
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis and bioactivities of microbial genotoxin colibactins
Jian-Wei
Tang†
ab,
Xin
Liu†
ab,
Wei
Ye
ad,
Zhong-Rui
Li
c and
Pei-Yuan
Qian
 *ab
*ab
aDepartment of Ocean Science, Hong Kong Branch of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), The Hong Kong University of Science and Technology, Kowloon, Hong Kong, China. E-mail: boqianpy@ust.hk
bSouthern Marine Science and Engineering Guangdong Laboratory, Guangzhou 511458, China
cDepartment of Chemistry, The Scripps Research Institute, La Jolla, CA 92037, USA
dState Key Laboratory of Applied Microbiology Southern China, Institute of Microbiology, Guangdong Academy of Sciences, Guangzhou 510070, China
First published on 15th March 2022
Abstract
Covering: up to 2021
Colibactin(s), a group of secondary metabolites produced by the pks island (clb cluster) of Escherichia coli, shows genotoxicity relevant to colorectal cancer and thus significantly affects human health. Over the last 15 years, substantial efforts have been exerted to reveal the molecular structure of colibactin, but progress is slow owing to its instability, low titer, and elusive and complex biosynthesis logic. Fortunately, benefiting from the discovery of the prodrug mechanism, over 40 precursors of colibactin have been reported. Some key biosynthesis genes located on the pks island have also been characterised. Using an integrated bioinformatics, metabolomics, and chemical synthesis approach, researchers have recently characterised the structure and possible biosynthesis processes of colibactin, thereby providing new insights into the unique biosynthesis logic and the underlying mechanism of the biological activity of colibactin. Early developments in the study of colibactin have been summarised in several previous reviews covering various study periods, whereas the two most recent reviews have focused primarily on the chemical synthesis of colibactin. The present review aims to provide an update on the biosynthesis and bioactivities of colibactin.
 Jian-Wei Tang | Jian-Wei Tang graduated from Dalian University of Technology with a bachelor's degree in pharmaceutical engineering. He obtained his PhD in medicinal chemistry in 2019 at Kunming Institute of Botany, Chinese Academy of Sciences. His doctoral research focused on structurally novel and bioactive natural products from plant-endophytic fungi and structural determination of complex molecules using quantum chemical calculations. He is currently working as a postdoc in Prof. Pei-Yuan Qian's group at the Hong Kong University of Science and Technology. His current research mainly focuses on omics-guided natural products discovery and the biosynthesis of novel compounds from marine bacteria. |
 Xin Liu | Xin Liu has a BSc in Applied Chemistry from the Department of Science, Nanchang University (Nanchang, China), and obtained her PhD in pharmacognosy from Sun Yat-sen University (Guangzhou, China) in 2017. After a one-and-a-half-years work at Shenzhen University as an associate researcher, she joined the group of Prof. Pei-Yuan Qian at the Hong Kong University of Science and Technology (Hong Kong, China) for her postdoc research in 2019. Her main research focuses on bioactive natural products from marine organisms, especially the application of compounds in neuroprotection. |
 Wei Ye | Wei Ye received his PhD degree from the School of Biology and Biological Engineering, South China University of Technology, in 2013, guided by Prof. XiaoNing Wang and Jufang Wang. He then worked at the Institute of Microbiology, Guangdong Academy of Sciences as an associate researcher in Weimin Zhang's group. He was a postdoc at Hong Kong University of Science Technology in Prof. Pei-Yuan Qian's group in 2021. His current research field focus on the biosynthesis of novel bioactive natural products from deep-sea fungi and Streptomyces. |
 Zhong-Rui Li | Zhongrui Li graduated from Zhejiang Ocean University with a bachelor's degree in pharmaceutical sciences. He then moved to the Hong Kong University of Science and Technology where he obtained his Master of Philosophy degree and conducted research in Prof. Pei-Yuan Qian's laboratory. After obtaining another master's degree in chemical engineering from the University of California at Berkeley, he is now pursuing his PhD degree at Scripps Research at La Jolla, California. He is conducting his research under the supervision of Prof. Chi-Huey Wong, studying glycosyltransferases from various natural product biosynthetic pathways. |
 Pei-Yuan Qian | Pei-Yuan Qian obtained his PhD degree in Zoology from the University of Alberta, Canada. He joined the Hong Kong University of Science and Technology as an assistant professor in 1993 after one year as a Killam Postdoc Fellow at the University of British Columbia, and currently is the David von Hansemann Professor of Science, Head and Chair Professor of Department of Ocean Science and Chair Professor in Division of Life Science. His work focuses on the interaction between marine microbes and animals, with a particular interest in marine natural products that mediate microbe–animal interactions. He holds many international patents on non-toxic antifouling compounds and drug leads. |
1. Introduction
The human gut microbiome comprises trillions of bacterial and fungal cells and virus particles, and thereby affects various physiological processes.1 An imbalance between the host and the microbiota could lead to human diseases.2 For instance, many studies have shown that gut microbial dysbiosis induced by pathogenic bacteria, such as Bacteroides fragilis and Escherichia coli,3,4 causes colorectal cancer (CRC).5,6 Considering that the interactions between the gut microbiome and its host are critically regulated by microbe-related metabolites, the characterisation of these mysterious molecules is a key step to understanding the molecular mechanisms underlying host–microbiome interactions.Colibactins represent a group of cryptic microbial metabolites.7 In 2006, Nougayrède and co-workers7 observed for the first time that E. coli strains harbouring the pks island (a 54 kb genomic island also known as the clb gene cluster) (Fig. 1) cause DNA double-strand breaks (DSBs), leading to cell-cycle arrest in the G2/M phase, megalocytosis, and even cell death. This gene cluster containing 19 genes (from clbA to clbS) encodes 3 nonribosomal peptide synthetases (NRPSs), 3 polyketide synthases (PKSs), 2 NRPS/PKS hybrid synthases, and 11 accessory and tailoring enzymes with respective functions for synthesising and modifying the genotoxin(s) (which are called colibactin(s)). Significantly, systematic mutagenesis studies of 18 genes (clbR was not tested in this research) revealed that these genes, except for clbM and clbS, are essential for the genotoxicity of colibactin(s).7 To identify the missing link between certain gut microbes and DNA DSBs and to reveal the possible mechanisms underlying colibactin-induced DNA damage, researchers have attempted to elucidate the molecular identity of genotoxic colibactin. They have made significant progress regarding the relationship between the pks island and the symptoms caused by DNA DSBs, and have further demonstrated that colibactin induces DSBs, leading to genomic instability, senescence, and apoptosis.7–10 However, the structural information of colibactin was seldom obtained from pks+ wild-type or heterologous strains until the discovery of the prodrug activation mechanism of colibactin,11–14 which paved the way for the analysis of the biosynthesis logic through the mutagenesis processes. Finally, several colibactins, especially genotoxic colibactin-645 (18) and colibactin-770 (19), have been characterised using integrated bioinformatics, metabolomics, and chemical synthesis.15–18
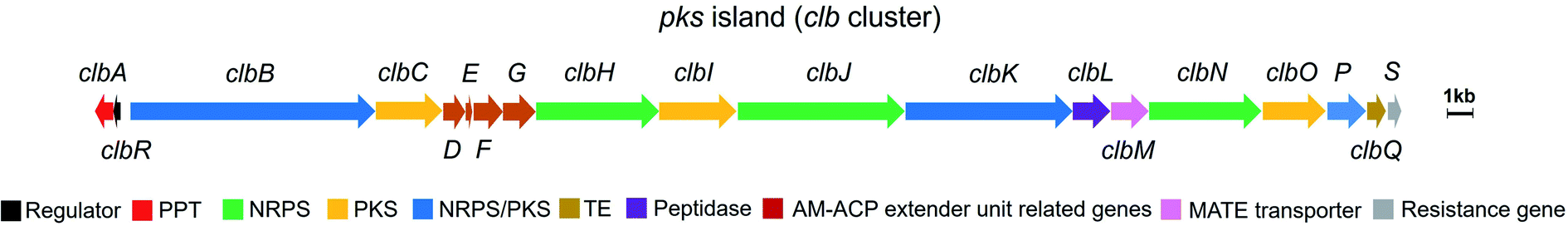 | ||
| Fig. 1 Organisation of the pks island with annotations (modified figure based on ref. 7, 19 and 20). PPT: phosphopantetheinyl transferase, NRPS: nonribosomal peptide synthetase, PKS: polyketide synthase, NRPS/PKS: hybrid nonribosomal peptide–polyketide synthase, TE: thioesterase, AM-ACP extender unit related genes include clbD and clbF (which encode two dehydrogenases), clbE (which encodes a free-standing acyl carrier protein), and clbG (which encodes a free-standing acyl transferase). | ||
The study of colibactin serves well as a significant example for natural product research. The instability of colibactin seriously impedes the traditional isolation-based strategy to access its bioactive products. Accordingly, methods such as bioinformatics and metabolomics are required to clarify this issue. Additionally, the biosynthesis logic of clb offers new insights into the mechanism of NRPS/PKS assembly lines. Most importantly, colibactin is critically involved in human health.21,22 Studies have reported that pks+ E. coli strains are commonly found in the human colon and can promote tumour formation in mouse models of CRC.3,8,22–24 Moreover, the colibactin-induced mutational signature is notably enriched in CRC patients.25,26 Thus, exposure to pks+ E. coli represents a great health risk.
Several previous reviews have highlighted the structures, biosynthesis, and genotoxicity of colibactin during their respective research periods,20,27,28 whereas two recent reviews have focused on the structure determination of colibactin through DNA adductomics coupled with chemical synthesis.29,30 In the present review, we focus on the biosynthesis logic of clb, summarise all currently known biosynthesis mechanisms, and discuss the possible mechanisms and undisclosed process(es) in late-stage biosynthesis. We also provide a summary of the biological activities of pks+ E. coli, especially the mechanism of colibactin-770 (19) and colibactin-645 (18)-induced DNA DSBs, as well as the potential relationship between colibactin(s) and CRC. Furthermore, this review discusses several questions: (1) What are the possible functions of ClbL and ClbQ in colibactin(s) biosynthesis? (2) Is the genotoxic colibactin(s) a single compound or a mixture? (3) Does the clb gene cluster specifically produce colibactin or simultaneously release other shunt metabolites to perform diverse functions? (4) What is the ecological function of clb for E. coli in natural settings? Moreover, the regulatory mechanism for activating colibactin(s) and their therapeutic potential are also discussed.
