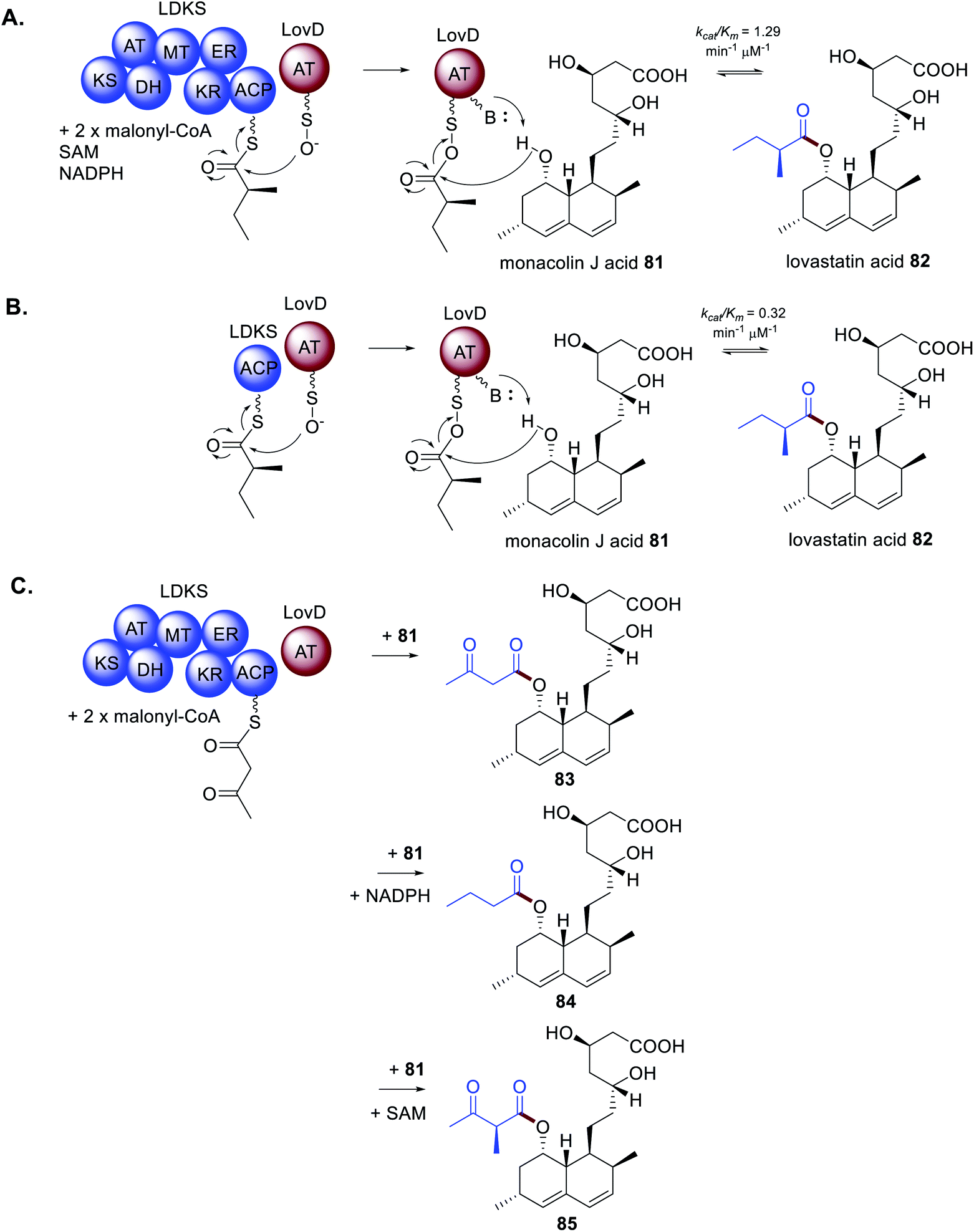 | ||
| Scheme 14 Characterization of the trans-acting acyltransferase LovD in diketide release from the HRPKS LDKS. (A) In vitro investigations of LDKS and LovD. (B) In vitro investigations of the ACP domain of LDKS with LovD. (C) In vitro investigation of LDKS and LovD missing key co-factors. | ||
Further investigation of the diketide transfer from LDKS by LovD through measuring reaction velocities and reaction kinetics demonstrated that LovD and LDKS interact by protein–protein interactions.5 The physical interaction was further confirmed by preparing the standalone holo-ACP domain from LDKS and measuring the catalytic efficiency of diketide transfer to LovD (Scheme 14B). The catalytic efficiency of transfer was comparable to that measured for LDKS and LovD, although it cannot be ruled out that LovD interacts with other regions of LDKS. However, when alternative ACP domains from bacterial PKS and FAS systems were substituted for the LDKS ACP no products were detected indicating that LovD is highly selective towards the ACP domain of LDKS.5
During investigations of the LDKS and LovD interactions the co-factors SAM and NADPH were not included in the reaction mixture essentially inactivating the MT and KR domains respectively. As expected, 82 was not observed, however analogous products 83–85 were (Scheme 14C).583 contains an unreduced, non-methylated diketide side chain due to the absence of both SAM and NADPH; 84 contains a fully reduced non-methylated diketide side chain due to the absence of SAM; and 85 contains a non-reduced methylated diketide side chain due to the absence of NADPH. Considering that these compounds were not observed under standard cultivation conditions it is speculated that the processing of the diketide occurs more rapidly within LDKS than by the trans-acting LovD. Taken together, these experiments demonstrate that protein–protein interactions are equally as important a consideration as substrate recognition.
The crystal structure of LovD contains an α,β-hydrolase fold, and the Ser76–Lys79–Tyr188 catalytic triad.158 The interface between the two domains forms a hollow cavity for the substrates with the serine active site at the base of the cleft (Fig. 19).158 Mutational studies demonstrated that conformational changes could be introduced to LovD that increased catalytic efficiency 11-fold and improved solubility and stability.158 Further in-depth studies using directed evolution generated a library of 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 779 LovD variants and led to identification of an optimized variant LovD9, 1000 times more active in vitro than the native enzyme.159
779 LovD variants and led to identification of an optimized variant LovD9, 1000 times more active in vitro than the native enzyme.159
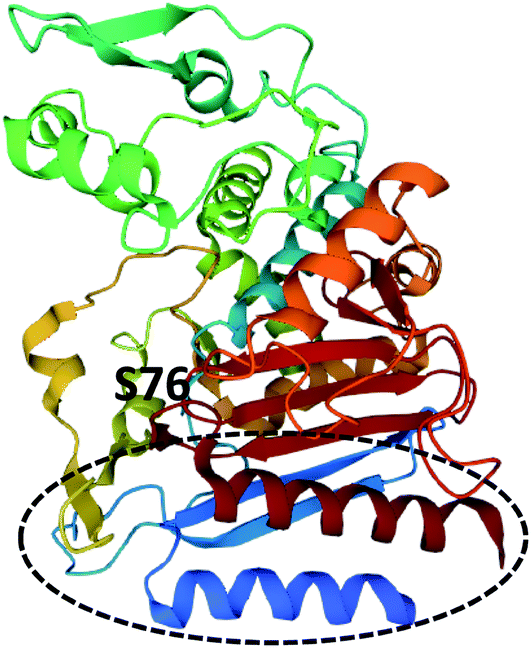 | ||
| Fig. 19 Structure of LovG (PDB ID = 3HLB).159 The catalytic serine residue, S76, is labelled and the ACP binding surface is circled. | ||
LovD9 contains 29 mutations at various positions around the enzyme and no longer requires an ACP partner enzyme.159 Specifically, the width of the active site is reduced, stabilizing catalysis and making it less accessible to partner enzymes i.e., the ACP domain of LDKS. Molecular dynamic simulations indicated that in the native LovD, protein–protein interactions with the ACP domain of LNKS cause conformational changes of the catalytic residues, enhances catalysis, and prevents alternate ACPs from other systems being used as the acyl donor.159 Furthermore, the ACP and phosphopantetheine binding surface was identified from the crystal structure (Fig. 19).
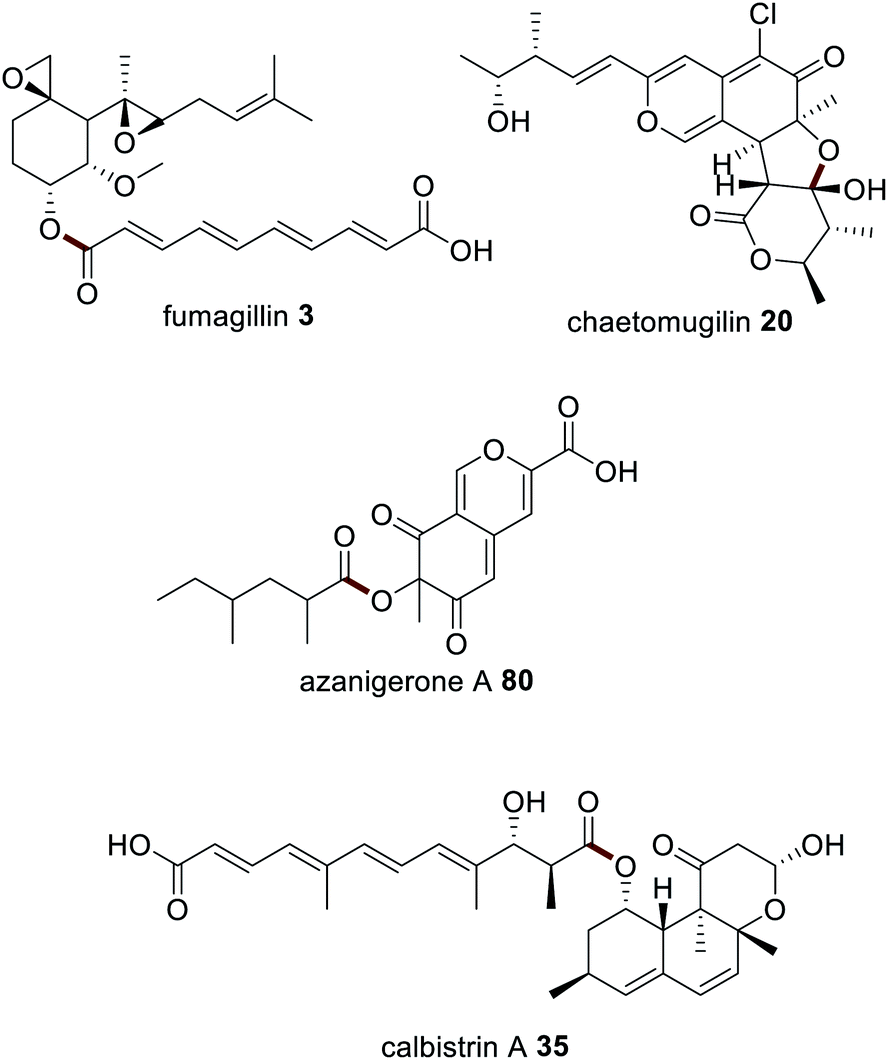
Subsequently polyketide release from a HRPKS by a collaborating acyltransferase enzyme has been confirmed in the biosynthesis of fumagillin 3,160 chaetomugilin 20,70 and calbistrin A 35 (ref. 113) (Fig. 20). CazE, the acyltransferase required in the biosynthesis of 20, was studied in vitro and shown to release a triketide from the HRPKS CazF.70 CazE is proposed to induce a conformational change in CazF to facilitate polyketide release, by protein–protein interactions.70
 | ||
| Fig. 20 Examples of fungal polyketides that utilize a trans-acting AT enzyme for polyketide release from a HRPKS. Chemical modifications introduced by the trans-acting AT enzymes are highlighted in brown. | ||
The biosynthesis of azanigerone 80 is also speculated to utilize the acyltransferase AzaD.154 However, an alternative scenario was proposed where the polyketide product of the HRPKS (AzaB) is released as a carboxylic acid and converted an acyl-CoA, similar to the biosynthesis of the tetraketide portion of 14,161 and various lipopeptides.162,163 The precise substrate for AzaB-mediated acylation has not yet been determined.