2. Discovery of precolibactins and colibactins
2.1. Prodrug mechanism in colibactin biosynthesis
The prodrug mechanism has already been described in detail by Balskus and co-workers28 in a previous review. Herein, this mechanism is briefly described to ensure that readers can easily grasp the following content.Prodrug activation is a general procedure in the biosynthesis of ribosomally synthesised and post-translationally modified peptides to afford mature peptides, but it is rare in nonribosomal peptide biosynthesis. Several exceptional cases do exist,31 such as those of amicoumacin,32 xenocoumacin,33 and zwittermicin,34 which use a membrane-located D-asparagine-specific peptidase (AmiB in amicoumacin biosynthesis, XcnG in xenocoumacin biosynthesis, and ZmaM in zwittermicin biosynthesis) to convert inactive precursors into bioactive compounds. In colibactin biosynthesis, ClbP is hypothesised to exhibit homology to XcnG and ZmaM in the phylogenetic analysis of the peptidase domain and to act as the colibactin-maturating enzyme based on analyses of crystal structure, mutagenesis experiments, and structure–function relationship.11,12 Later, inspired by the biosynthesis of xenocoumacin and zwittermicin, Balskus and coworkers13 proposed that the prodrug motif N-myristoyl-D-asparagine (1) could be synthesised at the initial NRPS module with C–A–T–E domains and transferred to another NRPS module, which should recognise the upstream D-amino acid to construct an amide bond as the eventual peptidase substrate. An NRPS ClbN with C–A–T–E domains and an NRPS-PKS hybrid ClbB with a DCL domain meet this requirement (Fig. 2). Subsequent in vitro experiments supported their hypothesis. ClbN specifically selects L-asparagine, performs the N-acylation of myristoyl-CoA to form N-acyl-asparagine, and subsequently epimerases in the E domain to form N-myristoyl-D-asparagine. The NRPS module of ClbB accepts N-myristoyl-D-asparagine and adds L-alanine (L-valine) to generate N-myristoyl-D-Asn-L-Ala. The peptidase ClbP could specifically cleave N-acyl-D-asparagine substrates to generate the prodrug motif. Notably, ClbP completely loses activity when cleaving the N-acyl-L-asparagine substrates, suggesting that the D-asparagine is the major component responsible for the specificity in the prodrug motif. Based on these discoveries, they proposed that the prodrug mechanism of colibactin biosynthesis is as follows: two NRPS modules (ClbN and ClbB) are critical for installing the prodrug motif on colibactin at the early stage, and the peptidase ClbP then cleaves N-myristoyl-D-asparagine–colibactin by releasing the prodrug motif at the final stage.13 Müller and co-workers also verified this mechanism with in vivo evidence by characterising the prodrug motif 1 in wild-type E. coli Nissle 1917 and analysing the metabolites of clb-containing heterologous E. coli and target-gene-deficient mutants for each gene.14
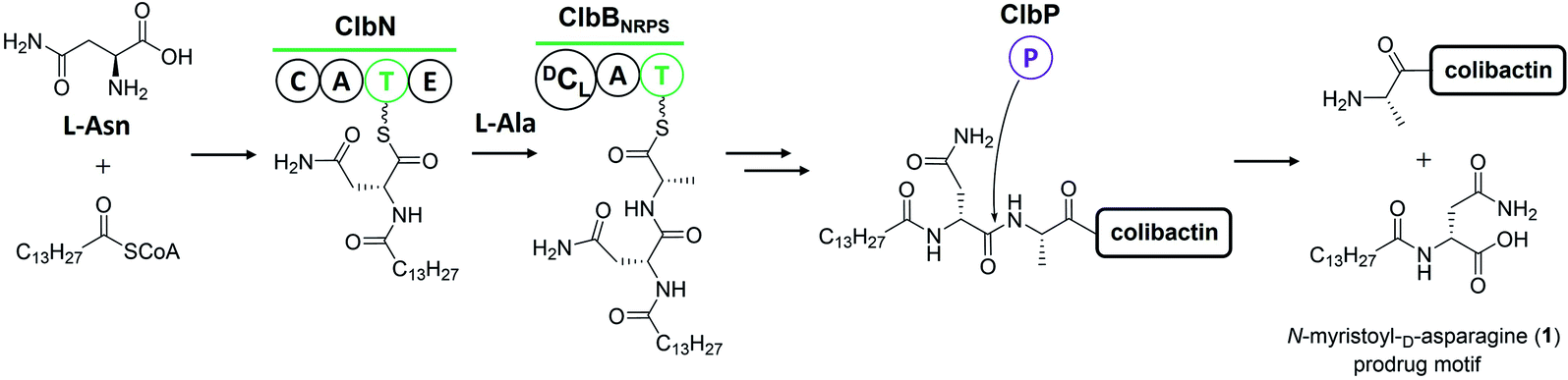 | ||
| Fig. 2 Illustration of the prodrug activation mechanism (modified figure based on ref. 13 and 28). ClbN and ClbBNRPS utilise L-Asn, L-Ala, and myristoyl-CoA to install the prodrug motif at the beginning of colibactin biosynthesis. At the final step, ClbP cleaves the prodrug motif to generate colibactin. | ||
2.2. Identification of various precolibactins
The nomenclature of precolibactins is relatively disorganised because authors do not provide scientific names for individual compounds. Instead, they label the relevant compounds with numbers. This practice is conventional in biosynthesis studies that illustrate the intermediates of the final product but is confusing for the identification of precolibactins. In our previous study, we used a precolibactin nomenclature of the form ‘precolibactin-molecular weight’ to name precolibactin-886 (14)35 and precolibactin-969 (16),18 which is much easier to follow (Fig. 3) and avoids confusion.
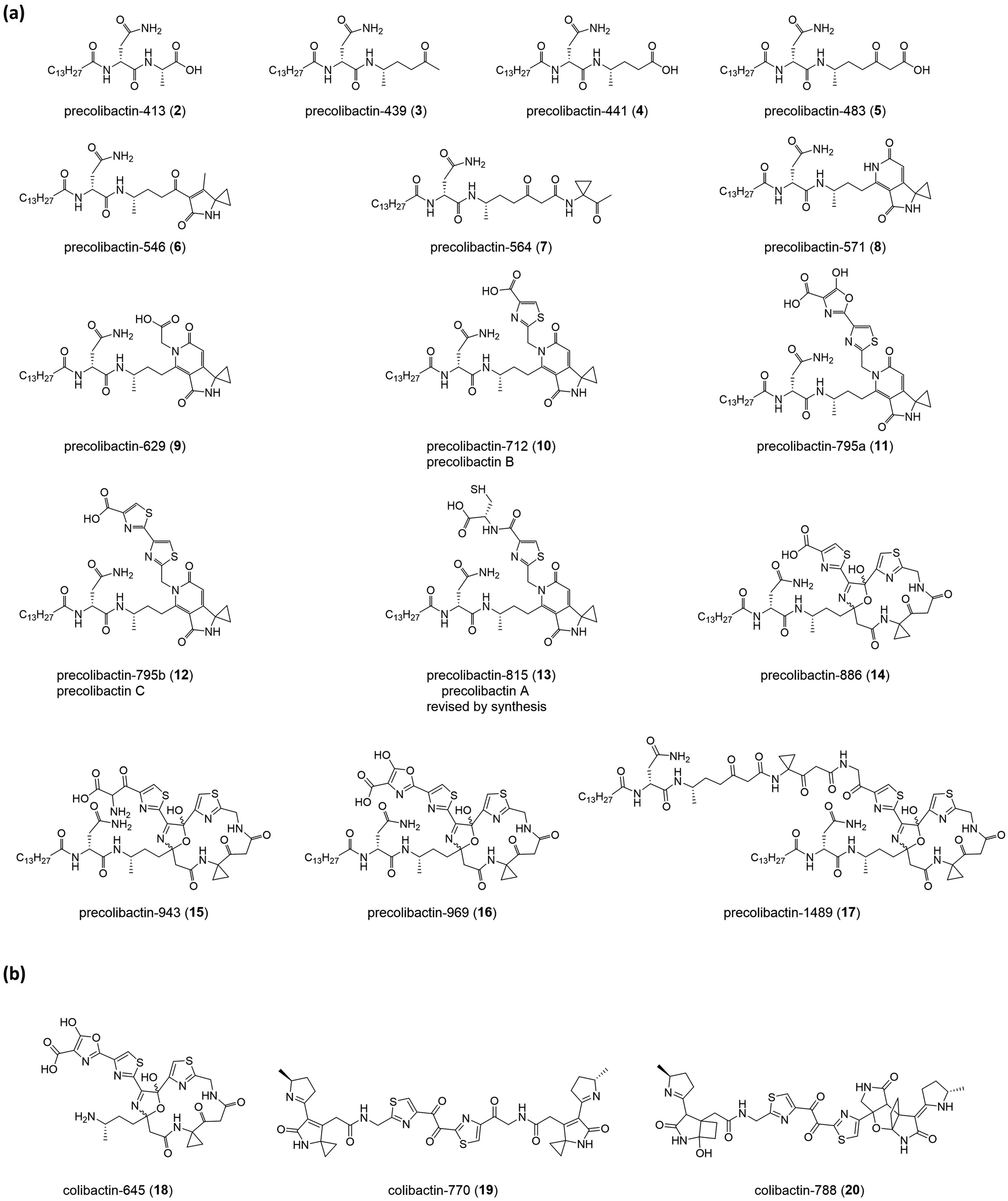
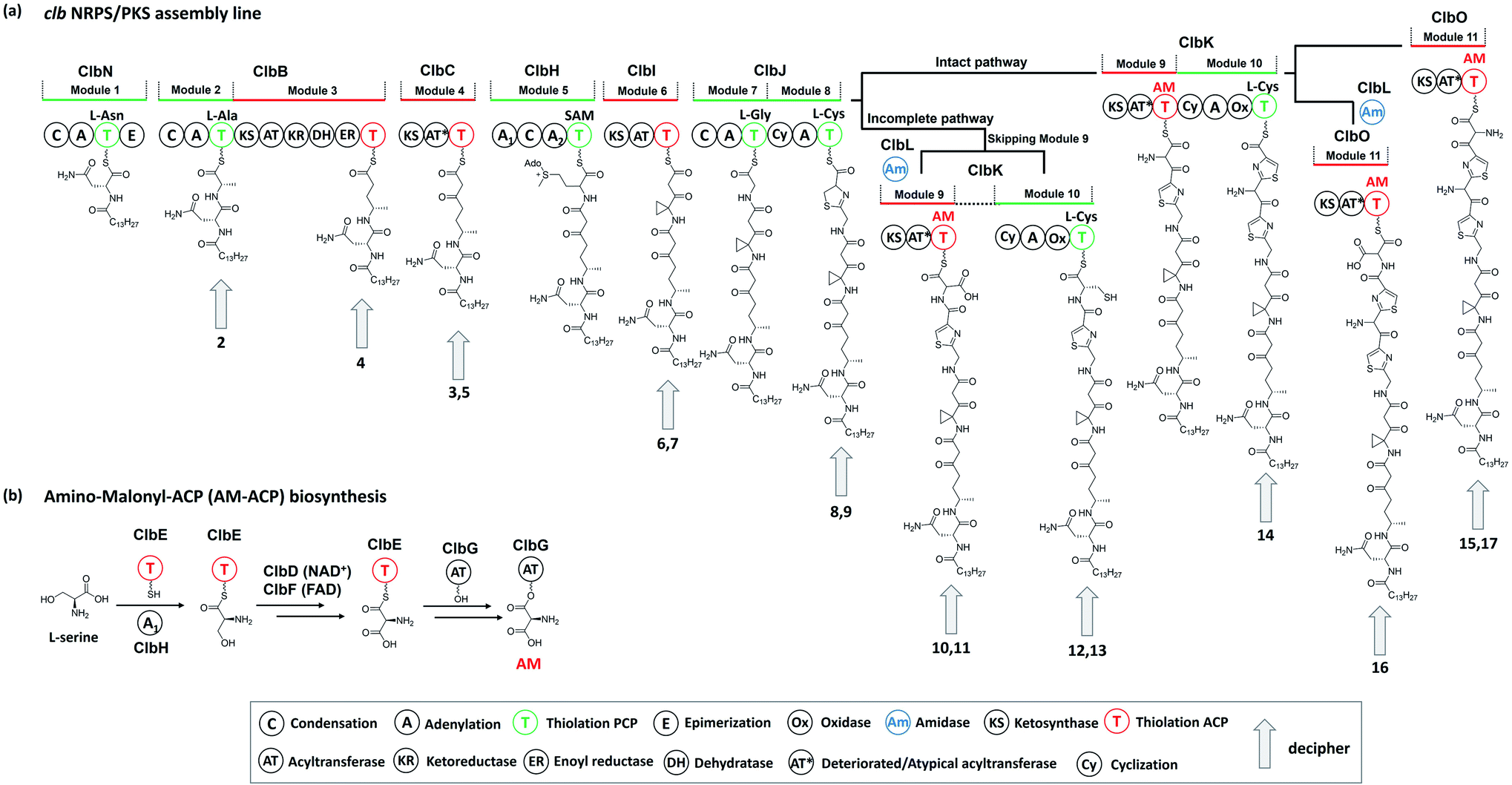 | ||
| Fig. 4 (a) Proposed biosynthesis of colibactin based on the discoveries of precolibactins (late-stage biosynthesis involving ClbL, ClbP, and ClbQ is not shown in this figure). (b) Biosynthesis of the unusual PKS extender unit AM-ACP (ClbH-A1 activates L-serine, which is then transferred to the holo-ClbE and finally forms AM-ACP-ClbE through the dehydrogenases ClbD and ClbF; ultimately, ClbG transfers AM to ClbKPKS and ClbO for subsequent biosynthesis). | ||
2.3. Identification of various colibactins
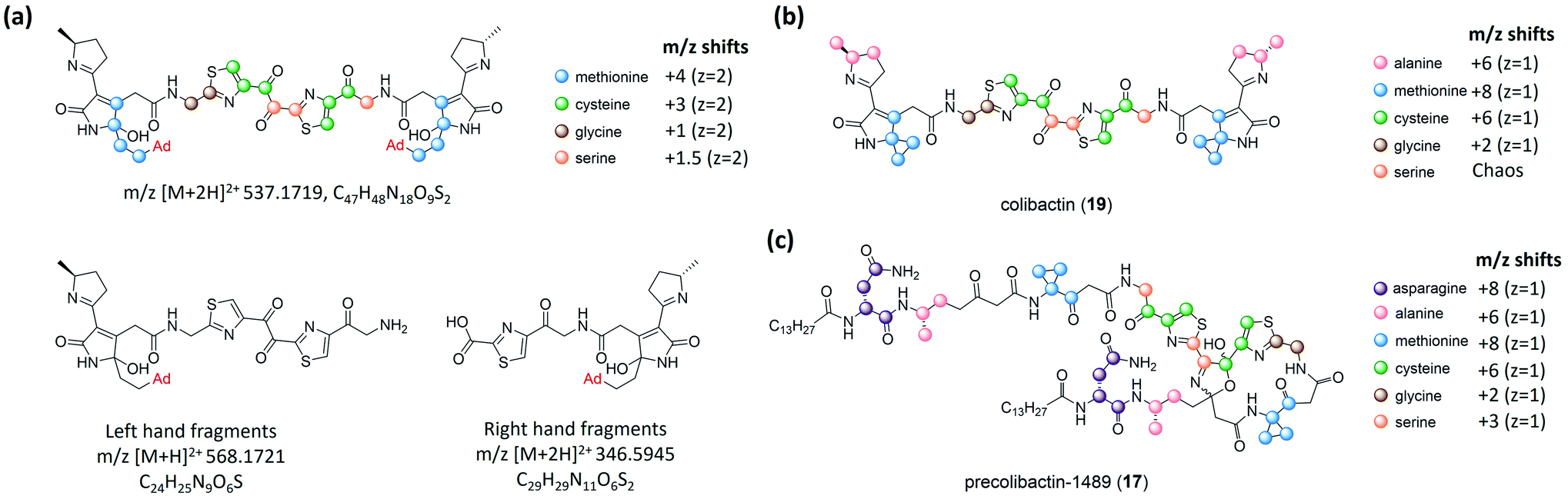 | ||
| Fig. 5 (a) Structure of the colibactin–bisadenine adduct with different isotope-labelling patterns and degenerated adenine adducts (left- and right-hand fragments). (b) Isotopic-labelling pattern of colibactin-770. (c) Isotopic-labelling pattern of precolibactin-1489 (17) (modified figure based on ref. 15). | ||
Balskus and coworkers16 established the structure of colibactin based on the characterisation of ClbL. In their analysis of the remaining uncharacterised components in colibactin biosynthesis, they focused on the function of ClbL, as bioinformatics analysis indicates that ClbL is an amidase containing a Ser-cis-Ser-Lys catalytic triad to hydrolyse the amide bond, and that the mutation of any active residues leads to the loss of genotoxicity in pks+ strains. Thus, comparison of the metabolites of the ClbL-expressing strain and of the ΔclbP/ΔclbL mutant provided the unhydrolysed precursor from the ΔclbP/ΔclbL mutant. However, they did not obtain any unique metabolite from the ΔclbP/ΔclbL mutant, but obtained precolibactin-728 (21) containing an indole-derivative moiety in the ClbL-expressing strain (Fig. 6a). They proposed that ClbL catalyses amide-bond formation, not the putative hydrolysis. In in vitro experiments, ClbL recognises mimics of the ClbC- and ClbJ-bound intermediates and appears to prefer the use of a mimic of the ClbI-bound thioester as an electrophile (Fig. 6b). For the nucleophile, α-aminoketone is crucial for ClbL recognition, and the AM-incorporated intermediates generate α-aminoketone through decarboxylation. Accordingly, they tested the reaction between the mimic of the ClbI-bound thioester and a mimic of the α-aminoketone derived from ClbO. The successful formation of an amide bond in the experiment verified that ClbL is a possible enzyme responsible for the final step of the formation of intact precolibactin. An additional labelling experiment using DNA adductomics53 enabled them to propose the structure of 19.
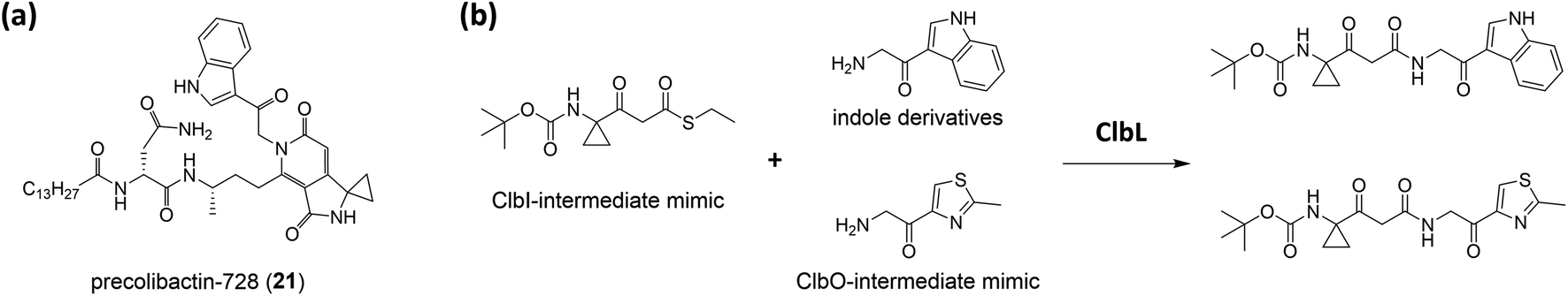 | ||
| Fig. 6 (a) Structure of precolibactin-728 (21). (b) Structure of the ClbI-bound thioester mimic, indole derivative, ClbO-bound thioester mimic, and their ClbL catalysis products. | ||
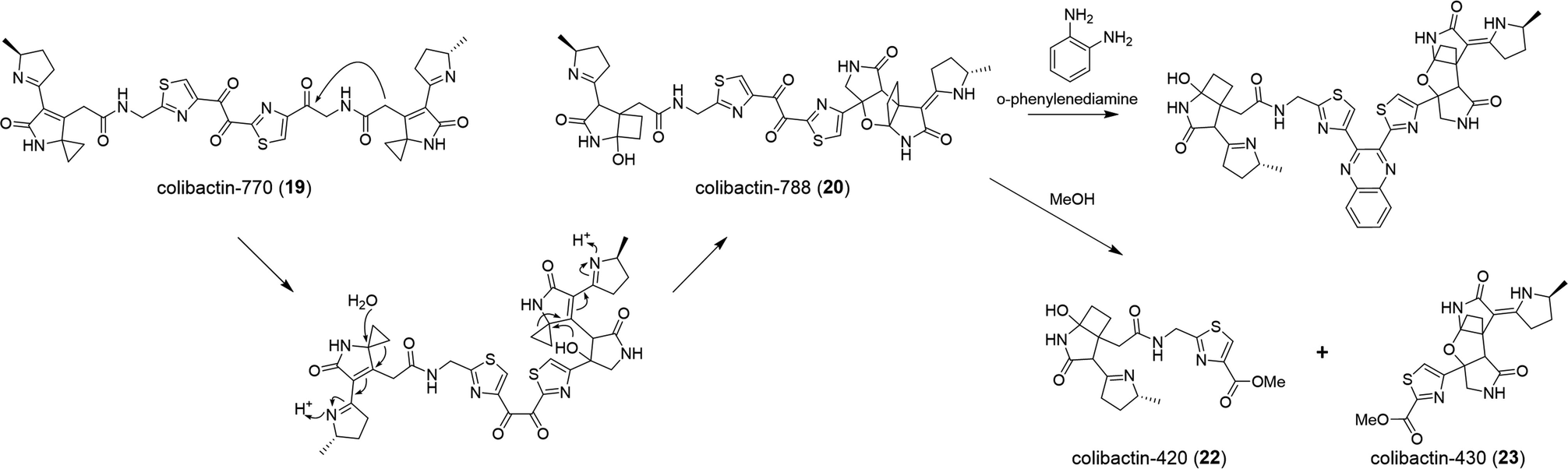 | ||
| Fig. 7 Biosynthesis of colibactin-788 (20) and its degenerated compounds colibactin-420 (22) and colibactin-430 (23) in MeOH. | ||
2.4. Late-stage colibactin biosynthesis
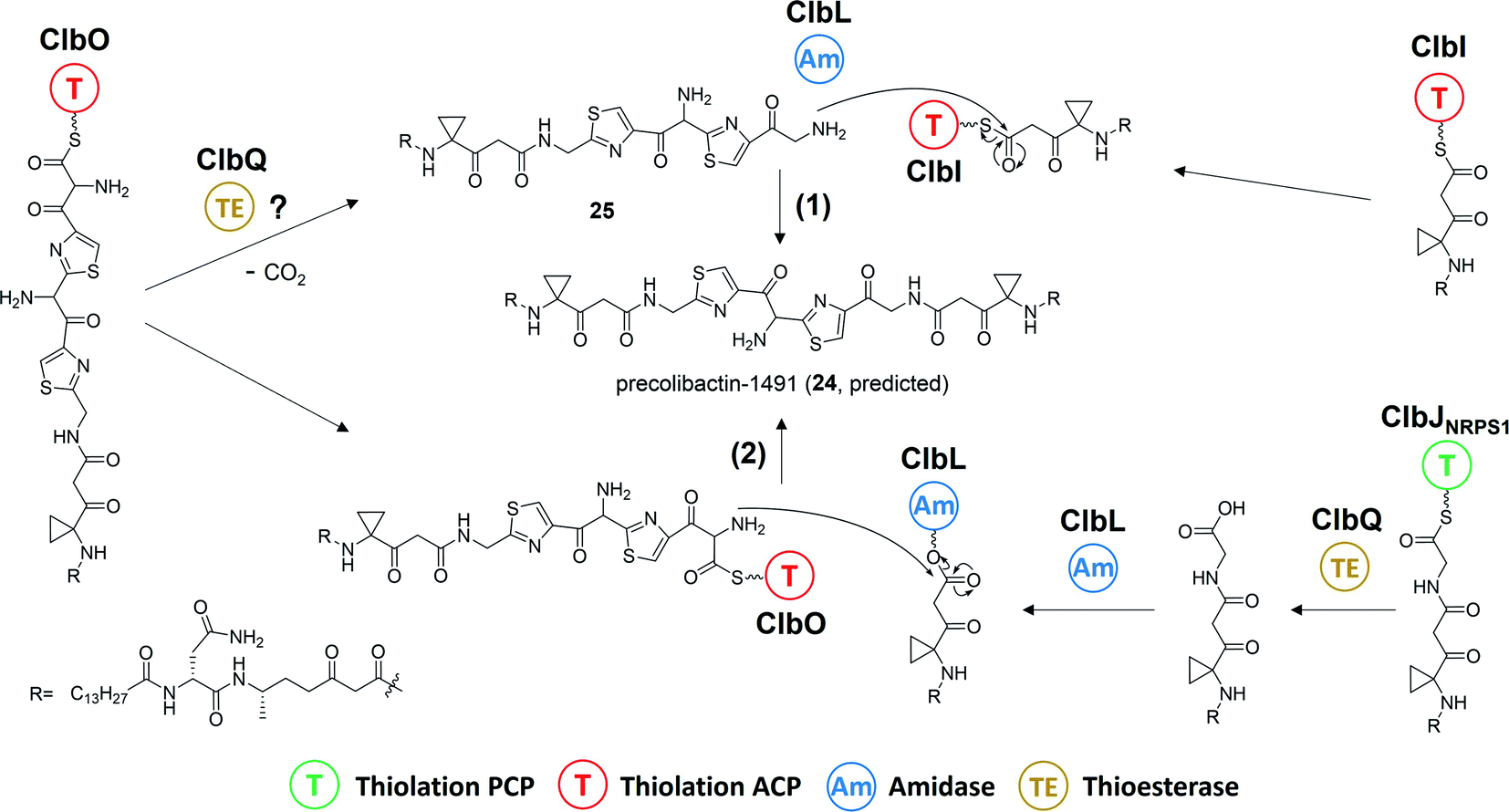 | ||
| Fig. 8 Proposed biosynthesis of predicted precolibactin-1491 (24): pathway (1) from the intermediates ClbO and ClbI was proposed by Balskus and co-workers, and pathway (2) from intermediates ClbO and ClbJNRPS1 was proposed by Crawford, Herzon, and co-workers. | ||
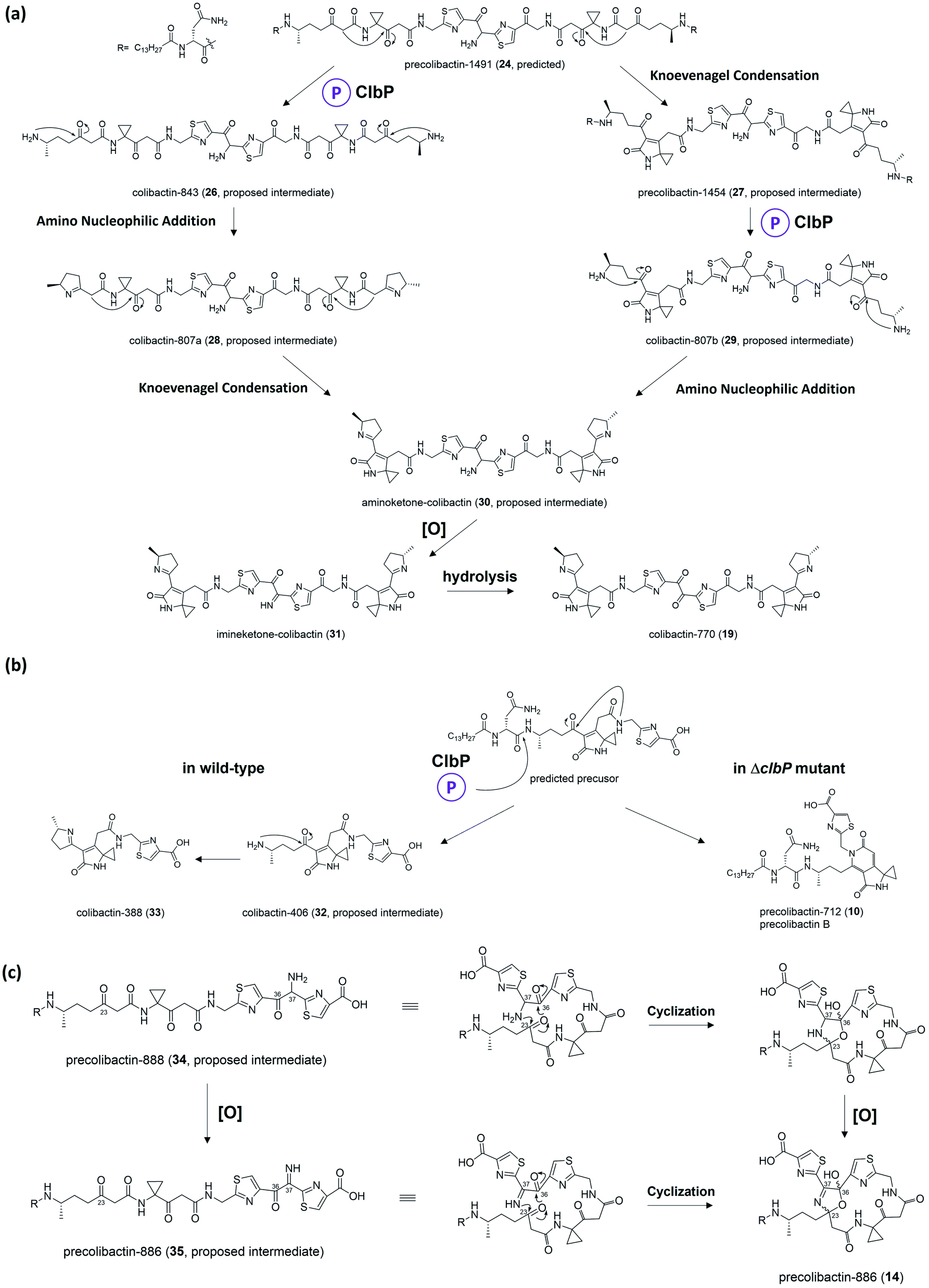 | ||
| Fig. 9 (a) Proposed biosynthesis from precolibactin-1491 (24) to colibactin-770 (19). (b) Proposed divergent biosynthesis with or without ClbP. (c) Generation mechanism of macrocyclic precolibactin. | ||
2.5. Degradation
Herzon, Crawford, and co-workers55 found that the C–C bond in the 1,2-diketone moiety in 19 or the α-imineketone moiety in 31 is readily broken by nucleophilic attack in the environment. Based on this discovery, they proposed that some precolibactins, such as precolibactin-712 (10) and precolibactin-815 (13), may be partly derived from the degradation of downstream products rather than solely from the clb assembly line offloading. The degradation dose provides another perspective on the biosynthesis of (pre)-colibactins. Taking 10 as an example, based on the knockout data in our early study,35 the production of 10 was found to be independent of ClbDEFG, indicating that 10 is likely to be derived from the ClbJ-bound intermediate (Fig. 10a). Thus, theoretically, the disruption of the subsequent ClbK and ClbO may increase or at least not decrease the production of 10. However, domain-targeted metabolomics56 showed that the titer of 10 in the ClbL and ClbP mutants is higher than that in mutants without ClbK domain function. This finding suggests that the degradation of downstream products, such as the offloaded product of ClbO, indeed affords 10. Furthermore, the titer difference is more pronounced for 13 (that of the ClbL mutant is significantly higher than that in the mutants without ClbK domain function and the ClbQ knockout mutant), which suggests that the degradation pathway is dominant in the biosynthesis of 13 rather than assembly line offloading. The degradation may have also occurred in macrocyclic (pre)-colibactins. Herzon and Crawford's research55 indicated that colibactin-562 (36), the ClbP-cleaved product of precolibactin-886 (14), can be spontaneously converted into colibactin-388 (37) through degradation (Fig. 10b). However, in our study,18 we could not detect any degraded compounds such as 37 from 36. This contradictory observation is difficult to explain, and may involve some imperceptible factors that could affect the stability of the macrocyclic precolibactins in these two independent studies. Therefore, more in-depth studies on degradation are required in the future to obtain a conclusive result.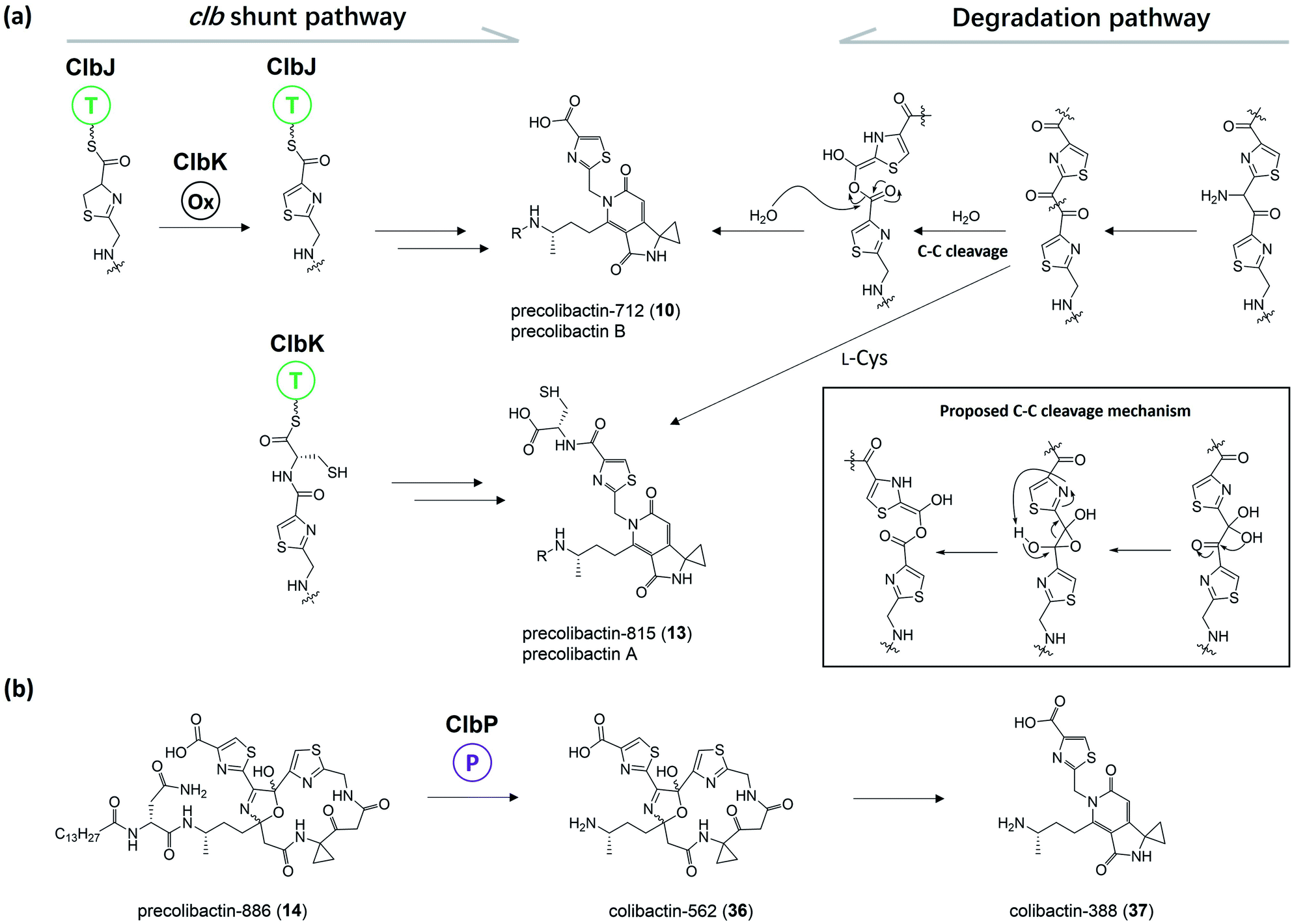 | ||
| Fig. 10 (a) Previously proposed biosynthesis pathway of 10 and 13 (from left to middle) and proposed degradation pathway of 10 and 13 (from right to middle); the C–C cleavage mechanism was proposed by Herzon and Crawford (modified illustration in black box based on ref. 55). (b) Observation of the conversion of 14 to 37 in Herzon and Crawford's research (modified figure based on ref. 55). | ||
3. Unique mechanisms in colibactin biosynthesis