5.3 Trans-acting reductases
Reductive release domains are well known in NRPKS, specifically in group VII,121 where polyketides are released as an aldehyde. In contrast, the HRPKS required for the biosynthesis of betaenone 85, Bet1, is one of the rare HRPKS that contains a terminal domain for product release; here a reductase (R) domain.164,165 Collaborating reductases have been proposed in the biosynthesis of sordarial 86,166 trichoxide 87,167 and flavoglaucin 88 (ref. 168) biosynthesis (Fig. 21) because the HRPKS required for their biosynthesis lack any release domains, and the resulting PKS products are proposed to arise from aldehyde intermediates. Investigations into the biosynthesis of 87 demonstrated that when the HRPKS VirA was heterologously expressed in A. nidulans an aldehyde product was detected, however when VirA was co-expressed with cupin-domain enzyme VirC relatively higher titers of the aldehyde HRPKS product were detected.167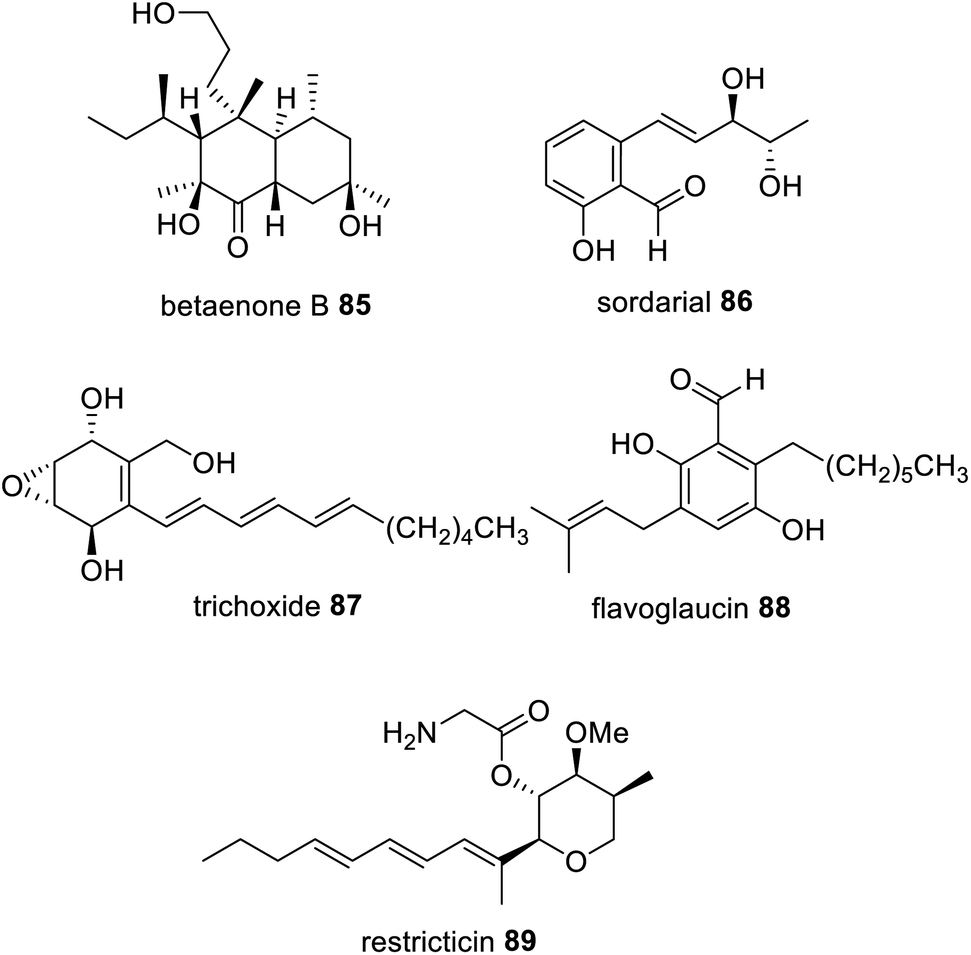 | ||
| Fig. 21 Examples of fungal polyketides that arise from an aldehyde intermediate synthesized via a HRPKS with an R domain or proposed to be the action of a trans-acting reductase. | ||
The biosynthesis of the lanosterol 14a-demethylase (CYP51) inhibitor, restricticin 89, is also proposed to arise from the action of a trans-acting short-chain dehydrogenase/reductase (SDR), Rstn4.169 Although considering that the biosynthesis of 89, was also investigated in A. nidulans, similar to the investigation of 87, reductases native to the heterologous host cannot be ruled out.
5.4 Trans-acting NRPS enzymes
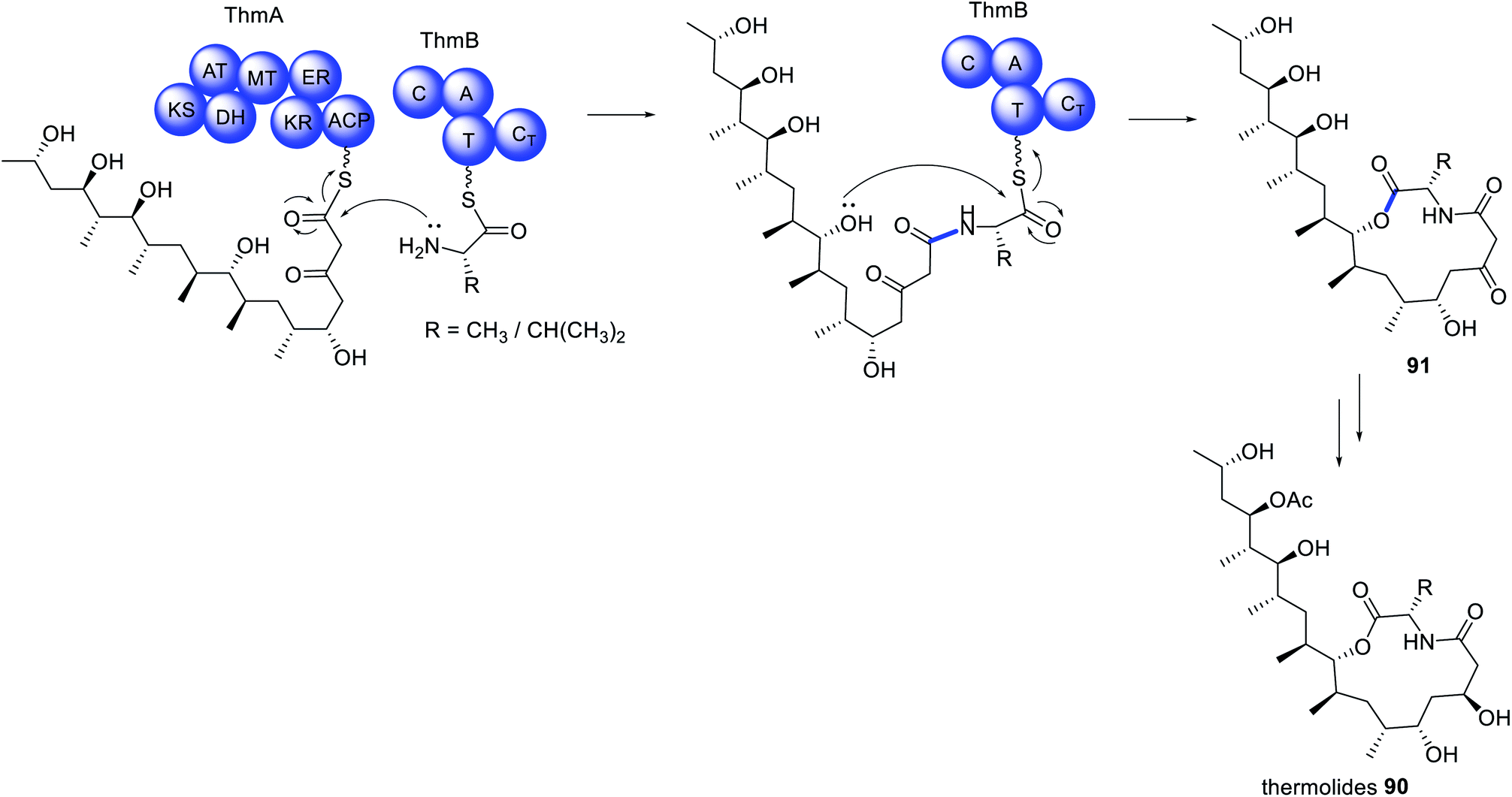
The initial report on the biosynthesis of the thermolides e.g.90 was attributed to a biosynthetic gene cluster encoding a PKS–NRPS enzyme and several accessory enzymes.170 However, a number of anomalies were noticed, leading to re-examination of the genome of Talaromyces thermophilus NRRL 2155. A second cluster encoding a PKS/NRPS was identified, however rather than discovering a HRPKS fused to a NRPS, two separate enzymes were predicted.171 Further examination of the NRPS indicated a domain organization that deviates from those typically found in fungal PKS–NRPS. Instead, a C–A–T–CT domain organization was observed, with an unusual terminal condensation (CT) domain,171 in contrast to the more common reductase (R) or Dieckmann cyclase (DKC) domains.The newly discovered Thm cluster was confirmed through heterologous expression in A. nidulans, leading to detection of 90.171 Co-expression of just the HRPKS (ThmA) and the NRPS (ThmB) in A. nidulans produced 91 (Scheme 15). The same compound 91 could also be obtained from recombinant production of ThmA and ThmB in yeast, incubated with acetyl-CoA, malonyl-CoA, SAM, NADPH, MgCl2, L-Ala/L-Val, indicating that these two enzymes collaborate to synthesize a PKS–NRPS derived product.171 Further investigation determined that the terminal CT domain of ThmB catalyzes a macrocyclization reaction, rare in fungal PKS–NRPS pathways.
 | ||
| Scheme 15 Biosynthesis of the thermolides requires the collaborative action of a HRPKS and a collaborating NRPS. | ||
Similar collaborating HRPKS and NRPS pairs have also been identified in the biosynthesis of acurin A 92 (Scheme 16).17292 was isolated from A. aculeatus and, based on its structural similarities to the fungal mycotoxin fusarin C, was proposed to be synthesized via a PKS–NRPS enzyme. The biosynthetic gene was cluster confirmed as being responsible for the biosynthesis of 92via gene knock out experiments. The cluster encodes separate HRPKS (AcrA) and NRPS (AcrB) enzymes separated by 31 kb of DNA sequence.172 Individual deletion of AcrA and AcrB by CRISPR/Cas9 led to loss of 92, and no intermediates were reported. Unlike the NRPS ThmB, AcrB contains a common terminal R domain, and homologs of the genes encoding AcrA and AcrB are found in nine other Aspergillus spp. indicating that the separation of these genes is not a result of errors in genome sequencing or annotation.172 The BGC encoding the biosynthesis of 92 also contains an α,β-hydrolase, AcrC, and disruption of the encoding gene did not reveal accumulation of intermediates, although this was observed for several other tailoring enzymes, therefore potential physical interactions between two or more tailoring enzymes are suggested.172
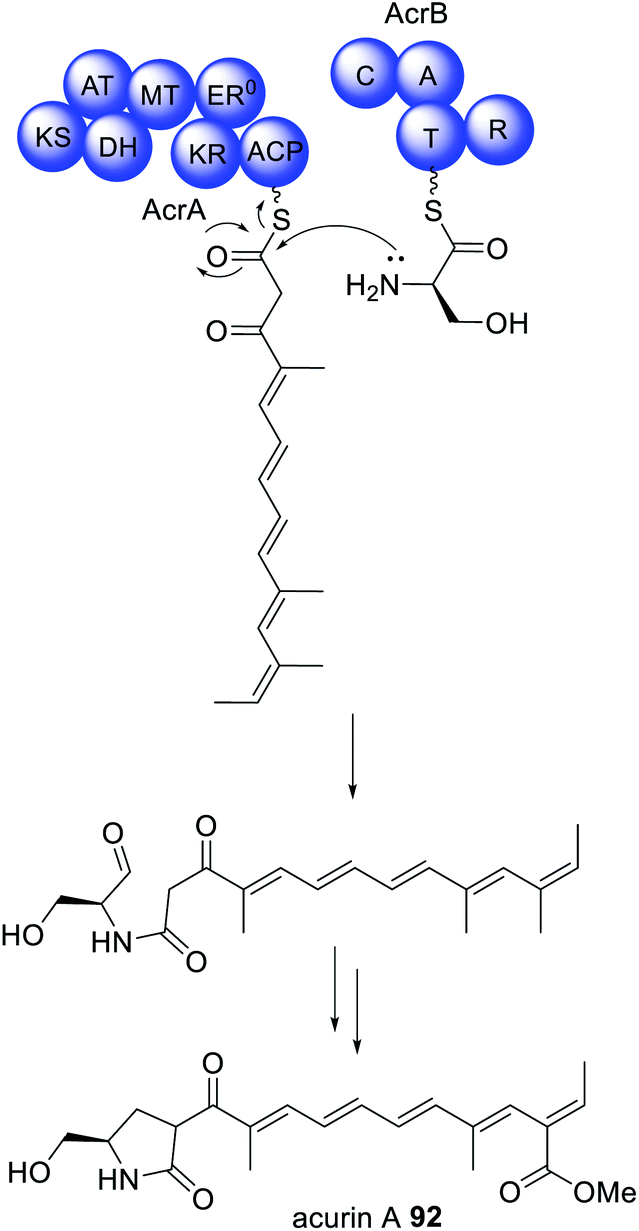 | ||
| Scheme 16 The biosynthesis of acurin A requires a trans-acting NRPS enzyme, AcrB. | ||
5.5 Trans-acting PLP-dependent enzymes