Unlike most modular PKS/NRPS assembly lines, colibactin biosynthesis involves many unique and rare biosynthesis mechanisms, such as the utilisation of a noncanonical PKS extender unit (AM-ACP), the existence of three functionless (deteriorated) cis-AT domains in the PKS module (ClbC, ClbKPKS, and ClbO), and the formation of a spiro-cyclopropane ring directly using SAM as the building block. Additionally, the freestanding type-II TE ClbQ is proposed to be responsible for the offloading of colibactin, but several independent studies have shown the release of intermediates without ClbQ. The amidase ClbL has been suggested to be responsible for amide-bond formation in the final step to generate precolibactin-1491 (21). However, ClbL is also involved in the biosynthesis of 11 and 16, thereby increasing the complexity of the biosynthesis logic in ClbK and ClbO. Therefore, it is necessary to specifically discuss these unique biosynthesis mechanisms of this intriguing NRPS/PKS assembly line.3.1. Unusual PKS extender unit AM-ACP and deteriorated AT domain in PKS
The PKS module in colibactin biosynthesis contains cis-AT and trans-AT domains, which are exclusive in most module PKSs.57,58 In 2014, Crawford and co-workers19 revised the annotation of the pks island and found an inactive AT domain in ClbC, ClbKPKS, and ClbO, which lacks the canonical GxSxG motif (the catalytically active site serine is responsible for covalently tethering the extender unit).59 They proposed that these inactive AT-containing PKSs are inactivated AT ancestral relics of cis-AT but have evolved to use a freestanding trans-AT domain.19,59 Given that ClbC lacks the active AT domain but remains responsible for the malonate extension, they suggested that ClbC may load malonyl-CoA by the help of other trans-AT PKS systems or by interacting with fatty acid biosynthesis.36 This hypothesis was later proven by Balskus and co-workers, who found that ClbC can load malonyl-CoA in the presence of ClbBPKS, ClbI, or the trans-AT domain FabD from fatty acid synthesis in vitro.44 The other two deteriorated AT PKSs, ClbKPKS and ClbO, were found to accept the unusual PKS building block AM. AM is rarely found in common PKS products. To the best of our knowledge, only zwittermicin,34,60 guadinomine61 and lumiquinone A62 involve AM as an extender unit in their biosynthesis. In 2015, Piel and co-workers discovered that ClbD and ClbF are homologous to ZmaG and ZmaI in zwittermicin biosynthesis and produce the AM-ACP extender unit.45 The biosynthesis of the AM unit occurs through the following steps: ClbH-A1 activates L-serine, which is then transferred to the holo-ClbE and finally forms AM-ACP-ClbE through the dehydrogenases ClbD and ClbF (Fig. 4b). Later, Balskus and co-workers characterised the ClbG responsible for transferring AM-ACP to multiple PKS modules. Moreover, their in vitro experiment showed that not only these deteriorated AT domains containing PKS (like ClbKPKS and ClbO), but also the cis-AT PKS ClbI, recognised the AM-ACP.443.2. ClbH uses SAM to form a spiro-cyclopropane ring
The cyclopropane ring is a very intriguing structure that is extensively present in terpenoids yet rarely present in NRPS/PSK products. Along with the identification of 6, Crawford and co-workers added L-[U-13C]-methionine and [2,2,3,3-D]-labelled ACC to distinguish the formation of cyclopropane through the direct or indirect incorporation of ACC.36 They observed a mass shift of 4 Dalton when adding the former, but no shift when adding the latter, suggesting that the ACC unit is involved in the formation of cyclopropane in an indirect way. Similar results were reported by Müller and his co-workers for feeding experiments with L-[U-13C,15N]-methionine and L-[methyl-D3]-methionine.38L-methionine is involved as a precursor in cyclopropane formation, possibly through an intramolecular SN2-type attack of a carbanion in ethylene biosynthesis, which requires ACC synthase to transform SAM into ACC.63 Thus, the deficiency of ACC synthases in E. coli as indicated by bioinformatics analysis inspired them to propose that ClbH acts as an ACC synthase. Later, the ClbH-A1 domain was found to activate L-serine and load it onto ClbE to participate in further AM-ACP synthesis; the remaining domain C-A2-T seems unable to function as an ACC synthase. Thus, Balskus and co-workers re-analysed the data and proposed that the ClbH-A2 domain activates an unusual nonproteinogenic amino acid, SAM, as the building block to form cyclopropane.39 Further biochemical experiments have been conducted to verify this hypothesis by testing the SAM activation and loading activity of ClbH, but no ACC-ClbH thioester has been detected in experiments. Thus, ClbH cannot work alone to generate cyclopropane. Results of in vivo experiments further indicate that ClbI may participate in malonate extension and cyclopropane formation. Interestingly, bioinformatics analysis of the KS domain in ClbI shows that the conserved active-site cysteine, which is responsible for transiently tethering the upstream intermediate and waiting for decarboxylation-driven Claisen condensation,42,64 was replaced by serine. A similar mutation has been found in the starter KSS domains, such as PksF65 and TaK,66 which perform the decarboxylation of the ACP-tethering extender unit. Thus, the KS domain in ClbI may not participate in malonate extension. Furthermore, mutating serine (S178) to alanine abolishes the downstream products. Based on these observations, they proposed two possible functions of the KS domain in ClbI that participated in cyclopropane ring formation: (1) the serine (S178) of ClbI potentially serves as a general base for promoting the cyclisation of the ClbH-tethered intermediate, and (2) the serine (S178) may perform a similar function to cysteine in the canonical KS domain to tether an intermediate onto ClbI during cyclopropane formation. The question of whether the malonate extension can be performed solely by ClbI or involves other KS domains in the assembly line, such as ClbB and ClbC, requires further study (Fig. 11).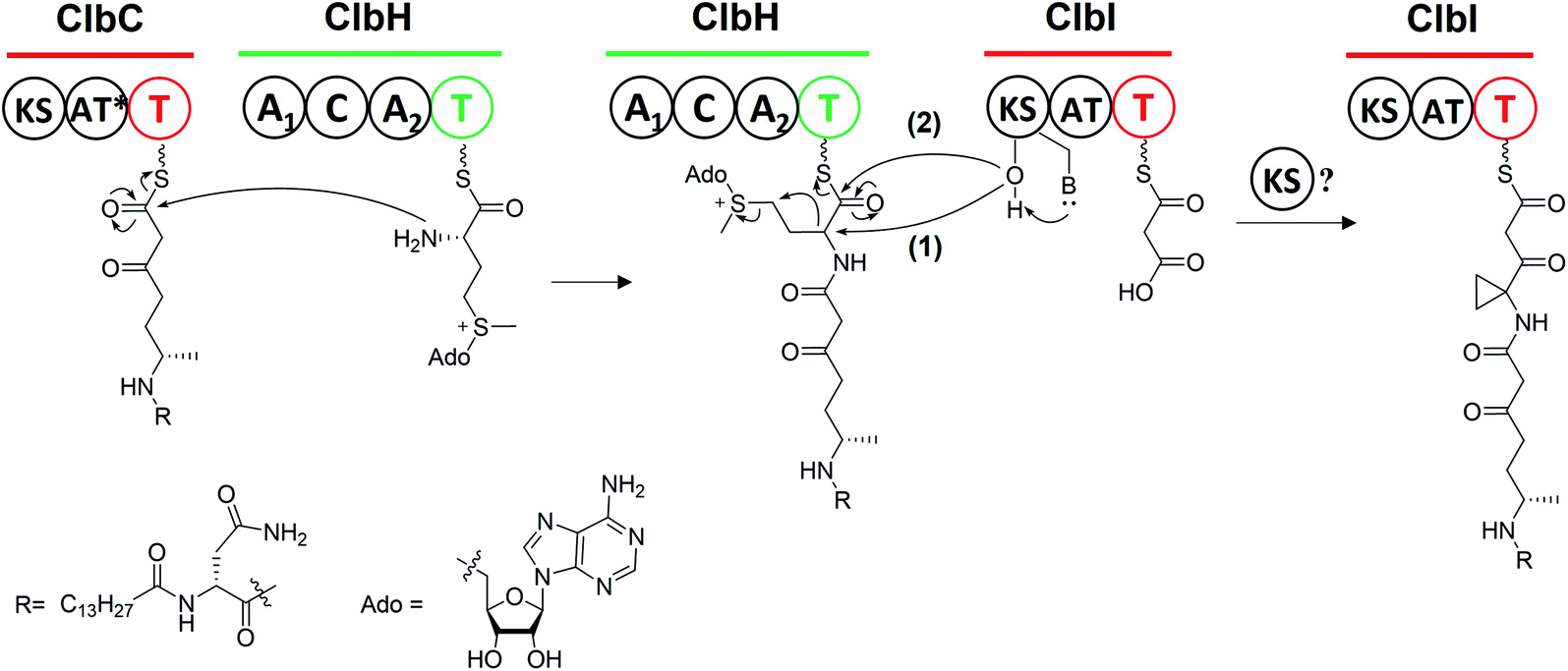 | ||