The infamous fumonisin mycotoxins derive from a polyketide fused to alanine, however not through amide bond formation nor by a PKS–NRPS enzyme.173 The biosynthesis requires a HRPKS enzyme, Fum1p, that lacks an obvious domain for polyketide release, similar to many other HRPKS (Scheme 17).174 Also contained within the fumonisin gene cluster is a gene encoding a protein that is a member of the 2-oxoamine synthase family, Fum8p. Fum8p is a pyridoxal 5′-phosphate (PLP)-dependent enzyme and, due to the diverse range of reactions catalyzed by PLP-dependent enzymes, was a candidate for being involved in decarboxylative condensation, and therefore polyketide release. Fum8p was heterologously expressed in yeast microsomes, incubated with recombinant Fum1p-ACP, PLP, C18-CoA, L-alanine, and shown to catalyze the PLP-dependent release of the C-18 chain from Fum1p-ACP to form 93.175 The assay also showed some tolerance of Fum8p towards C-16 and C-14 polyketide chains, although Fum8p clearly preferred the C-18 substrate.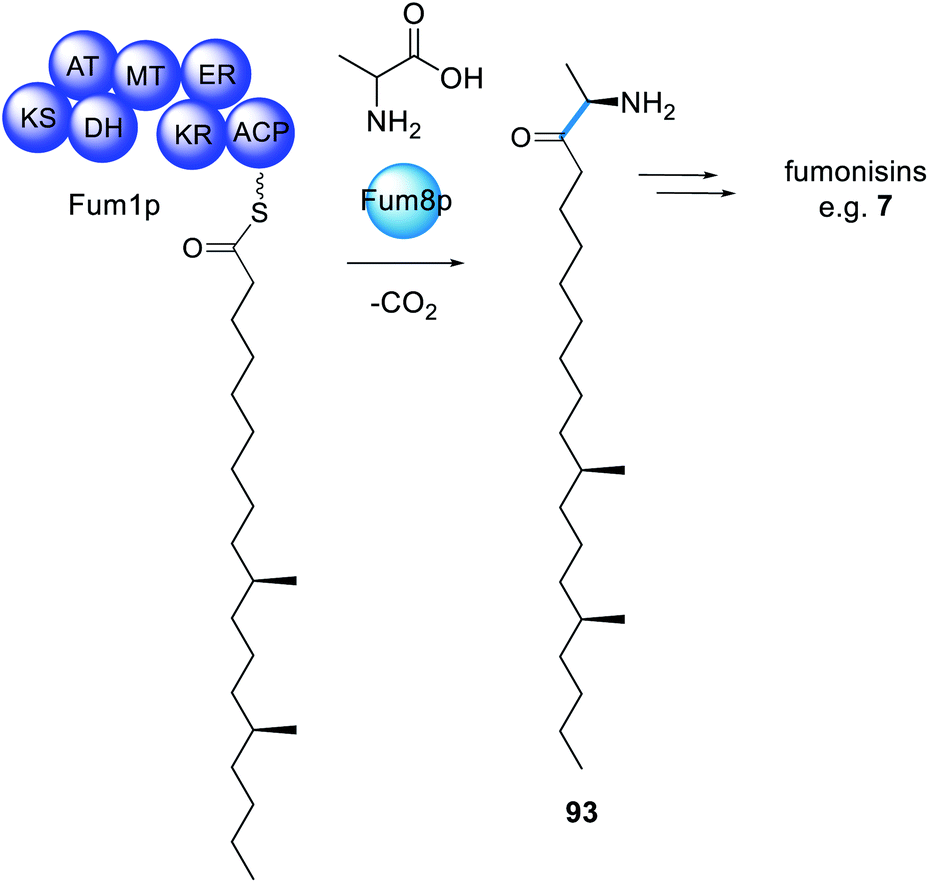 | ||
| Scheme 17 The biosynthesis of fumonisins requires a trans-acting PLP-dependent enzyme for polyketide extension and release. | ||
Similar to fumonisins, AAL-toxins e.g.94 (Fig. 22) also derive from a HRPKS condensed with an amino acid, however AAL-toxins comprise of a C-16 polyketide chain, synthesized by Alt1p, fused with glycine. The AAL-toxin gene cluster reportedly also contains an α-oxoamine synthase type enzyme, Alt4p, proposed to function in a similar mechanism to Fum1p.176 The addition of an amino acid to a highly reduced polyketide chain shares similarities with the coupling of serine with palmitic acid in sphingosin biosynthesis.177
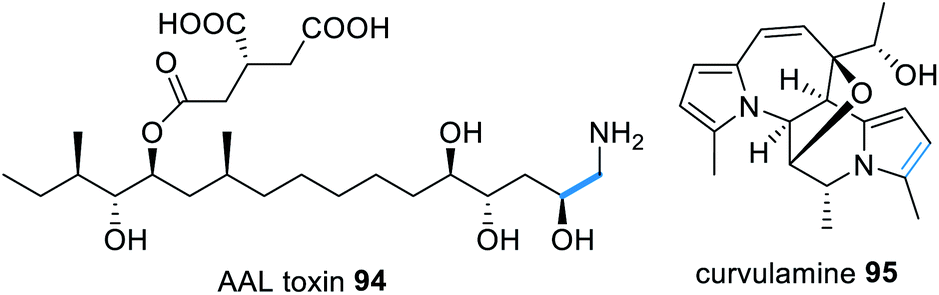 | ||
| Fig. 22 Examples of natural products that require a trans-acting PLP-dependent enzyme for polyketide release from a HRPKS. | ||
The biosynthesis of the indolizidine alkaloid curvulamine 95 (Fig. 22) also requires a trans-acting PLP-dependent enzyme, CuaB, for condensation with alanine, resulting in decarboxylative condensation.178 However CuaB is a bi-functional aminotransferase and also catalyzes α-hydroxlation of alanine. Using CuaB, fungal genome databases were mined identifying 10 BGCs suspected of encoding indolizidine alkaloids.178 Over-expression of a transcription factor within one of these cryptic clusters in Bipolaris maydis led to the identification of nine new indolizidine alkaloids, named bipolamines A–I.178
5.6 Trans-acting cyclases
The aurovertins e.g.96 (Fig. 23) are ATP inhibitors derived from a polyketide polyene α-pyrone. A BGC was discovered in Calcarisporium arbuscula that encodes the HRPKS (AurA) and a number of tailoring enzymes, including a protein, AurE, with sequence homology to bacterial aromatic polyketide cyclases.179 The cluster was investigated through gene knockout and heterologous expression in yeast. Inactivating AurE reduced the titers of polyketides obtained, whilst including AurE in heterologous expression experiments increased the rate of product release via pyrone formation. Although an exact function of AurE was not yet determined it seems likely that it collaborates with AurA to facilitate polyketide release.179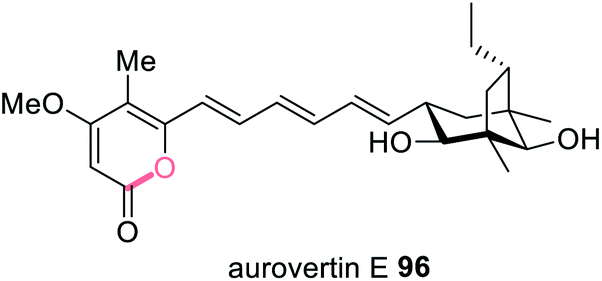 | ||
| Fig. 23 Structure of the ATP inhibitor aurovertin E 96 proposed to require an unusual collaborating cyclase. | ||
6. Trans-acting oxidases
In addition to trans-acting enzymes that appear to introduce functionality reminiscent of missing or inactive PKS domains, are trans-acting tailoring enzymes that introduce specific functionality that PKS enzymes are unable to. For example, in the biosynthesis of TAN-1612 97, a tetracycline-like compound synthesized by the NRPKS AdaA, two trans-acting enzymes are required; the trans-acting TE, AdaB, and the flavin-dependent monooxygenase (FMO), AdaC.180 When AdaA and AdaB were co-expressed without AdaC, a tricyclic anthracenone 98 was observed (Scheme 18). 98 is an off-pathway shunt intermediate arising from hydrolysis that is not converted to the final metabolite 97. Similarly, when AdaA and AdaC were co-expressed only trace amounts of intermediate 98 was detected. The biosynthesis of 97 requires FMO AdaC to hydroxylate the C-2 position of the polyketide intermediate while it is still tethered to AdaA and this site-specific hydroxylation enables the trans-acting TE, AdaB, to function correctly to release 99.180 Finally, the O-methyltransferase AdaD acts on the released polyketide product 99 to complete the biosynthesis of 97 (Scheme 18).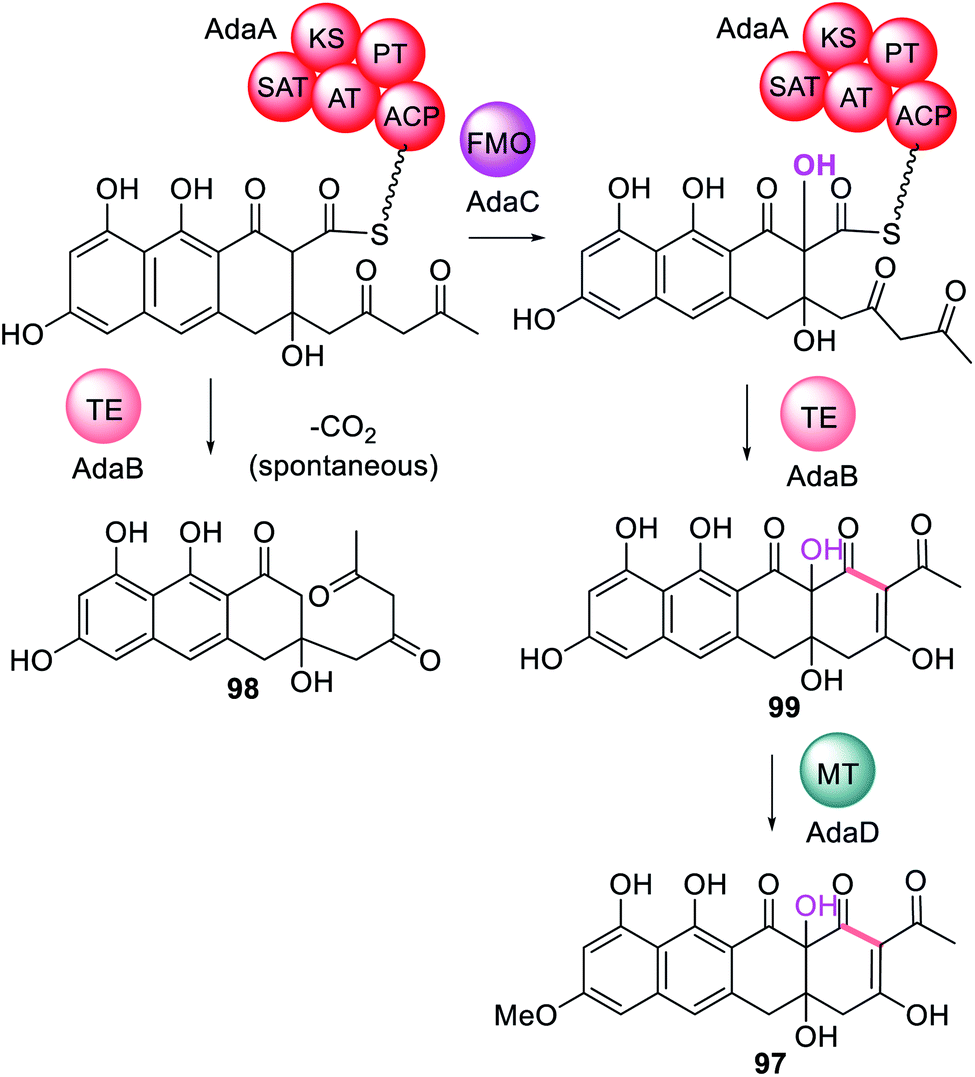 | ||
| Scheme 18 The biosynthesis of TAN-1612 97 requires the collaborative action of the trans-acting FMO, AdaC, followed by release via the trans-acting TE, AdaB. Only chemical modifications introduced by the trans-acting enzymes are highlighted in colour. | ||
The three gene cassette encoding an NRPKS, FMO and TE has also been identified in other fungi that have been confirmed as producing polycyclic polyketide products with a C-2 hydroxylation including asperthecin 60 and viridicatumtoxin 13 (Fig. 24).180 Currently no structural information is available to understand the potential protein–protein interactions between these three enzymes.