| Fig. 11 Biosynthesis machinery of the cyclopropane ring (ClbH utilises a non-proteinogenic amino acid SAM as the building block, with subsequent nucleophilic attack of the upstream acyl-S-T-ClbC to afford ClbH-tethered intermediates; later, the serine (S178) of the KS domain in ClbI (1) potentially acts as a general base to promote the transformation from SAM to cyclopropane or (2) to tether an intermediate onto ClbI during cyclopropane formation; the intermediate is then catalysed by itself or other KS domains to finish another round of malonate extension). | ||
3.3. ClbQ, a type-II TE, is responsible for the offloading procedure
There are two types of thioesterase (TE) in NRPS/PKS. Type-I TE is located at the terminus of the final module of PKS and NRPS megasynthase to mediate the offloading of the final product, whereas type-II TE is free-standing and is in charge of diverse functions.67,68 Type-II TE is commonly used as a corrective cleaner through hydrolysis to remove incorrect acyl or peptidyl groups from misprimed PKS and/or NRPS assembly lines.69–71 The NRPS/PKS assembly line of colibactin does not contain intrinsic type-I TE. Type-II TE ClbQ is assumed to be in charge of the offloading of colibactin. In an early work by our group, a ΔclbP/ΔclbQ mutant showed dramatically decreased amounts of upstream intermediates and a 22-fold increase in the amount of precolibactin-886 (14).35 Our in vitro experiments have also shown that ClbQ readily hydrolyses the N-acetylcysteamine thioester (SNAC) derivatives of upstream precolibactins (Fig. 12). Similar conclusions have been drawn by the Brunauer laboratory.72 They also found that ClbQ shows low specificity but a strong preference for upstream intermediates. Additionally, they found that ClbQ offloads the AM unit from AM-ACP-ClbE. Accordingly, they provided another explanation for the increase in the production of 14 in the ΔclbQ mutant, i.e., it may be due to not only the release of upstream products caused by the presence of ClbQ, but also the stable existence of the AM unit ensured by the absence of ClbQ, thereby increasing the yield of 14. Furthermore, many enzyme-bound thioester intermediates in the assembly line were observed to be released in the absence of ClbQ in the domain-targeted metabolomics analysis conducted by Crawford and co-workers.56 Interestingly, their analysis shows that 14 is independent of ClbQ, but the titer has significant differences (a slightly decreased yield in the ClbQ inactivation mutant) compared with our data. This finding may be attributed to the difference in the site-mutagenesis methods used by Crawford and co-workers and the gene knockout in our study. Nevertheless, these findings suggest that ClbQ releases early-stage intermediates of clb and that some of the downstream intermediates may be released independently. Notably, the domain-targeted metabolomics analysis of precolibactin-1489 (17) shows that 17 is ClbQ dependent.15 Additionally, we detected precolibactin-943 (15), the offloaded product of intermediate-ClbO, in the ΔclbP/ΔclbQ mutant, suggesting that the offloading of intermediate-ClbO may be ClbQ independent.18 These observations indicate that ClbQ may participate in the final-stage biosynthesis of colibactin-770 (19) through an uncharacterised process(es). The function(s) of ClbQ deserves more in-depth study.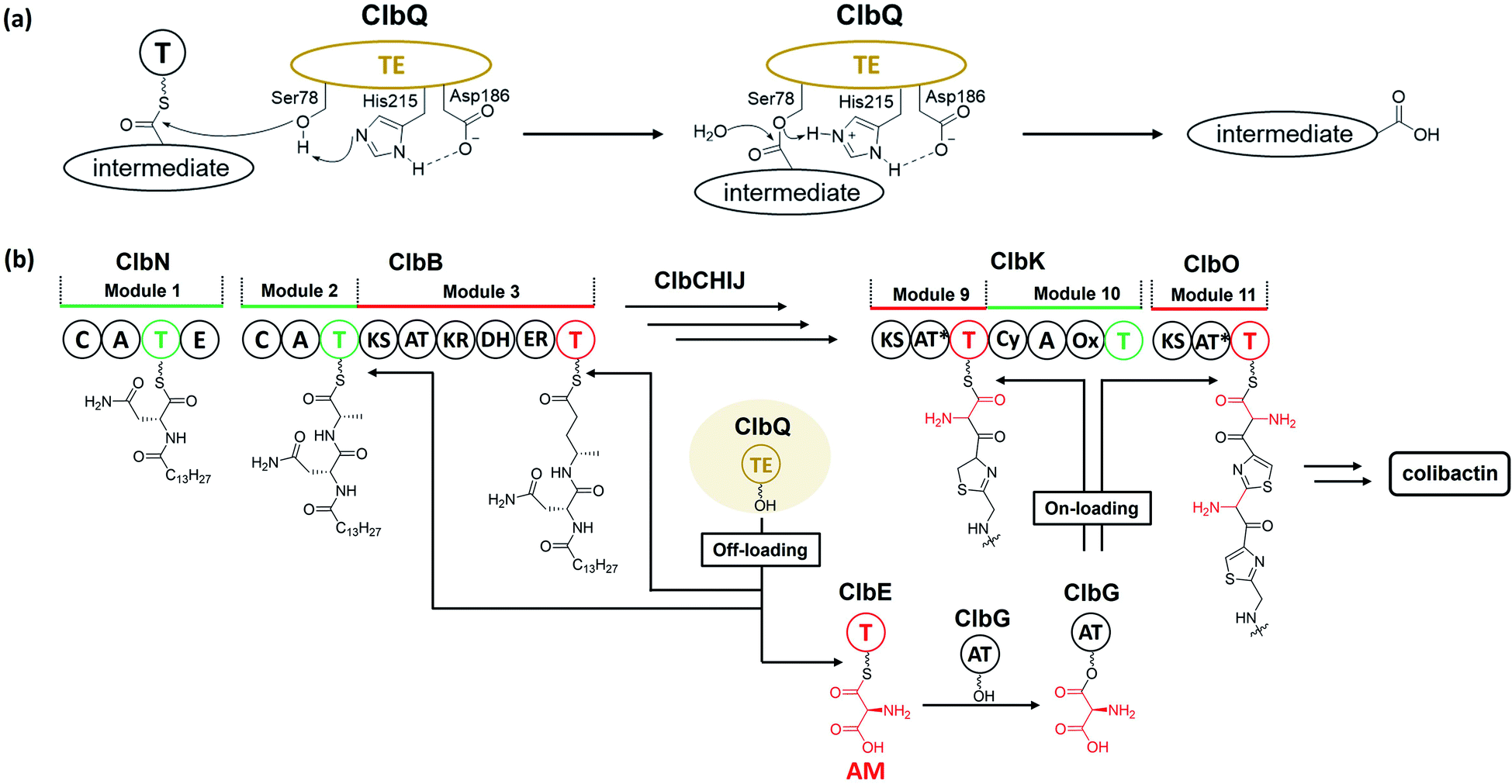 | ||
| Fig. 12 (a) Offloading mechanism of ClbQ (the nucleophilic oxygen atom of Ser78 is activated by His215 and Asp186, and then attacks the thioester bond of the thiolation domain to yield intermediate-O-Ser78; later, other nucleophiles such as H2O attack intermediate-O-Ser78 to finally afford the hydrolysed intermediate). (b) Verified functions of ClbQ (ClbQ offloads the early-stage intermediates; it also offloads the AM unit, a key building block, to interrupt the biosynthesis of late-stage intermediates). | ||
3.4. ClbL, an amidase, is responsible for amide-bond formation