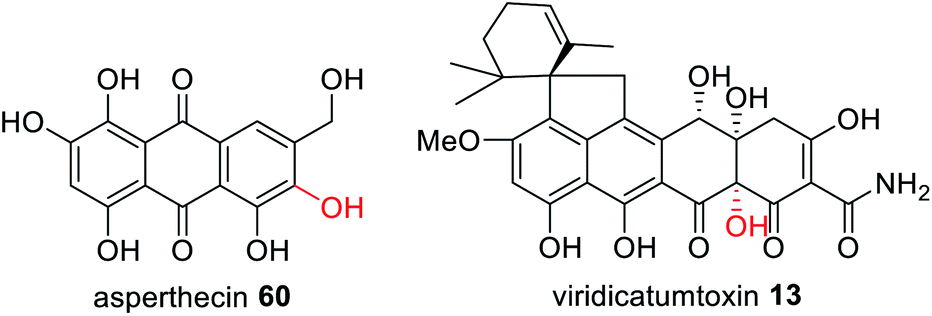 | ||
| Fig. 24 Examples of fungal polyketides that utilize a trans-acting FMO for correct polyketide release from the NRPKS. | ||
The biosynthesis of oxaleimide 100 requires the action of the trans-acting cytochrome P450 (PoxM) to hydroxylate the polyketide intermediate tethered to the HRPKS (PoxF) prior to release of the carboxylic acid product 101.181 This was confirmed via heterologous expression in A. nidulans since expression of PoxF alone did not yield any detectable product. PoxM is an iterative P450 and further oxidizes 101 to 102 after release from PoxF (Scheme 19).181
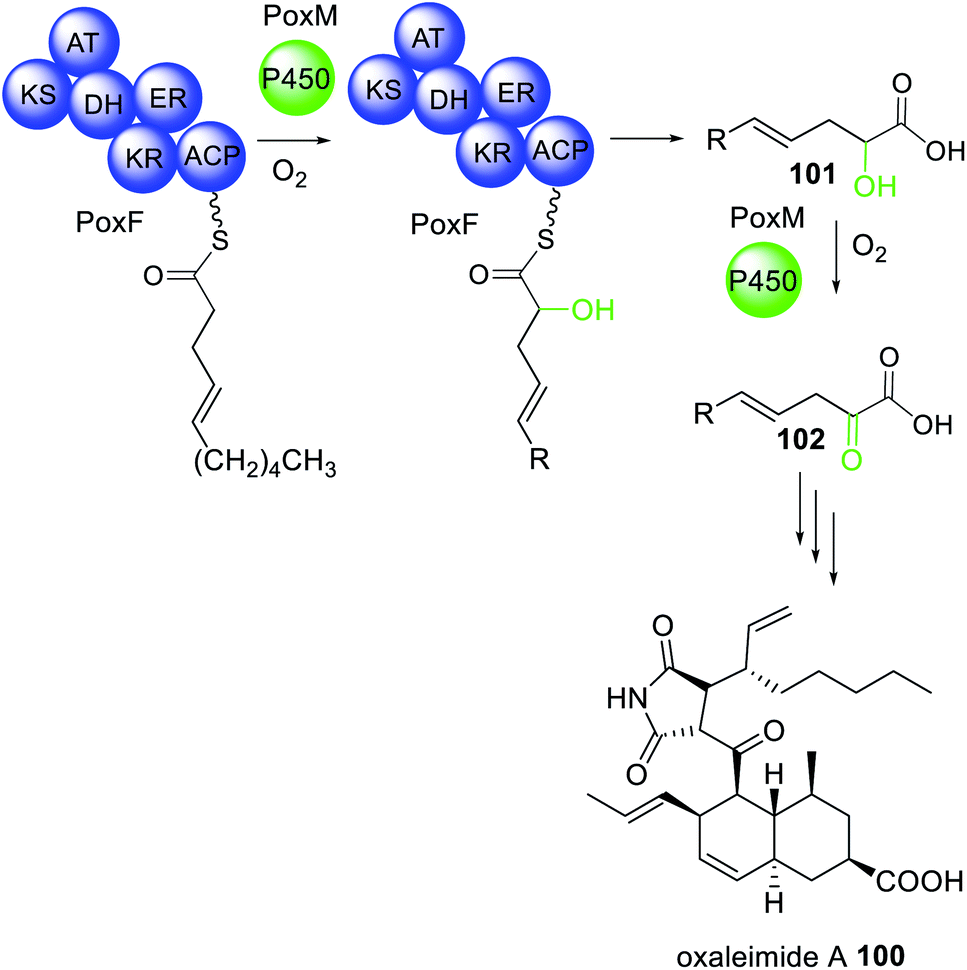 | ||
| Scheme 19 The biosynthesis of oxaleimide 100 requires the action of the trans-acting P450 PoxM. Chemical modifications introduced by the trans-acting P450 are highlighted in light green. | ||
A trans-acting P450 has recently been proposed in the biosynthesis of the C4-alkylated dihydroisocoumarin 103.167 The gene cluster responsible for the biosynthesis of 103 encodes: an NRPKS (AcreC), a P450 (AcreB), an O-methyltransferase (AcreA), and a hydrolase (AcreD). Heterologous expression of AcreA–D in A. nidulans resulted in formation of 103, as expected (Scheme 20).182 Investigating AcreA, B and D in vitro revealed that AcreB did not hydroxylate the NRPKS product 104. To exclude the possibly that AcreB was inactive/misfolded, 104 was fed to an A. nidulans strains expressing only AcreB, however no conversion to 105 was detected. As a control, AcreA expressed in A. nidulans could convert 104 to 106, confirming that 104 can penetrate the cell wall, but is not a substrate for AcreB. Therefore, the P450, AcreB, appears to act in-trans hydroxylating the polyketide chain still tethered to AcreC.182 Genome mining identified gene clusters encoding homologous NRPKS, P450, and hydrolase in other fungi, however the polyketide products are unknown.182
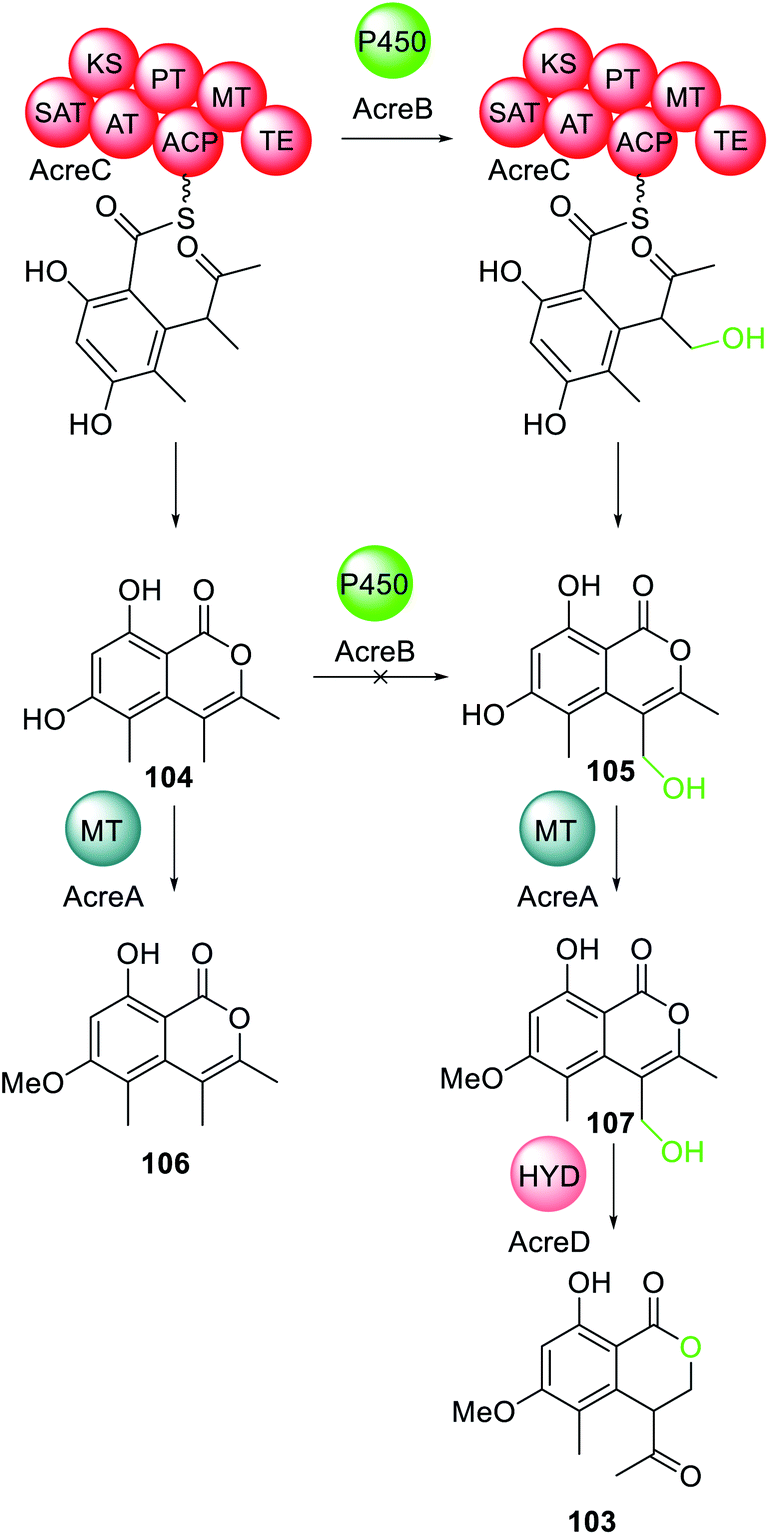 | ||
| Scheme 20 The biosynthesis of 103 requires the action of the trans-acting P450 AcreB. Chemical modifications introduced by the trans-acting P450 is highlighted in light green. | ||
These examples of oxidases hydroxylating intermediates tethered to the PKS are similar to the proposal of the role of the P450 in the biosynthesis of brefeldin 67. Therefore, this type of collaboration between a PKS and oxidases may be more frequent than anticipated. Although the oxidase is not involved in chain extension or chain release, it introduces a functional group that is essential for biosynthesis to proceed as expected and demonstrates the ever-evolving nature of these pathways.
7. Biosynthetic pathways utilizing multiple trans-acting and collaborating enzymes
With an increasing number of fungal biosynthetic pathways being fully interrogated, using both in vivo and in vitro approaches, it is clear that trans-acting and collaborating enzymes are a recurrent feature and often more than one trans-acting enzyme can be found in a specific pathway. For example, the biosynthesis of TAN-1612 97 (Scheme 18) requires hydroxylation by the trans-acting FMO (AdaC) to facilitate release of the correct polyketide intermediate by the trans-acting TE (AdaB).180 As might be expected, where biosynthetic pathways require more than one PKS megasynthase that collaborate with trans-acting partners the number of protein–protein interactions increase significantly.The biosynthesis of lovastatin 1 requires two HRPKS, LNKS and LDKS, and three trans-acting enzymes, LovC, LovG and LovD (Scheme 21A).5,77,133,183 Without the trans-ER, LovC, LNKS is unable to synthesize the required polyketide intermediates and without the trans-acting thiolesterase, LovG, LNKS is unable to release the polyketide product 25. Similarly, LDKS requires the trans-acting AT to transfer its diketide intermediate to 81 resulting in formation of lovastatin acid 82.
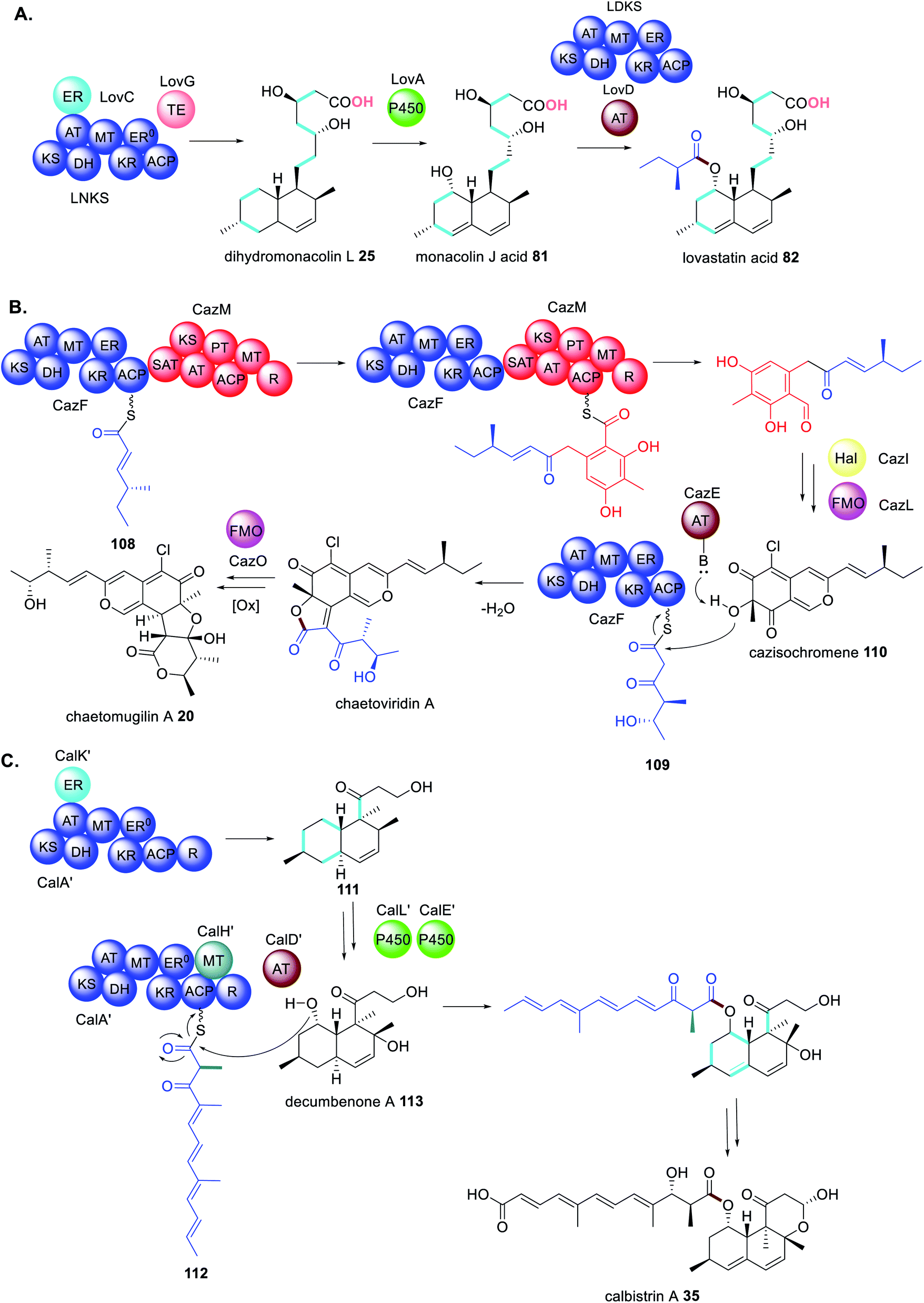 | ||
| Scheme 21 Biosynthetic pathways using multiple trans-acting or collaborating enzymes (A) lovastatin acid 82, (B) chaetomugilin A 20 and (C) calbistrin A 35. Chemical modifications introduced by the trans-acting enzymes are highlighted in colour. | ||
Likewise, the biosynthesis of chaetomugilin A 20 requires the collaborating HRPKS and NRPKS enzymes, CazF and CazM, in addition to a trans-acting AT (CazE).70,184 However, the main reason for the complexity in this pathway is that CazF is capable of synthesizing two different triketides 108 and 109; 108 is transferred to CazM, and 109 is transferred by CazE to the modified product of CazF/CazM cazisochromene 110 (Scheme 21B).169 This is a revealing example of collaborating enzymes competing for substrates and altering the product profile of a megasynthase.