ClbL, an amidase containing a conserved Ser179-Ser155-Lys80 catalytic triad,73,74 is involved in the biosynthesis of 11 and 16 and is responsible for the final step of precolibactin-1491 (24) formation. Amidases commonly function as hydrolases, consistent with the observation that ClbL cleaves the amide bond of asparagine in vitro.30 It also exhibits amide-/acid-/acyl-transferase activity,75 which is assumed to be due to the fact that the nucleophilicity of different nucleophiles towards the intermediate-O-Ser is stronger than that of oxygen in H2O. However, the catalytic machinery of ClbL remains obscure. The proposed role of ClbL in the biosynthesis of 24 is illustrated in Section 2.4.1. Regarding the role of ClbL in the biosynthesis of 11 and 16, a systematic mutagenesis experiment18 revealed that ClbL catalyses the amide-bond formation between the amino of the AM unit and the carbonyl of intermediate-ClbJNRPS2 and intermediate-ClbK (Fig. 13). Knocking out ClbK and ClbO also abolishes the production of 11 and 16, respectively, suggesting that the AM unit originates from AM-ACP on the PKS module instead of the free-standing AM-ACP-ClbG. Moreover, considering that 11 and 16 do not undergo malonate extension in ClbK and ClbO, based on PKS chemistry logic, it is unclear whether the biosynthesis of 11 and 16 requires the KS domain in ClbK and ClbO, respectively. Nevertheless, on the basis of these discoveries, we hypothesise that the ClbL-mediated amide-bond formation of 11 and 16 may be a shunt reaction during the amino-malonate extension. Taking the biosynthesis of 11 as example, when the KS domain transiently binds the upstream intermediate of ClbJNRPS2, decarboxylation-driven Claisen condensation (C–C bond formation) and ClbL-driven amide bond formation occur competitively. The amino-malonate extension product is subjected to the action of downstream enzymes, ClbKNRPS module and ClbO, to afford 15 and 16. The amide extension product cyclises under mediation by ClbL with two proposed mechanisms: (1) ClbL again acts as a general base to remove the proton of NH and promote cyclisation in the ClbKPKS domain; and (2) ClbL acts as a TE to offload the intermediate, Lys80 deprives the proton of intramolecular NH with subsequent electron transfer, and oxygen attacks the acyl-O-Ser179 to yield 11. These hypotheses require further testing through site-mutagenesis and in vitro experiments in the future.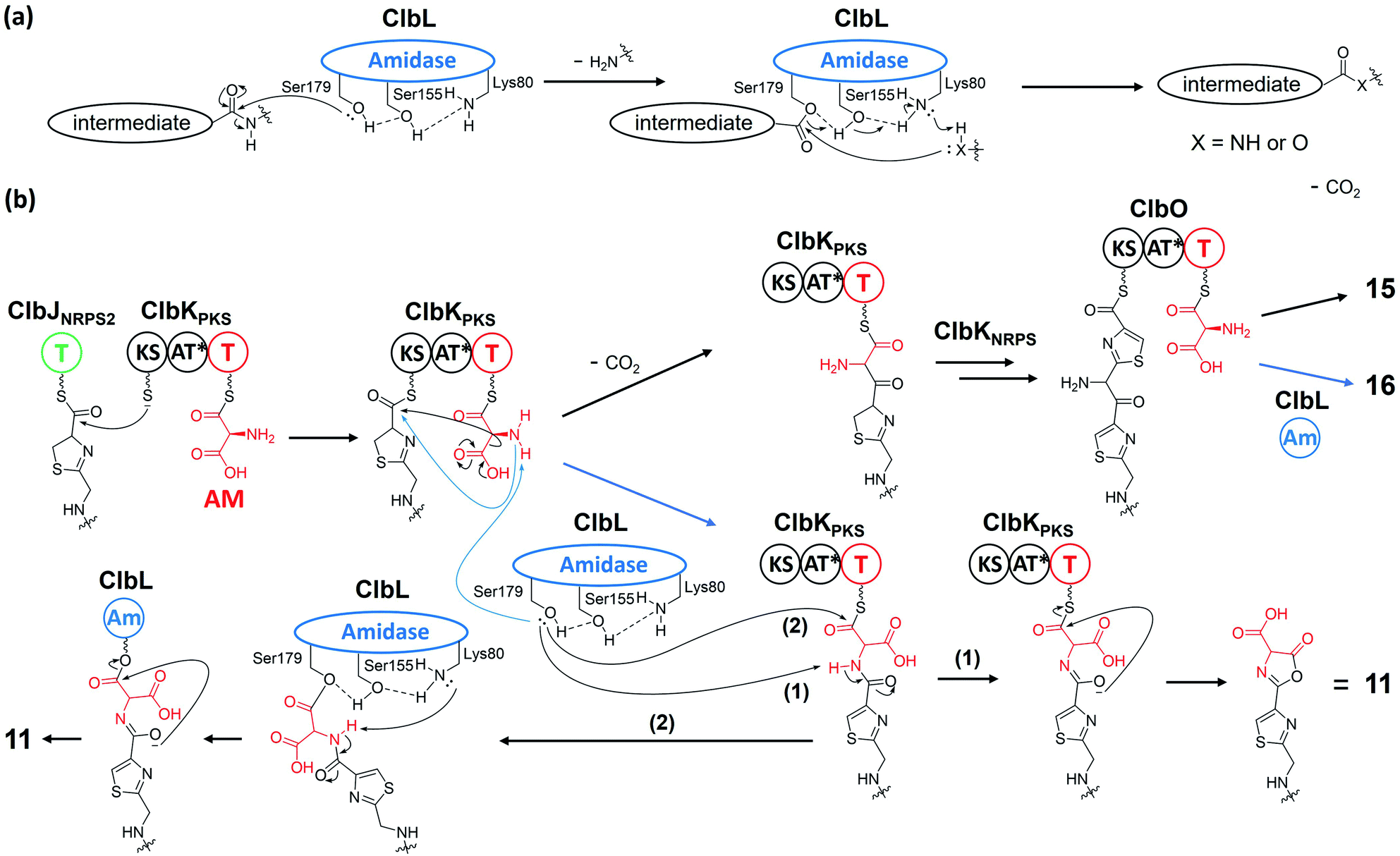 | ||
| Fig. 13 (a) Proposed mechanisms of ClbL acting as a hydrolase or amide-/acid-/acyl-transferase (the nucleophilic oxygen atom of Ser179 is activated by Ser155 and Lys80 before attacking the carbonyl group to yield intermediate-O-Ser179; later, nucleophiles such as H2O, NH2-, and OH-attack the carbonyl of intermediate-O-Ser179 to yield the respective products). (b) Proposed mechanisms of ClbL for the amide-bond formation of precolibactin-795a (11) and precolibactin-969 (16) (the nucleophilicity of the amino group of AM is increased by ClbL; then, during the subsequent decarboxylation-derived malonate extension, the activated amino competitively attacks the transiently bound acyl-S-KS). | ||
3.5. Self-resistance mechanism and MATE transporter ClbM
pks+ E. coli uses a combination strategy to avoid self-toxicity. At the onset of NRPS/PKS assembly, ClbN and ClbB install the prodrug motif on colibactin,13,14 thereby preventing the formation of the bioactive imine moiety. pks+ E. coli also produces ClbS,46–49 a cyclopropane hydrolase that opens the genotoxic cyclopropane in these offloaded intermediates. Furthermore, the multidrug and toxic compound extrusion (MATE) inner-membrane transporter ClbM pumps intermediates into the periplasm.76,77 Notably, in Jobin and co-workers' research,78 crystallographic analysis of ClbM indicated that the binding pocket spans nearly 40 Å. Based on this discovery, they hypothesised that ClbM specifically transports 700–900 Da precolibactins, which is obviously inconsistent with the size of the colibactin precursor precolibactin-1491 (24). To be clear, knocking out clbM does not lead to a loss of genotoxicity, indicating that precolibactin-1491 (24) can also be transported to the periplasm by other means. However, given that ClbM is the only natural transporter on clb, the fact that the size of its binding pocket is incapable of satisfying the final product raises two questions: (1) Is the hypothesis of specifically transporting 700–900 Da precolibactins rigorous? (2) If the hypothesis is correct, should ClbM work for the ‘shunt products’ or the ‘real clb natural product’ rather than the ‘final product (like 24)’ of the clb assembly line?4. Biological activities of clb metabolites
4.1. DNA-damage activity of colibactins and the underlying mechanisms
Colibactin-770 (19) and colibactin-645 (18) are two mature colibactins with different skeletons that induce DNA DSBs. Intriguingly, they inflict DNA damage through different mechanisms. DNA DSBs are induced by 19 through DNA alkylation and crosslinking and by 18 through copper-mediated oxidative cleavage.The DNA damage-inducing mechanism of linear colibactin 19 was elucidated based on its structure, i.e., 19 contains two ‘warheads’ with the azaspiro[2.4]bicyclic-ring substructure. The α,β-unsaturated imines of this substructure render the cyclopropane rings electrophilic, thereby facilitating the formation of colibactin–DNA adducts. Thus, the generation of DNA interstrand crosslinks (ICLs) is initiated by twofold cyclopropane ring opening. At present, one of the two warheads is well-known to react at the N3 of adenine, whereas the second alkylation site remains an open question. Notably, precolibactin-546 (6) with a single warhead also causes ICLs in vitro in the presence of dithiothreitol or β-mercaptoethanol, which suggests the involvement of cyclopropane ring opening and Michael addition.36 However, the DNA crosslinking activity of 6 is relatively weak, and a reducing agent is necessary, indicating that it cannot be the mode of ICL formation observed in pks+ E. coli-infected cells.