The biosynthesis of calbistrin A 35 also demonstrates how competing trans-acting enzymes can alter the product profile of a megasynthase and generate highly complex polyketide products.113 Unlike chaetomugilin A 20, calbistrin A 35 requires only a single HRPKS CalA′. If CalA′ collaborates with the trans-acting ER CalK′ a decalin product 111 is formed. However, if CalA′ collaborates with the trans-acting MT CalH′ this abolishes the collaboration with CalK′ and a shorter polyketide with a different methylation 112 pattern is synthesized (Scheme 21C).113 Intriguingly the reductase (R) domain of CalA′ that is clearly active, no longer functions when CalH′ acts and instead the trans-acting AT CalD′ releases the polyene polyketide product, coupling it with the modified polyketide product 113 of CalA′/CalK′.113
In the biosynthesis of oxaleimide 100 a HRPKS (PoxF) collaborates with the trans-acting P450 (PoxM) to synthesize the precursor 102 of the amino acid substrate 114 utilized by the PKS–NRPS (PoxE) (Scheme 22A).181 As is common for PKS–NRPS PoxE collaborates with the trans-acting ER (PoxP) and it appears that the Diels-Alderase PoxQ, is able to act on the polyketide chain 115 still tethered to PoxE.181
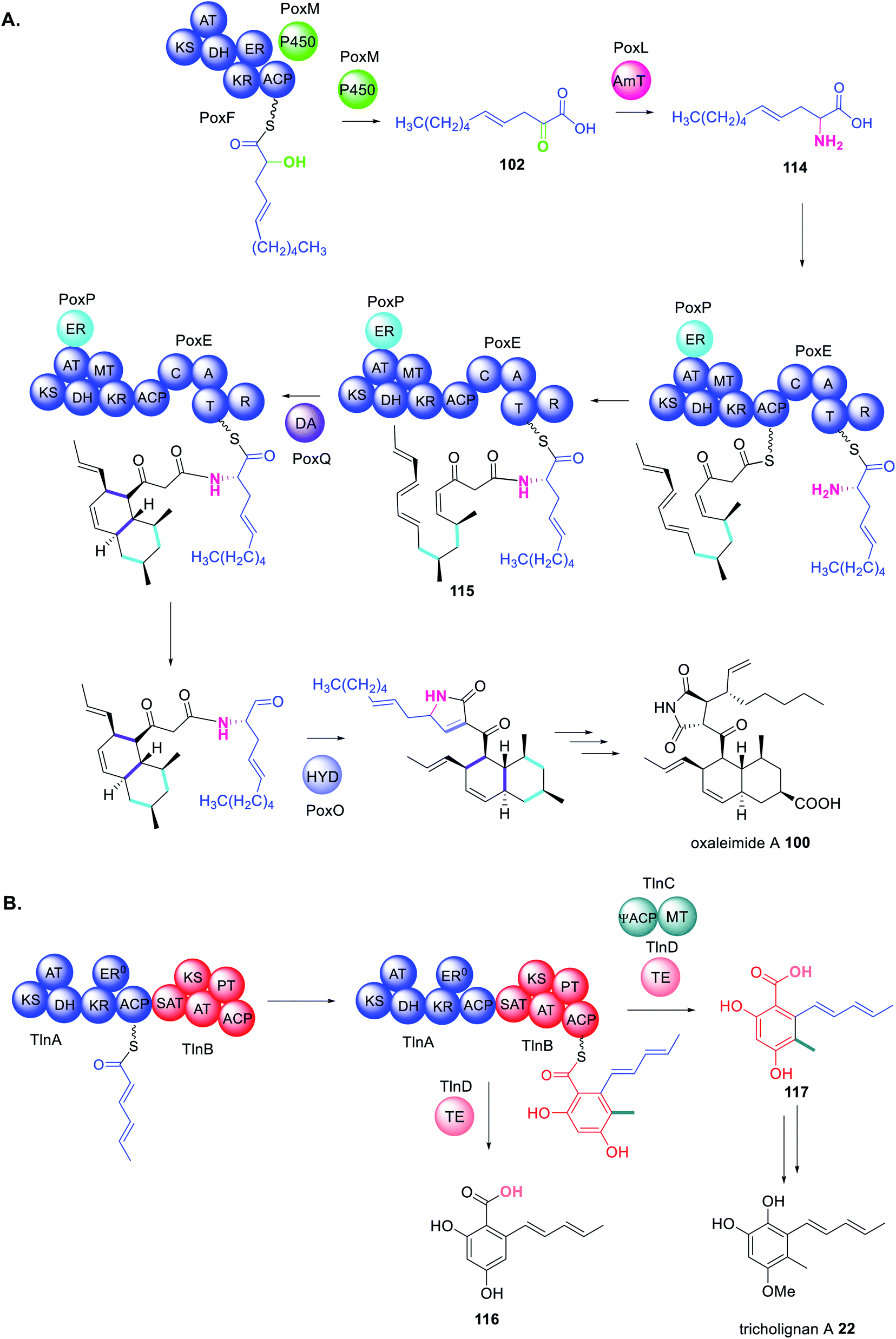 | ||
| Scheme 22 Biosynthetic pathways using multiple trans-acting or collaborating enzymes (A) oxaleimide A 100 and (B) tricholognan A 22. Chemical modifications introduced by the trans-acting enzymes are highlighted in colour. | ||
Finally, the biosynthesis of tricholignan A 22 requires a HRPKS and NRPKS pair, TlnA and TlnB, that collaborate with trans-acting MT and TE domains, TlnC and TlnC respectively, in a highly unusual biosynthetic pathway (Scheme 22B).72 TlnC is a didomain protein consisting of an inactive ACP (ψACP) fused to a MT domain, reminiscent of the organization of a truncated NRPKS. Investigating the stand alone TlnC-MT domain in vitro indicated that the TlnC-ψACP is required to increase the efficiency of the methylation step.72 When TlnC-ψACP is activated by introducing the active site Ser residue a non-methylated pentaketide 116 is observed, instead of 117, indicating that an active TlnC-ACP inhibits TlnC-MT. Together these findings indicate protein–protein interactions between TlnC and TlnB to facilitate methylation during the third round of chain extension i.e. on the tetraketide intermediate.72 Domain deconstruction experiments using TlnB–SAT–KS–AT0, TlnB–ACP, TlnC, and a tetraketide mimic demonstrated that higher concentrations of TlnC inhibited chain elongation. The TlnC-ψACP domain contains a number of negatively charged residues throughout the first half of its sequence, similar to other ACPs.72
TlnC is therefore proposed to form electrostatic protein–protein interactions with the KS domain of TlnB enabling TlnC to intercept the tetraketide and methylate before the tetraketide is shuttled to the KS domain for elongation.72
These examples reveal the high complexity of these pathways and the precise co-ordination between multiple enzymes. They also demonstrate how thoroughly fungal biosynthetic pathways may need to be interrogated in order to fully understand how some of the first enzyme-free intermediates are produced. Although the pathway genes encoding the biosynthetic enzymes are clustered, the genes encoding trans-acting enzymes are not always adjacent to each other within the BGC. This review is intended to categorize the many examples of trans-acting enzymes and act as a guide when considering pathway engineering.
8. Biosynthetic pathways engineering opportunities
Due to the importance of natural products in the pharmaceutical industry, and the declining rate of discovery of novel antibiotics entering the market, methods to engineer analogues of bioactive molecules with improved properties may be desirable. Without a thorough understanding of the elaborate network of collaborating megasynthases and trans-acting accessory enzymes, this may be difficult to achieve. Furthermore, the process will be neither quick nor easy as firstly BGCs have to be fully dissected/reassembled, potential collaborations identified and interrogated, and eventually novel collaborations engineered.Because there are so few structures of fungal PKS and collaborating enzymes reported, compared to the number of fungal polyketides discovered, the majority of engineering attempts have been curiosity driven in trying to understand the enzymes' tolerance rather than the nature of the collaboration specifically. For example, when studying how HRPKS collaborate with NRPKS during benzenediol lactone biosynthesis using phylogenetically diverse donors, it was determined that generally the NRPKS enzyme as a whole could tolerate unnatural starter units leading to product diversity (Scheme 23A).185 However, in many cases the SAT domain of the NRPKS acceptor needed to be swapped to ensure efficient substrate transfer,186 indicating that protein–protein interactions are just as important a consideration as the substrate tolerance of the enzyme being investigated.
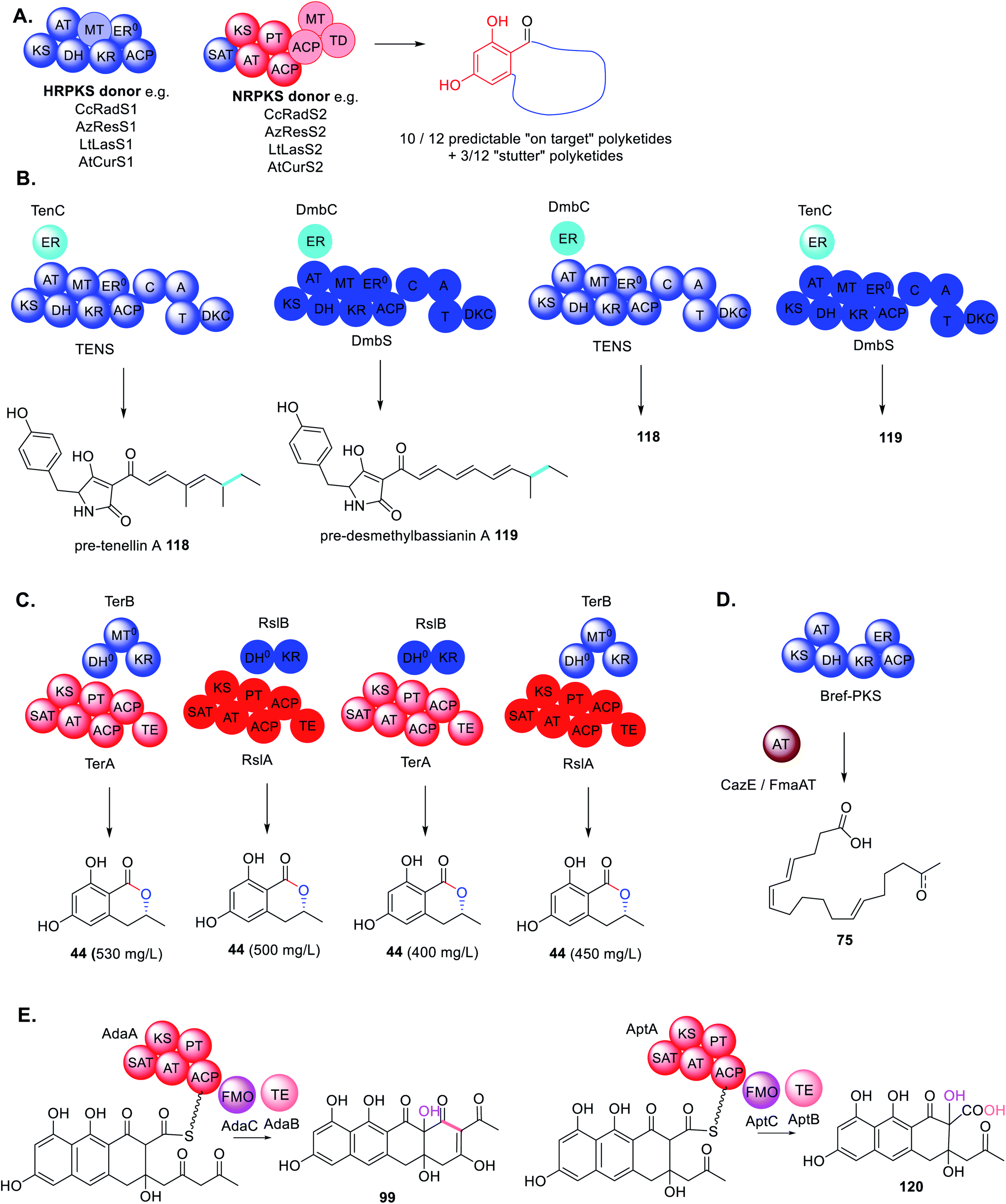 | ||
| Scheme 23 Summary of results of combinations of trans-acting enzymes with non-native PKS enzymes. (A) The investigation of HRPKS and NRPKS shuffling; (B) trans-ER swaps in closely related PKS–NRPS; (C) swaps between PKS-like enzymes TerB and RslB; (D) substituting trans-acting thiolhydrolase BrefTH with trans-acting acyltransferases; (E) shuffling trans-acting TE and FMOs with NRPKS that produce related polyketides but with different chain lengths. | ||
Similarly, when investigating the programming of PKS–NRPS and their trans-ER partners, it was demonstrated that trans-ER enzymes could be swapped between closely related BGCs (∼90% sequence identity).187 The swaps had no effect on the structure of the final molecules 118 and 119 as both trans-ER catalyzed the same chemistry on the same substrate (Scheme 23B). However, in the absence of the trans-ER a shunt product was observed confirming the essential role of this trans-acting enzyme in PKS programming.76
Likewise, when investigating the trans-acting KR enzyme involved biosynthesis of 44, homologous enzymes from the terrein and roussoellatide pathways could be swapped without adverse effects on titers 44 (Scheme 23C).118 Although not necessarily surprising due to these enzymes catalyzing identical chemistry, the enzymes are quite different in that RslB is 300aa shorter that TerB and does not contain the MT0 domain.118
To understand Bref-TH in the biosynthesis of 75 and determine its precise role in chain length control, the acyltransferases CazE and Fma-AT, from the chaetomugilin A 20 and fumagillin 3 pathways respectively, were substituted for Bref-TH (Scheme 23D).139 Both of these acyltransferases are trans-acting and confirmed as being involved in polyketide chain release from their partner HRPKS.70,160 However, the major products remained nonaketides, similar to Bref-PKS alone. Considering that only Bref-TH is able to compete with the KS domain demonstrates highly specific protein–protein interactions with Bref-PKS.
The roles of the trans-acting oxidase (AdaC) and thioesterase (AdaB) in the biosynthesis of 97 were studied by combinatorial biosynthesis with the homologous enzymes required for the biosynthesis of asperthecin 60, AptC and AptB, respectively (Scheme 23E).180 The precursors to 97 and 60, 99 and 120 respectively, differ in that 99 is a decaketide and 120 is a nonaketide, however both molecules are hydroxylated at position C-2. Combination of AptA with AdaB and AdaC led to formation of the expected product 120 in similar titers to the natural combination of AptABC. In contrast combination of AdaA with AptB and AptC also formed the expected product 120 but with significantly lower titers, indicating a less efficient collaboration.180
Finally, there are several reports of deconstructed NRPKS domains that are able to interact with type II PKS enzymes from bacteria (Scheme 24).188 For example, PKS4 from Gibberella fujikuroi synthesizes the aromatic polyketide SMA76a 121 in E. coli (Scheme 24A) yet when the TE domain is removed the product profile changes to SMA93 122 (Scheme 24B).189 Constructing an artificial PKS (PKS_WJ) consisting of unnatural fused KS–AT–ACP domains enabled the synthesis of napthopyrone 123 (Scheme 24C) which was the identical product of the three domains individually expressed in E. coli.188
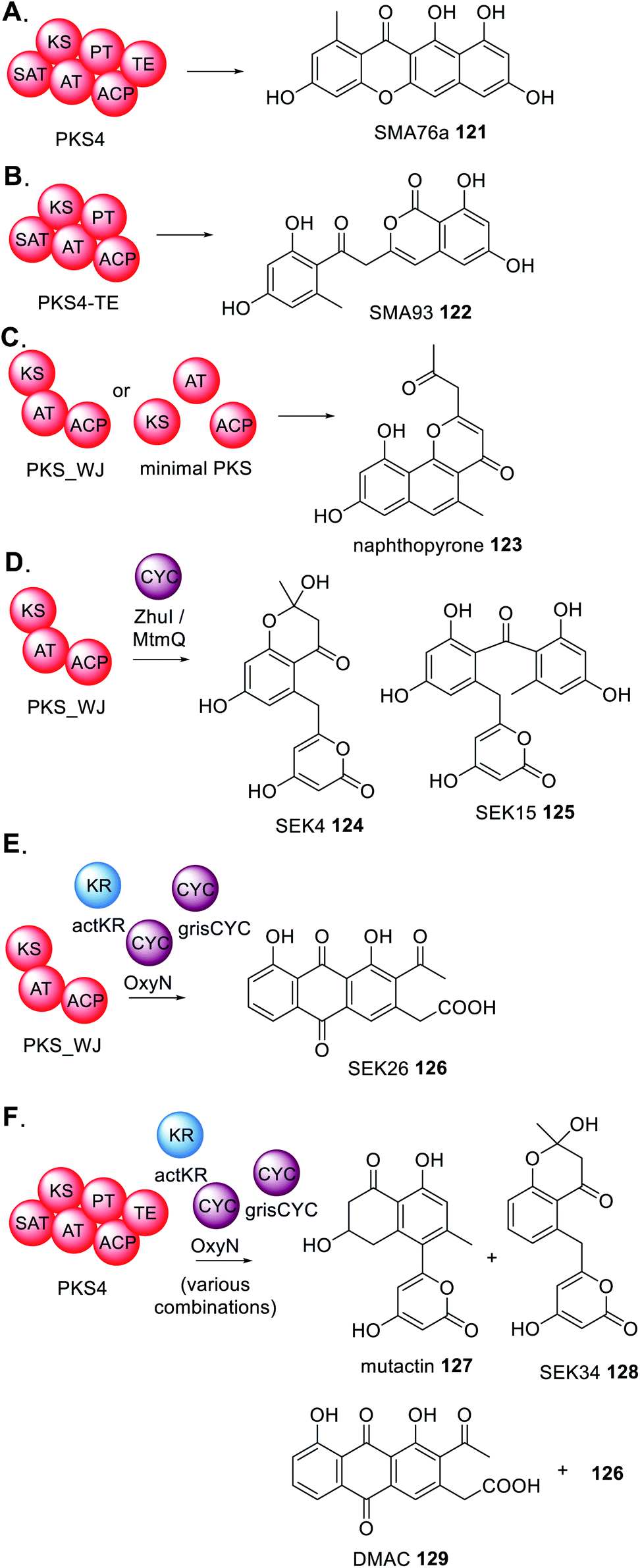 | ||
| Scheme 24 Summary of experiments investigating the potential collaborations between fungal PKS and PKS modification enzymes from bacteria. | ||
The addition of bacterial cyclases (CYC) e.g. ZhuI or MtmQ, caused a significant change in product profile to 124 and 125 (Scheme 24D) indicating that these cyclases can collaborate with the fungal enzymes and introduce programmed cyclization and re-direct the biosynthesis to compounds typically found in bacteria.189 The study was expanded to include cyclase enzymes from different bacteria that introduce different modes of cyclization, and a trans-acting KR. Again, these bacterial enzymes collaborated with PKS-WJ, this time synthesizing SEK26 126 (Scheme 24E).189 Furthermore, by studying the complete PKS4 with various combinations of bacterial accessory enzymes, it was demonstrated that the bacterial enzymes are capable of competing with the functional PT domain of PKS4 and diverting the product profile to generate mutactin 127, SEK34 128, DMAC 129 and 126, polyketides typically found in bacteria (Scheme 24F).189
Although these PKS enzyme are very distantly related, the range of substrate flexibility tolerated by the PKS enables successful engineering. However, in all examples given, the driving factor appears to be appropriate protein–protein interactions that orientate both enzymes close enough that they have access to the polyketide intermediates. A similar observation has been made when engineering chimeric modular PKS enzymes; protein–protein interactions between domains is a more significant factor to consider than enzyme–substrate recognition.190
9. Conclusions and future prospects
For the future investigation and engineering of fungal PKS enzymes, including combinatorial biosynthesis, researchers must consider that these multifaceted systems may require considerable forethought into the variables. This review collates the various classes of collaborating and trans-acting enzymes that have been identified in fungal polyketide biosynthesis demonstrating some common themes and also some rare examples. As more pathways are interrogated we will gain an understanding of which collaborations are the “rule” and which are the exceptions.Of the collaborations discovered, still very little is known about how these enzymes interact. For example, it is unknown whether all interactions are a result of protein–protein interactions or mere physical proximity of enzymes. As more information is gained from pathway elucidation combined with structural biology approaches it may be possible to rationally engineer these complex systems to generate novel bioactive molecules with improved properties.
10. Conflicts of interest
There are no conflicts to declare.11. Acknowledgements
Protein structures were visualized and manipulated using the Mol* Viewer.19112. References
- R. J. Cox and E. J. Skellam, Fungal Non-Reducing Polyketide Synthases, Comprehensive Natural Products III: Chemistry and Biology, 2020, vol 1, pp. 266–309 Search PubMed.
- J. C. Frisvad, T. Isbrandt and T. O. Larsen, Fungal Partially Reducing Polyketides and Related Natural Products From Aspergillus, Penicillium, and Talaromyces, Comprehensive Natural Products III: Chemistry and Biology, 2020, vol. 1, pp. 313–332 Search PubMed.
- E. B. Go and Y. Tang, Fungal Highly Reducing Polyketide Synthases and Associated Natural Products, Comprehensive Natural Products III: Chemistry and Biology, 2020, vol. 1, pp. 333–364 Search PubMed.
- H. Li, T. J. Booth and Y.-H. Chooi, Fungal Polyketide-Non-ribosomal Peptide Synthetases and Their Associated Natural Products, Comprehensive Natural Products III: Chemistry and Biology, 2020, vol. 1, pp. 415–442 Search PubMed.
- X. Xie, M. J. Meehan, W. Xu, P. C. Dorrestein and Y. Tang, J. Am. Chem. Soc., 2009, 131, 8388–8389 CrossRef CAS PubMed.
- C.-S. Yun, T. Motoyama and H. Osada, Nat. Commun., 2015, 6, 8758 CrossRef CAS PubMed.
- F. Luo, S. Hong, B. Chen, Y. Yin, G. Tang, F. Hu, H. Zhang and C. Wang, ACS Chem. Biol., 2020, 15, 2476–2484 CrossRef CAS PubMed.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2020–2026 Search PubMed.
- Y.-H. Chooi and Y. Tang, J. Org. Chem., 2012, 77, 9933–9953 CrossRef CAS PubMed.
- Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS PubMed.
- N. Funa, T. Awakawa and S. Horinouchi, J. Biol. Chem., 2007, 282, 14476–14481 CrossRef CAS PubMed.
- J. C. Navarro-Munoz and J. Collemare, Front. Microbiol., 2020, 10, 3018 CrossRef PubMed.
- T. Moriguchi, Y. Kezuka, T. Nonaka, Y. Ebizuka and I. Fujii, J. Biol. Chem., 2010, 285, 15637–15643 CrossRef CAS PubMed.
- D. A. Herbst, C. A. Townsend and T. Maier, Nat. Prod. Rep., 2018, 35, 1046–1069 RSC.
- J. M. Crawford, B. C. R. Dancy, E. A. Hill, D. W. Udwary and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16728–16733 CrossRef CAS PubMed.
- J. M. Crawford, T. P. Korman, J. W. Labonte, A. L. Vagstad, E. A. Hill, O. Kamari-Bidkorpeh, S.-C. Tsai and C. A. Townsend, Nature, 2009, 461, 1139–1143 CrossRef CAS PubMed.
- T. P. Korman, J. M. Crawford, J. W. Labonte, A. G. Newman, J. Wong, C. A. Townsend and S.-C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6246–6251 CrossRef CAS PubMed.
- I. Fujii, A. Watanabe, U. Sankawa and Y. Ebizuka, Chem. Biol., 2001, 8, 189–197 CrossRef CAS PubMed.
- A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, Chem. Commun., 2007, 4053–4055 RSC.
- J. T. Kealey, L. Liu, D. V. Santi, M. C. Betlach and P. J. Barr, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 505–509 CrossRef CAS PubMed.
- B. Li, Y. Chen, Y. Zong, Y. Shang, Z. Zhang, X. Xu, X. Wang, M. Long and S. Tian, Environ. Microbiol., 2019, 21, 1124–1139 CrossRef CAS PubMed.
- Y.-H. Chooi, C. Krill, R. A Barrow, S. Chen, R. Trengove, R. P. Oliver and P. S. Solomon, Appl. Environ. Microbiol., 2015, 81, 177–186 CrossRef PubMed.
- J. Wang, J. Liang, L. Chen, W. Zhang, L. Kong, C. Peng, C. Su, Y. Tang, Z. Deng and Z. Wang, Nat. Commun., 2021, 12, 867 CrossRef CAS PubMed.
- T. J. Simpson, Nat. Prod. Rep., 2014, 31, 1247–1252 RSC.
- E. Skellam, Trends Biotechnol., 2019, 37, 416–427 CrossRef CAS PubMed.
- J. M. Winter, D. Cascio, D. Dietrich, M. Sato, K. Watanabe, M. R. Sawaya, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 9885–9893 CrossRef CAS PubMed.
- P. Wattana-amorn, C. Williams, E. Ploskon, R. J. Cox, T. J. Simpson, J. Crosby and M. P. Crump, Biochem, 2010, 49, 2186–2193 CrossRef CAS PubMed.
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 1315–1322 CrossRef CAS PubMed.
- J. K. Stoops, M. J. Arslanian, Y. H. Oh, K. C. Aune, T. C. Vanaman and S. J. Wakil, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 1940–1944 CrossRef CAS PubMed.
- A. R. Hawkins, J. D. Moore and A. M. Adeokun, Biochem. J., 1993, 296, 451–457 CrossRef CAS PubMed.
- A. Nivina, K. P. Yuet, J. Hsu and C. Khosla, Chem. Rev., 2019, 119, 12524–12547 CrossRef CAS PubMed.
- H. S. T. Bukhari, R. P. Jakob and T. Maier, Structure, 2014, 22, 1775–1785 CrossRef CAS PubMed.
- H. Jenke-Kodama, A. Sandmann, R. Mueller and E. Dittmann, Mol. Biol. Evol., 2005, 22, 2027–2039 CrossRef CAS PubMed.
- J. M. Crawford and C. A. Townsend, Nat. Rev. Microbiol., 2010, 12, 879–889 CrossRef PubMed.
- P. A. Srere, Trends Biochem. Sci., 1985, 10, 109–110 CrossRef.
- P. A. Srere, Annu. Rev. Biochem., 1987, 56, 89–124 CrossRef CAS PubMed.
- B. Bulutoglu, K. E. Garcia, F. Wu, S. D. Minteer and S. Banta, ACS Chem. Biol., 2016, 11, 2847–2853 CrossRef CAS PubMed.
- S. Shuib, I. Ibrahim, M. M. Mackeen, C. Ratledge and A. A. Hamid, Sci. Rep., 2018, 8, 3077 CrossRef PubMed.
- K. S. F. e Silva, R. M. Lima, L. C. Baeza, P. de Sousa Lima, T. de Moura Cordeiro, S. Charneau, R. A. da Silva, C. M. de Almeida Soares and M. Pereira, Front. Microbiol., 2019, 10, 1537 CrossRef PubMed.
- F. Wu and S. Minteer, Angew. Chem., Int. Ed., 2015, 54, 1851–1854 CrossRef CAS PubMed.
- Y. Zhang, K. F. M. Beard, C. Swart, S. Bergmann, I. Krahnert, Z. Nikoloski, A. Graf, R. G. Ratcliffe, L. J. Sweetlove, A. R. Fernie and T. Obata, Nat. Commun., 2017, 8, 15212 CrossRef CAS PubMed.
- T. Laursen, J. Borch-Jensen, C. Knudsen, K. Bavishi, F. Torta, H. J. Martens, D. Silvestro, N. S. Hatzakis, M. R. Wenk, T. R. Dafforn, C. E. Olsen, M. S. Motawia, B. Hamberger, B. L. Moller and J.-E. Bassard, Science, 2016, 354, 890–893 CrossRef CAS PubMed.
- T. Nakayama, S. Takahashi and T. Waki, Front. Plant Sci., 2019, 10, 821 CrossRef PubMed.
- S. Mucha, S. Heinzlmeir, V. Kriechbaumer, B. Strickland, C. Kirchhelle, M. Choudhary, N. Kowalski, R. Eichmann, R. Hueckelhoven, E. Grill, B. Kuster and E. Glawischnig, Plant Cell, 2019, 31, 2697–2710 CAS.
- M. Gou, X. Ran, D. W. Martin and C.-J. Liu, Nat. Plants, 2018, 4, 299–310 CrossRef CAS PubMed.
- J. Beld, E. C. Sonnenschein, C. R. Vickery, J. P. Noel and M. D. Burkart, Nat. Prod. Rep., 2104, 31, 61–108 RSC.
- H. Oberegger, M. Eisendle, M. Schrettl, S. Graessle and H. Haas, Curr. Genet., 2003, 44, 211–215 CrossRef CAS PubMed.
- J.-M. Kim, H.-Y. Song, H.-J. Choi, K.-K. So, D.-H. Kim, K.-S. Chae, D.-M. Han and K.-Y. Jahng, J. Microbiol., 2015, 53, 21–31 CrossRef CAS PubMed.
- D. Keszenman-Pereyra, S. Lawrence, M.-E. Twfieg, J. Price and G. Turner, Curr. Genet., 2003, 43, 186–190 CrossRef CAS PubMed.
- O. Marquez-Fernandez, A. Trigos, J. L. Ramos-Balderas, G. Viniegra-Gonzalez, H. B. Deising and J. Aguirre, Eukaryotic Cell, 2007, 6, 710–720 CrossRef CAS PubMed.
- J. M. Crawford, A. L. Vagstad, K. C. Ehrlich, D. W. Udwary and C. A. Townsend, ChemBioChem, 2008, 9, 1559–1563 CrossRef CAS PubMed.
- C. M. H. Watanabe and C. A. Townsend, Chem. Biol., 2002, 9, 981–988 CrossRef CAS PubMed.