Colibactin can induce intracellular DNA ICLs and DSBs, whereas extracellular DNA exposed to pks+ E. coli exhibits DNA ICLs instead of DSBs,36 suggesting that the DSBs are derived from the repair pathway of DNA ICLs (Fig. 14). HeLa cells infected with pks+ E. coli respond to the replication stress induced by ICLs through the Ataxia Telangiectasia Mutated-and Rad3-related (ATR) signalling pathway, which increases the phosphorylation of ATR, Chk1, and RPA.51 Subsequently, the Fanconi anaemia (FA) repair pathway is recruited to repair the damage.51 The FANCD2 is monoubiquitinylated and colocalises with γH2AX, permitting incisions of ICLs followed by DSBs. Finally, the homologous recombination (HR) pathway is activated. The HR pathway is characterised by increased phosphorylation of ATM, H2AX, and Chk2, together with the formation of p53-binding protein 1 (53BP1) foci.7,51
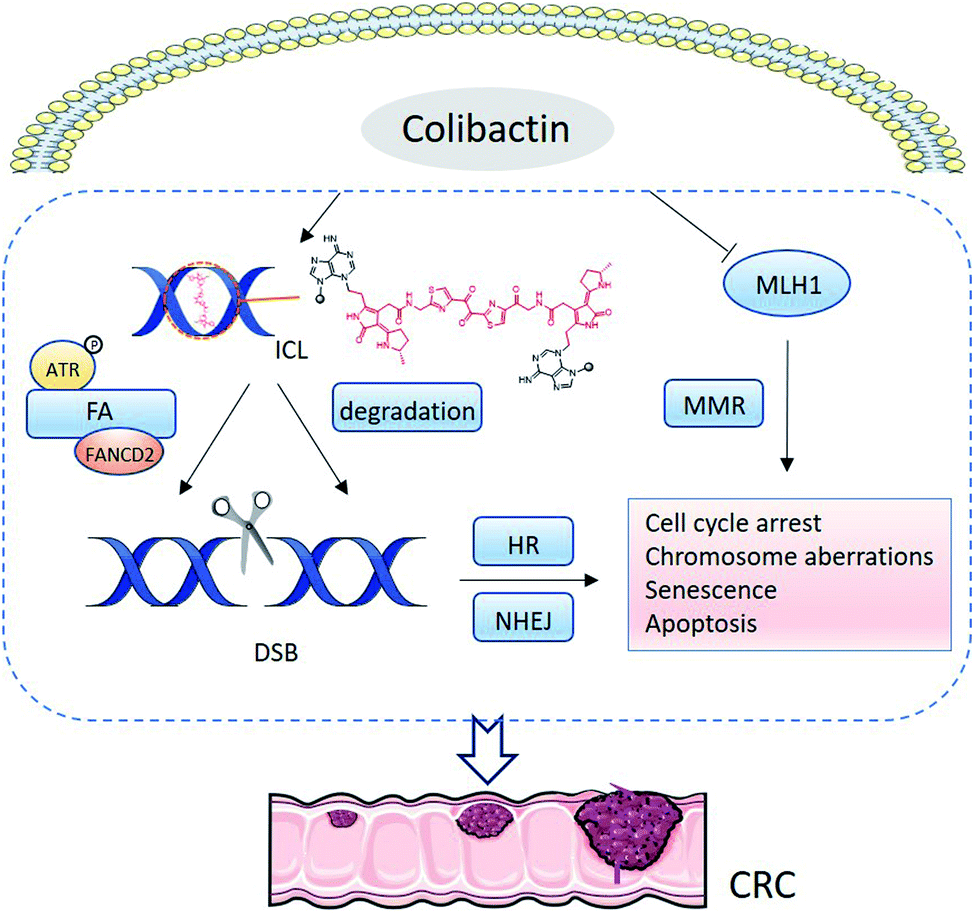 | ||
| Fig. 14 Mechanism of conventional colibactin-induced DNA damage. Dual electrophilic cyclopropane warheads are adducted with adenine residues by ring opening, forming ICLs. In mammalian cells, ICL-induced replication stress activates ATR signalling and recruits the FA pathway to repair the damage. In the repair process, the co-localisation of FANCD2 with γH2AX permits incisions of the ICLs, resulting in DSBs, which are repaired by the HR pathway. Additionally, colibactin-derived ICLs can be spontaneously degraded through depurination and elimination in 3′-phosphate, generating DSBs. This phenomenon likely activates the NHEJ repair pathway. Colibactin can also inhibit MLH1, disturbing MMR. DNA damage arises from DNA DSBs, and the inhibition of MMR leads to cell-cycle arrest and chromosome aberration. When cell repair fails, the cell undergoes senescence or apoptosis. The accumulation of DNA damage results in CRC. | ||
In addition to the FA repair pathway, DNA DSBs also arise from ICL degradation. Colibactin-derived ICLs are reportedly unstable in depurination and 3′-phosphate elimination, and repeating the degeneration at the remaining intact alkylated nucleotide leads to a DSB.79 HR and nonhomologous-end joining (NHEJ) are the two main DNA DSB repair pathways. Cells deficient in Ku80 are sensitive to pks+ E. coli, suggesting the involvement of NHEJ in DNA repair.8 FA is linked only to the HR pathway, so DSBs generated from the spontaneous depurination are proposed to activate the NHEJ repair pathway. Furthermore, pks+ E. coli can induce ROS and inhibit the mismatch repair (MMR) protein mutL homologue 1 (MLH1), further leading to DNA damage (Fig. 14).80
Colibactin-645 (18) with a macrocyclic skeleton exerts direct DNA-DSB activity through copper-mediated oxidative cleavage (Fig. 15) rather than through the formation of ICLs.18 In the presence of Cu(II) in vitro, 18 causes significant DNA DSBs. HeLa cells show the formation and co-localisation of foci derived from γH2AX and 53BP1 when treated with 18 or pks+ wild-type E. coli. The addition of a Cu-sequestering agent significantly decreases the levels of DNA damage. Copper-mediated oxidative DNA cleavage is generally proposed to involve copper-complex-induced ROS or reactive metal-oxo species (RMOS). Subsequently, the ROS or RMOS attack the DNA and initiate DNA cleavage. A suitable reductant or photo-irradiation is initially required to reduce Cu(II) to Cu(I), which then reacts with O2 or H2O2 to generate ROS. In this case, the macrocyclic scaffold of colibactin can serve as a reductant and a binding site. The colibactin·Cu(II) complex, the structure of which is proposed in Fig. 15a, is reduced by the ligand to form colibactin·Cu(I). The addition of mannitol, dimethylsulfoxide, or superoxide dismutase (SOD) (the former two are hydroxyl radical scavengers and the latter one catalyses the conversion of the superoxide radical into O2 and H2O2) does not influence the activity of precolibactin-969.18 Conversely, the H2O2 blockers potassium iodide and catalase significantly inhibit the cleavage reaction, suggesting that H2O2 is involved in mediating DNA cleavage in vitro.29 This is consistent with the previous observation that the fluorescence of H2-DCFDA (which acts as a sensor of hydroxyl and peroxyl radicals, and of hydrogen peroxide production) increases significantly in cells treated with colibactin-producing E. coli.9 The colibactin·Cu(I) complex is proposed to coordinate with O2 to generate colibactin·Cu(II)–OOH, which can be converted into HO˙ and colibactin·Cu(II)–O˙ (Fig. 15b). Colibactin·Cu(II)–O˙ is the active species responsible for DNA carbon–hydrogen bond activation.81 This mechanism is akin to the proposed mechanism for the generation of activated bleomycin in vivo,82 with differences in the metal usage and the intrinsic metal-reduction activity of the compounds.
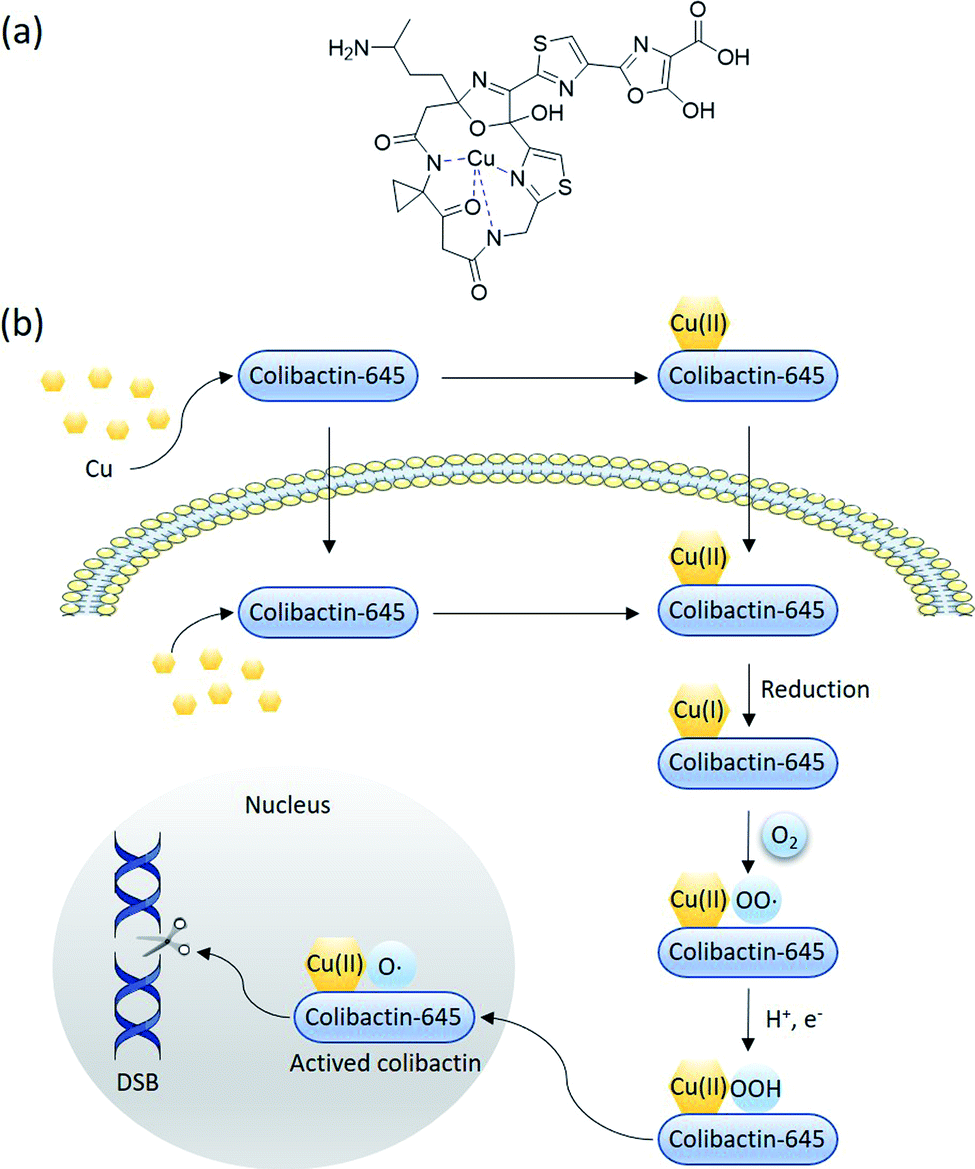 | ||
| Fig. 15 Proposed mechanism of macrocyclic colibactin-induced oxidative DNA cleavage (modified figure based on ref. 18). (a) The proposed structure of the colibactin·Cu(II) complex. (b) Colibactin-645 binds to copper in the intestinal lumen to form the colibactin·Cu(II) complex, which is quickly transported into epithelial cells. Colibactin-645 may also quickly enter the cell after being secreted whilst forming the colibactin·Cu(II) complex with intracellular copper. Colibactin-645 reduces this complex to colibactin·Cu(I), which then coordinates with O2 to generate colibactin·Cu(II)–OO˙. In the presence of H+ and e−, colibactin·Cu(II)–OO˙ is converted to colibactin·Cu(II)–OOH, which can in turn be converted into HO˙ and colibactin·Cu(II)–O˙. The latter is the active species responsible for DNA carbon–hydrogen bond activation. | ||
Based on structure–activity relationship analyses, we hypothesise that the loss of the N-terminal fatty acyl–asparagine residue helps mature colibactins to enter epithelial cells; the ‘active colibactin’ intercalates into DNA through the thiazole/5-hydroxyoxazole tail. However, further investigation should be conducted to verify this hypothesis and to confirm the mechanism of copper-mediated oxidative DNA cleavage.
The pks island produces multiple active metabolites with various modes of action. They may work synergistically to achieve DNA-damage activity. Thus, the elucidation of the mechanisms by which various colibactins induce DNA damage will provide insight into the development of specific gene-therapy strategies for CRC.
Despite the different DNA-damage-inducing modes of the two colibactins, various colibactin-producing E. coli strains can initiate DNA damage. Short-term exposure to pks+ E. coli leads to the formation of anaphase bridges and chromosomal abnormalities in dividing cells,9 whereas persistent exposure promotes the accumulation of mutations.8 To maintain genome integrity, cells respond to DNA damage by activating the DNA-repair pathway. Nevertheless, improper DNA repair triggers the senescence or apoptosis of the target cells. Colibactin induces DNA DSBs in lymphocytes, followed by cell-cycle arrest and cell death by apoptosis.10 Infection with pks+ E. coli leads to chronic DSBs, prolonged cell-cycle arrest, and cellular senescence.9pks+ E. coli upregulates miR-20a-5p expression through the c-Myc transcription factor in infected cells. The binding of miR-20a-5p to SUMO Specific Peptidase 1 (SENP1) mRNA 3′-UTR triggers the transcriptional silencing of SENP1, leading to the accumulation of SUMO-conjugated p53, thereby resulting in cellular senescence.83 Moreover, the senescence is transmissible and promotes tumour-cell growth through the secretion of hepatocyte growth factor (HGF) in vitro and in vivo.9,83 In short, pks+ E. coli exposure can also promote the progress of CRC through senescence.
4.2. Other bioactivities of clb metabolites
In addition to the DNA-damage genotoxicity inflicted by colibactin-producing E. coli, pks+ E. coli also has cytotoxic, antibacterial, inflammation-promoting, anti-inflammation, and analgesia bioactivities. These diverse bioactivities are associated with the different moieties of colibactin, precolibactins, and other bactins produced by various pks+ E. coli.The pks island is also related to anti-inflammatory activity. Compared with wild-type Nissle 1917, ΔclbA Nissle (an isogenic mutant of Nissle 1917) does not show DNA-damage activity in eukaryotic cells, whereas its anti-intestinal inflammation effect is abrogated.75 The DNA-damage and anti-inflammatory activities are restored when the wild-type clbA gene is reintroduced to the chromosome of Nissle ΔclbA (Nissle ΔclbA + clbA). The abolition of the ACP activator ClbA possibly results in the abrogation of the colibactin, precolibactin, and colibactin moieties, which may contribute to the anti-inflammation activity. However, anti-inflammation activity of these colibactin moieties cannot be observed in the pathogenic pks+ E. coli strains CFT073 and M1/5. We cannot exclude the possibility of ClbA involvement in the syntheses of other anti-inflammatory products because clbA participates in siderophore biosynthesis. Therefore, the functional pks island appears to be a double-edged sword in terms of inflammation in various pks+ E. coli strains with diverse genomes.
5. Relationship between colibactin and CRC formation
5.1. Colibactin plays a carcinogenic role in CRC
Previous studies have shown a high prevalence of pks+ E. coli in CRC.21,22 Around 55–67% of CRC patients carry pks+ E. coli, whereas less than 20% of healthy people carry pks+ E. coli.21,22 In animal experiments, pks+ E. coli enhances tumourigenesis in FAP and CRC models and promotes invasive carcinoma in azoxymethane (AOM)-treated interleukin-10-deficient mice (IL10−/−).14 The deletion of the pks island decreases tumour multiplicity and invasion.22 Multiple intestinal neoplasia (Min) mice infected with pks+ E. coli 11G5, a strain isolated from a human CRC biopsy, display a remarkably increased number of visible colonic polyps after seven weeks of injection.3 Data obtained from an AOM/dextran sodium sulphate (DSS) colon-cancer mouse model,83 xenograft model,83,91 and other models92 also support the carcinogenic properties of colibactin. However, these models are essentially tumour models, so the development of cancer may be attributed to genetic susceptibility. Whether colibactin initiates or promotes carcinogenesis remains unclear.Recent evidence shows that colibactin plays a causative role in CRC. The first and most important clue is that the mutational fingerprint of colibactin is detected in CRC patients.25,26 Whole-genome sequencing or whole-exome sequencing analyses reveal the mutant signature of colibactin. Mammalian cells exposed to pks+ E. coli show increased single-base substitutions (SBSs) and the induction of a characteristic small indel signature (ID-pks).25 SBSs show a preference for T > N substitutions within AT-rich DNA regions, which is defined as a pks-specific SBS signature (SBS-pks), whereas ID-pks is characterised by single T deletions at T homopolymers.25 The substitutions in specific AT-rich motifs are consistent with the sites at which colibactin adducts with adenine by alkylation. Moreover, the same mutational signature is detected in human cancer genomes.25,26 Out of 5876 CRC samples, SBS-pks and ID-pks are enriched in around 6.5% and 7.1% of patients, respectively, whereas 4.65% of samples are high in SBS-pks and ID-pks.25 Furthermore, APC is the most frequently mutated gene in CRC,93 in which 5.3% of cancer-driven mutations match the motifs induced by colibactin.25 In short, the high level of colibactin-induced mutational fingerprints in CRC patients suggests that colibactin plays a direct role in CRC.
Studies using models without genetic susceptibility and carcinogens provide another important evidence. Short-term pks+ E. coli infection inflicts DNA damage to healthy primary colon epithelial cells, resulting in genomic instability.24 Short-term exposure is closer to the dynamic process of crypt regeneration in the colon. Organoids from primary murine colon epithelial cells exposed to pks+ E. coli for 3 h exhibit DNA damage and Wnt independence. Wnt-independent organoids exhibit higher organoid-forming capacity and proliferation, which is highly similar to the character of CRC cells.24 Furthermore, infection with pks+ E. coli triggers the formation of invasive colonic tumours in a DSS-induced chronic inflammation mouse model.23
Overall, the high abundance of pks+ E. coli in CRC patients and the mutant fingerprint suggest the carcinogenic role of colibactin in CRC development. However, the colibactin mutational signature is also found in healthy human colon crypts,25,26 suggesting that colibactin may