- P. Mohr and C. Tamm, Tetrahedron, 1981, 37, 201–212 CrossRef.
- H. Tomoda, N. Tabata, Y. Nakata, H. Nishida, T. Kaneko, R. Obata, T. Sunazuka and S. Omura, J. Org. Chem., 1996, 61, 882–886 CrossRef CAS.
- Y.-H. Chooi, R. Cacho and Y. Tang, Chem. Biol., 2010, 17, 483–494 CrossRef CAS PubMed.
- N. Liu, Y.-S. Hung, S.-S. Gao, L. Hang, Y. Zou, Y.-H. Chooi and Y. Tang, Org. Lett., 2017, 19, 3560–3563 CrossRef CAS PubMed.
- Z. Iqbal, L.-C. Han, A. M. Soares-Sello, R. Nofiani, G. Thormann, A. Zeeck, R. J. Cox, C. L. Willis and T. J. Simpson, Org. Biomol. Chem., 2018, 16, 5524–5532 RSC.
- J. M. Crawford, A. L. Vagstad, K. P. Whitworth and C. A. Townsend, ChemBioChem, 2008, 9, 1019–1023 CrossRef CAS PubMed.
- C. M. H. Watanabe, D. Wilson, J. E. Linz and C. A. Townsend, Chem. Biol., 1996, 3, 463–469 CrossRef CAS.
- J. Foulke-Abel and C. A. Townsend, ChemBioChem, 2012, 13, 1880–1884 CrossRef CAS PubMed.
- J. M. Crawford, A. L. Vagstad, K. C. Ehrlich and C. A. Townsend, Bioorg. Chem., 2008, 36, 16–22 CrossRef CAS PubMed.
- I. Gaffoor and F. Trail, Appl. Environ. Microbiol., 2006, 72, 1793–1799 CrossRef CAS PubMed.
- Y.-T. Kim, Y.-R. Lee, J. Jin, K.-H. Han, H. Kim, J.-C. Kim, T. Lee, S.-H. Yun and Y.-W. Lee, Mol. Microbiol., 2005, 58, 1102–1113 CrossRef CAS PubMed.
- H. Zhou, K. Qiao, Z. Gao, M. J. Meehan, J. W.-H. Li, X. Zhao, P. C. Dorrestein, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2010, 132, 4530–4531 CrossRef CAS PubMed.
- C. D. Reeves, Z. Hu, R. Reid and J. T. Kealey, Appl. Environ. Microbiol., 2008, 74, 5121–5129 CrossRef CAS PubMed.
- S. Wang, Y. Xu, E. A. Maine, E. M. K. Wijeratne, P. Espinosa-Artiles, A. A. L. Gunatilaka and I. Molnar, Chem. Biol., 2008, 15, 1328–1338 CrossRef CAS PubMed.
- H. Zhou, K. Qiao, Z. Gao, J. C. Vederas and Y. Tang, J. Biol. Chem., 2010, 285, 41412–41421 CrossRef CAS PubMed.
- Y. Xu, P. Espinosa-Artiles, V. Schubert, Y.-M. Xu, W. Zhang, M. Lin, A. A. L. Gunatilaka, R. Suessmuth and I. Molnar, Appl. Environ. Microbiol., 2013, 79, 2038–2047 CrossRef CAS PubMed.
- Y.-M. Chiang, E. Szewczyk, A. D. Davidson, N. Keller, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2009, 131, 2965–2970 CrossRef CAS PubMed.
- J. M. Winter, M. Sato, S. Sugimoto, G. Chiou, N. K. Garg and Y. Tang, J. Am. Chem. Soc., 2012, 134, 17900–17903 CrossRef CAS PubMed.
- L. Kahlert, E. F. Bassiony, R. J. Cox and E. J. Skellam, Angew. Chem., Int. Ed., 2020, 59, 5816–5822 CrossRef CAS PubMed.
- M. Chen, Q. Liu, S.-S. Gao, A. E. Young, S. E. Jacobsen and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5499–5504 CrossRef CAS PubMed.
- R. V. K. Cochrane, R. Sanichar, G. R. Lambkin, B. Reiz, W. Xu, Y. Tang and J. C. Vederas, Angew. Chem., Int. Ed., 2016, 55, 664–668 CrossRef CAS PubMed.
- W. Bunnak, P. Wonnapinij, A. Sriboonlert, C. M. Lazarus and P. Wattana-Amorn, Org. Biomol. Chem., 2019, 17, 374–379 RSC.
- Y. Xu, T. Zhou, P. Espinosa-Artiles, Y. Tang, J. Zhan and I. Molnar, ACS Chem. Biol., 2014, 9, 1119–1127 CrossRef CAS PubMed.
- D. A. Herbst, C. R. Huitt-Roehl, R. P. Jakob, J. M. Kravetz, P. A. Storm, J. R. Alley, C. A. Townsend and T. Maier, Nat. Chem. Biol., 2018, 14, 474–479 CrossRef CAS PubMed.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS PubMed.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS PubMed.
- U. Bat-Erdene, D. Kanayama, D. Tan, W. C. Turner, K. N. Houk, M. Ohashi and Y. Tang, J. Am. Chem. Soc., 2020, 142, 8550–8554 CrossRef CAS PubMed.
- S. Bergmann, J. Schuemann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS PubMed.
- L. Liu, J. Zhang, C. Chen, J. Teng, C. Wang and D. Luo, Fungal Genet. Biol., 2015, 81, 191–200 CrossRef CAS PubMed.
- J. W. Cary, V. Uka, Z. Han, D. Buyst, P. Y. Harris-Coward, K. C. Ehrlich, Q. Wei, D. Bhatnagar, P. F. Dowd, S. L. Martens, A. M. Calvo, J. C. Martins, L. Vanhaecke, T. Coenye, S. De Saeger and J. D. Di Mavungu, Fungal Genet. Biol., 2015, 81, 88–97 CrossRef CAS PubMed.
- T. Ugai, A. Minami, K. Gomi and H. Oikawa, Tetrahedron Lett., 2016, 57, 2793–2796 CrossRef CAS.
- Z. Zhang, C. S. Jamieson, Y.-L. Zhao, D. Li, M. Ohashi, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2019, 141, 5659–5663 CrossRef CAS PubMed.
- L. Li, M.-C. Tang, S. Tang, S. Gao, S. Soliman, L. Hang, W. Xu, T. Ye, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2018, 140, 2067–2071 CrossRef CAS PubMed.
- L. Li, P. Yu, M.-C. Tang, Y. Zou, S.-S. Gao, Y.-S. Hung, M. Zhao, K. Watanabe, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2016, 138, 15837–15840 CrossRef CAS PubMed.
- T. B. Kakule, S. Zhang, J. Zhan and E. W. Schmidt, Org. Lett., 2015, 17, 2295–2297 CrossRef CAS PubMed.
- D. Tan, C. S. Jamieson, M. Ohashi, M.-C. Tang, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2019, 141, 769–773 CrossRef CAS PubMed.
- N. Kato, T. Nogawa, R. Takita, K. Kinugasa, M. Kinai, M. Uchiyama, H. Osada and S. Takahashi, Angew. Chem., Int. Ed., 2018, 57, 9754–9758 CrossRef CAS PubMed.
- S. Janevska, B. Arndt, L. Baumann, L. H. Apken, L. M. M. Marques, H.-U. Humpf and B. Tudzinski, Toxins, 2017, 9, 126 CrossRef PubMed.
- Y. Abe, T. Suzuki, C. Ono, K. Iwamoto, M. Hosobuchi and H. Yoshikawa, Mol. Genet. Genomics, 2002, 267, 636–646 CrossRef CAS PubMed.
- K. Qiao, Y.-H. Chooi and Y. Tang, Metab. Eng., 2011, 13, 723–732 CrossRef CAS PubMed.
- Z. Song, W. Bakeer, J. W. Marshall, A. A. Yakasai, R. M. Khalid, J. Collemare, E. Skellam, D. Tharreau, M.-H. Lebrun, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Sci., 2015, 6, 4837–4845 RSC.
- M. L. Nielsen, T. Isbrandt, L. M. Petersen, U. H. Mortensen, M. R. Andersen, J. B. Hoof and T. O. Larsen, PLoS One, 2016, 11, e0161199 CrossRef PubMed.
- K. Ishiuchi, T. Nakazawa, F. Yagishita, T. Mino, H. Noguchi, K. Hotta and K. Watanabe, J. Am. Chem. Soc., 2013, 135, 7371–7377 CrossRef CAS PubMed.
- C. Wang, V. Hantke, R. J. Cox and E. Skellam, Org. Lett., 2019, 21, 4163–4167 CrossRef CAS PubMed.
- H. Li, H. Wei, J. Hu, E. Lacey, A. N. Sobolov, K. A. Stubbs, P. S. Solomon and Y.-H. Chooi, ACS Chem. Biol., 2020, 15, 226–233 CrossRef CAS PubMed.
- S. C. Heard, G. Wu and J. M. Winter, J. Antibiot., 2020, 73, 803–807 CrossRef CAS PubMed.
- M. Hashimoto, H. Kato, A. Katsuki, S. Tsukamoto and I. Fujii, ChemBioChem, 2018, 19, 535–539 CrossRef CAS PubMed.
- M. Ohashi, T. B. Kakule, M.-C. Tang, C. S. Jamieson, M. Liu, Y.-L. Zhao, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2021, 143, 5605–5609 CrossRef CAS PubMed.
- X.-L. Yang, T. Awakawa, T. Wakimoto and I. Abe, ChemBioChem, 2014, 15, 1578–1583 CrossRef CAS PubMed.
- Y. Zhu, J. Wang, P. Mou, Y. Yan, M. Chen and Y. Tang, Org. Biomol. Chem., 2021, 19, 1985–1990 RSC.
- H. Li, C. L. M. Gilchrist, H. J. Lacey, A. Crombie, D. Vuong, J. I. Pitt, E. Lacey, Y.-H. Chooi and A. M. Piggott, Org. Lett., 2019, 21, 1287–1291 CrossRef CAS PubMed.
- Y. Hai and Y. Tang, J. Am. Chem. Soc., 2018, 140, 1271–1274 CrossRef CAS PubMed.
- B. D. Ames, C. Nguyen, J. Bruegger, P. Smith, W. Xu, S. Ma, E. Wong, S. Wong, X. Xie, J. W.-H. Li, J. C. Vederas, Y. Tang and S.-C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11144–11149 CrossRef CAS PubMed.
- D. Komagata, S. Fujita, N. Yamashita, S. Saito and T. Morino, J. Antibiot., 1996, 49, 958–959 CrossRef CAS PubMed.
- D. Rubanova, P. Dadova, O. Vasicek and L. Kubala, Int. J. Mol. Sci., 2021, 22, 1938 CrossRef CAS PubMed.
- S. Maiya, A. Grundmann, X. Li, S.-M. Li and G. Turner, ChemBioChem, 2007, 8, 1736–1743 CrossRef CAS PubMed.
- P. Wiemann, C.-J. Guo, J. M. Palmer, R. Sekonyela, C. C. C. Wang and N. P. Keller, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17065–17070 CrossRef CAS PubMed.
- Y. Tsunematsu, M. Fukutomi, T. Saruwatari, H. Noguchi, K. Hotta, Y. Tang and K. Watanabe, Angew. Chem., Int. Ed., 2014, 53, 8475–8479 CrossRef CAS PubMed.
- Y. Zou, W. Xu, Y. Tsunematsu, M. Tang, K. Watanabe and Y. Tang, Org. Lett., 2014, 16, 6390–6393 CrossRef CAS PubMed.
- S. Kishimoto, Y. Tsunematsu, T. Matsushita, K. Hara, H. Hashimoto, Y. tang and K. Watanabe, Biochemistry, 2019, 58, 3933–3937 CrossRef CAS PubMed.
- H. Tao, T. Mori, X. Wei, Y. Matsuda and I. Abe, Angew. Chem., Int. Ed., 2021, 60, 8851–8858 CrossRef CAS PubMed.
- Z. Z. Zhou, H. J. Zhu, L. P. Lin, X. Zhang, H. M. Ge, R. H. Jiao and R. X. Tan, Chem. Sci., 2019, 10, 73–82 RSC.
- L. Kahlert, M. Villanueva, R. J. Cox and E. J. Skellam, Angew. Chem., Int. Ed., 2021, 60, 11423–11429 CrossRef CAS PubMed.
- C. Zaehle, M. Gressler, E. Shelest, E. Geib, C. Hertweck and M. Brock, Chem. Biol., 2014, 21, 719–731 CrossRef CAS PubMed.
- T. Ugai, A. Minami, S. Tanaka, T. Ozaki, C. Liu, H. Shigemori, M. Hashimoto and H. Oikawa, ChemBioChem, 2020, 21, 360–367 CrossRef CAS PubMed.
- L. Kahlert, D. Bernardi, M. Hauser, L. P. Ioca, R. G. S. Berlinck, E. J. Skellam and R. J. Cox, Chem.–Eur. J., 2021, 27, 11895–11903 CrossRef CAS PubMed.
- L. Hang, M.-C. Tang, C. J. B. Harvey, C. G. Page, J. Li, Y.-S. Hung, N. Liu, M. E. Hillenmeyer and Y. Tang, Angew. Chem., Int. Ed., 2017, 56, 9556–9560 CrossRef CAS PubMed.
- M.-C. Tang, C. R. Fischer, J. V. Chari, D. Tan, S. Suresh, A. Chu, M. Miranda, J. Smith, Z. Zhang, N. K. Garg, R. P. St. Onge and Y. Tang, J. Am. Chem. Soc., 2019, 141, 8198–8206 CrossRef CAS PubMed.
- L. Liu, Z. Zhang, C.-L. Shao and C.-Y. Wang, Front. Microbiol., 2017, 8, 1685 CrossRef PubMed.
- N. Lenfant, T. Hotelier, E. Velluet, Y. Bourne, P. Marchot and A. Chatonnet, Nucleic Acids Res., 2013, 41, D423–D429 CrossRef CAS PubMed.
- T. Awakawa, K. Yokota, N. Funa, F. Doi, N. Mori, H. Watanabe and S. Horinouchi, Chem. Biol., 2009, 16, 613–623 CrossRef CAS PubMed.
- Y.-H. Chooi, J. Fang, H. Liu, S. G. Filler, P. Wang and Y. Tang, Org. Lett., 2013, 15, 780–783 CrossRef CAS PubMed.
- Y.-M. Chiang, E. Szewczyk, A. D. Davidson, R. Entwistle, N. P. Keller, C. C. C. Wang and B. R. Oakley, Appl. Environ. Microbiol., 2010, 76, 2067–2074 CrossRef CAS PubMed.
- E. Szewczyk, Y.-M. Chiang, C. E. Oakley, A. D. Davidson, C. C. C. Wang and B. R. Oakley, Appl. Environ. Microbiol., 2008, 74, 7607–7612 CrossRef CAS PubMed.
- K. Throckmorton, F. Y. Lim, D. P. Kontoyiannis, W. Zheng and N. P. Keller, Environ. Microbiol., 2016, 18, 246–259 CrossRef CAS PubMed.
- M. T. Nielsen, J. B. Nielsen, D. C. Anyaogu, D. K. Holm, K. F. Nielsen, T. O. Larsen and U. H. Mortensen, PLoS One, 2013, 8, e72871 CrossRef CAS PubMed.
- A. J. Szwalbe, K. Williams, Z. Song, K. De Mattos-Shipley, J. L. Vincent, A. M. Bailey, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Sci., 2019, 10, 233–238 RSC.
- S. Griffiths, C. H. Mesarich, B. Saccomanno, A. Vaisberg, P. J. G. M. De Wit, R. Cox and J. Collemare, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6851–6856 CrossRef CAS PubMed.
- L. Neubauer, J. Dopstadt, H.-U. Humpf and P. Tudzynski, Fungal Biol. Biotechnol., 2016, 3, 2 CrossRef PubMed.
- C. Greco, K. de Mattos-Shipley, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Sci., 2019, 10, 2930–2939 RSC.
- W. Xu, Y.-H. Chooi, J. W. Choi, S. Li, J. C. Vederas, N. A. Da Silva and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 6472–6475 CrossRef CAS PubMed.
- K. Williams, A. J. Szwalbe, N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Angew. Chem., Int. Ed., 2016, 55, 6784–6788 CrossRef CAS PubMed.
- A. Droce, W. Saei, S. H. Jorgensen, R. Wimmer, H. Giese, R. D. Wollenberg, T. E. Sondergaard and J. L. Sorensen, Molecules, 2016, 21, 1710 CrossRef PubMed.
- Y. Hai, M. Chen, A. Huang and Y. Tang, J. Am. Chem. Soc., 2020, 142, 19668–19677 CrossRef CAS PubMed.
- K. M. J. de Mattos-Shipley, C. E. Spenser, C. Greco, D. M. Heard, D. E. O'Flynn, T. T. Dao, Z. Song, N. P. Mulholland, J. L. Vincent, T. J. Simpson, R. J. Cox, A. M. Bailey and C. L. Willis, Chem. Sci., 2020, 11, 11570–11578 RSC.
- R. Fujii, Y. Matsu, A. Minami, S. Nagamine, I. Takeuchi, K. Gomi and H. Oikawa, Org. Lett., 2015, 17, 5658–5661 CrossRef CAS PubMed.
- A. O. Zabala, Y.-H. Chooi, M. S. Choi, H.-S. Lin and Y. Tang, ACS Chem. Biol., 2014, 9, 1576–1586 CrossRef CAS PubMed.
- F. Trenti, K. E. Lebe, E. Adelin, J. Ouazzani, C. Schotte and R. J. Cox, RSC Adv., 2020, 10, 27369–27376 RSC.
- Y. Morishita, Y. Aoki, M. Ito, D. Hagiwara, K. Torimaru, D. Morita, T. Kuroda, H. Fukano, Y. Hoshino, M. Suzuki, T. Taniguchi, K. Mori and T. Asai, Org. Lett., 2020, 22, 5876–5879 CrossRef CAS PubMed.
- M. F. Grau, R. Entwistle, Y.-M. Chiang, M. Ahuja, C. E. Oakley, T. Akashi, C. C. C. Wang, R. B. Todd and B. R. Oakley, ACS Chem. Biol., 2018, 13, 3193–3205 CrossRef CAS PubMed.
- Y. Morishita, T. Sonohara, T. Taniguchi, K. Adachi, M. Fujita and T. Asai, Org. Biomol. Chem., 2020, 18, 2813–2816 RSC.
- Y. Morishita, H. Zhang, T. Taniguchi, K. Mori and T. Asai, Org. Lett., 2019, 21, 4788–4792 CrossRef CAS PubMed.
- D.-W. Gao, C. S. Jamieson, G. Wang, Y. Yan, J. Zhou, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2021, 143, 80–84 CrossRef CAS PubMed.
- K. Kobayashi and T. Ui, Tetrahedron Lett., 1975, 47, 4119–4122 CrossRef.
- W.-G. Wang, H. Wang, L.-Q. Du, M. Li, L. Chen, J. Yu, G.-G. Cheng, M.-T. Zhan, Q.-F. Hu, L. Zhang, M. Yao and Y. Matsuda, J. Am. Chem. Soc., 2020, 142(18), 8464–8472 CrossRef CAS PubMed.
- X. Wei, L. Chen, J.-W. Tang and Y. Matsuda, Front. Microbiol., 2020, 11, 562063 CrossRef PubMed.
- M.-C. Tang, Y. Zou, D. Yee and Y. Tang, AIChE J., 2018, 64, 4182–4186 CrossRef CAS PubMed.
- H. Zhang, V. Hantke, P. Bruhnke, E. J. Skellam and R. J. Cox, Chem.–Eur. J., 2021, 27, 3106–3113 CrossRef CAS PubMed.
- T. Yamamoto, Y. Tsunematsu, H. Noguchi, K. Hotta and K. Watanabe, Org. Lett., 2015, 17, 4992–4995 CrossRef CAS PubMed.
- Y. He and R. J. Cox, Chem. Sci., 2016, 7, 2119–2127 RSC.
- P. A. Storm and C. A. Townsend, Chem. Commun., 2017, 54, 50–53 RSC.
- A. O. Zabala, W. Xu, Y.-H. Chooi and Y. Tang, Chem. Biol., 2012, 19, 1049–1059 CrossRef CAS PubMed.
- K. Williams, C. Greco, A. M. Bailey and C. L. Willis, ChemBioChem, 2021, 22, 3027–3036 CrossRef CAS PubMed.
- A. L. Vagstad, S. B. Bumpus, K. Belecki, N. L. Kelleher and C. A. Townsend, J. Am. Chem. Soc., 2012, 134, 6865–6877 CrossRef CAS PubMed.
- A. G. Newman, A. L. Vagstad, P. A. Storm and C. A. Townsend, J. Am. Chem. Soc., 2014, 136, 7348–7362 CrossRef CAS PubMed.
- X. Gao, X. Xie, I. Pashkov, M. R. Sawaya, J. Laidman, W. Zhang, R. Cacho, T. O. Yeates and Y. Tang, Chem. Biol., 2009, 16, 1064–1074 CrossRef CAS PubMed.
- G. Jimenez-Oses, S. Osuns, X. Gao, M. R. Sawaya, L. Gilson, S. J. Collier, G. W. Huisman, T. O. Yeates, Y. Tang and K. N. Houk, Nat. Chem. Biol., 2014, 10, 431–436 CrossRef CAS PubMed.
- H.-C. Lin, Y.-H. Chooi, S. Dhingra, W. Xu, A. M. Calvo and Y. Tang, J. Am. Chem. Soc., 2013, 135, 4616–4619 CrossRef CAS PubMed.
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC.
- M. B. Lukassen, W. Saei, T. E. Sondergaard, A. Tamminen, A. Kumar, F. Kempken, M. G. Wiebe and J. L. Sorensen, Mar. Drugs, 2015, 13, 4331–4343 CrossRef CAS PubMed.
- Y.-M. Chiang, E. Szewczyk, T. Nayak, A. D. Davidson, J. F. Sanchez, H.-C. Lo, W.-Y. Ho, H. Simityan, E. Kuo, A. Praseuth, K. Watanabe, B. R. Oakley and C. C. C. Wang, Chem. Biol., 2008, 15, 527–532 CrossRef CAS PubMed.
- T. Ugai, A. Minami, R. Fujii, M. Tanaka, H. Oguri, K. Gomi and H. Oikawa, Chem. Commun., 2015, 51, 1878–1881 RSC.
- H. Li, J. Hu, H. Wei, P. S. Solomon, K. A. Stubbs and Y.-H. Chooi, Chem.–Eur. J., 2019, 25, 15062–15066 CrossRef CAS PubMed.
- Z. Zhao, Y. Ying, Y.-S. Hung and Y. Tang, J. Nat. Prod., 2019, 82, 1029–1033 CrossRef CAS PubMed.
- L. Liu, M.-C. Tang and Y. Tang, J. Am. Chem. Soc., 2019, 141, 19538–19541 CrossRef CAS PubMed.
- J. Nies, H. Ran, V. Wohlgemuth, W.-B. Yin and S.-M. Li, Org. Lett., 2020, 22, 2256–2260 CrossRef CAS PubMed.
- N. Liu, E. D. Abramyan, W. Cheng, B. Perlaati, C. J. B. Harvey, G. F. Bills and Y. Tang, J. Am. Chem. Soc., 2021, 143, 6043–6047 CrossRef CAS PubMed.
- X. Niu, L. Chen, Q. Yue, B. Wang, J. Zhang, C. Zhu, K. Zhang, G. F. Bills and Z. An, Org. Lett., 2014, 16, 3744–3747 CrossRef CAS PubMed.
- J.-M. Zhang, H.-H. Wang, X. Liu, C.-H. Hu and Y. Zou, J. Am. Chem. Soc., 2020, 142, 1957–1965 CrossRef CAS PubMed.
- P. B. Wolff, M. L. Nielsen, J. C. Slot, L. N. Andersen, L. M. Petersen, T. Isbrandt, D. K. Holm, U. H. Mortensen, C. S. Nodvig, T. O. Larsen and J. B. Hoof, Fungal Genet. Biol., 2020, 139, 103378 CrossRef CAS PubMed.
- N. J. Alexander, R. H. Proctor and S. P. McCormick, Toxin Rev., 2009, 28, 198–215 CrossRef CAS.
- R. S. Bojja, R. L. Cerny, R. H. Proctor and L. Du, J. Agric. Food Chem., 2004, 52, 2855–2860 CrossRef CAS PubMed.
- R. Gerber, L. Lou and L. Du, J. Am. Chem. Soc., 2009, 131, 3148–3149 CrossRef CAS PubMed.
- X. Zhu, C. Vogeler and L. Du, J. Nat. Prod., 2008, 71, 957–960 CrossRef CAS PubMed.
- P. J. Harrison, T. M. Dunn and D. J. Campopiano, Nat. Prod. Rep., 2018, 35, 921–954 RSC.
- G. Z. Dai, W. B. Han, Y. N. Mei, K. Xu, R. H. Jiao, H. M. Ge and R. X. Tan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1174–1180 CrossRef CAS PubMed.
- X.-M. Mao, Z.-J. Zhan, M. N. Grayson, M.-C. Tang, W. Xu, Y.-Q. Li, W.-B. Yin, H.-C. Lin, Y.-H. Chooi, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2015, 137, 11904–11907 CrossRef CAS PubMed.
- Y. Li, Y.-H. Chooi, Y. Sheng, J. S. Valentine and Y. Tang, J. Am. Chem. Soc., 2011, 133, 15773–15785 CrossRef CAS PubMed.
- M. Sato, J. E. Dander, C. Sato, Y.-S. Hung, S.-S. Gao, M.-C. Tang, L. Hang, J. M. Winter, N. K. Garg, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2017, 139, 5317–5320 CrossRef CAS PubMed.
- Y. Cai, L. Rao and Y. Zou, Org. Lett., 2021, 23, 2337–2341 CrossRef CAS PubMed.
- J. Barriuso, D. T. Nguyen, J. W.-H. Li, J. N. Roberts, G. MacNevin, J. L. Chaytor, S. L. Marcus, J. C. Vederas and D. K. Ro, J. Am. Chem. Soc., 2011, 133, 8078–8081 CrossRef CAS PubMed.
- M. Sato, J. M. Winter, S. Kishimoto, H. Noguchi, Y. Tang and K. Watanabe, Org. Lett., 2016, 18, 1446–1449 CrossRef CAS PubMed.
- Y. Xu, T. Zhou, S. Zhang, P. Espinosa-Artiles, L. Wang, W. Zhang, M. Lin, A. A. L. Gunatilaka, J. Zhan and I. Molnar, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12354–12359 CrossRef CAS PubMed.
- J. Bai, Y. Lu, Y.-M. Xu, W. Zhang, M. Chen, M. Lin, A. A. Gunatilaka, Y. Xu and I. Molnar, Org. Lett., 2016, 18, 1262–1265 CrossRef CAS PubMed.
- M. N. Heneghan, A. A. Yakasai, K. Williams, K. A. Kadir, Z. Wasil, W. Bakeer, K. M. Fisch, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, Chem. Sci., 2011, 2, 972–979 RSC.
- W. Zhang, Y. Li and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(52), 20683–20688 CrossRef CAS PubMed.
- S. M. Ma, J. Zhan, X. Xie, K. Watanabe, Y. Tang and W. Zhang, J. Am. Chem. Soc., 2008, 130, 38–39 CrossRef CAS PubMed.
- M. Klaus, M. P. Ostrowski, J. Austerjost, T. Robbins, B. Lowry, D. E. Cane and C. Khosla, J. Biol. Chem., 2016, 291, 16404–16415 CrossRef CAS PubMed.
- D. Sehnal, S. Bittrich, M. Deshpande, R. Svobodova, K. Berka, V. Bazgier, S. Velankar, S. K. Burley, J. Koca and A. S. Rose, Nucleic Acids Res., 2021, 49, W431–W437 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |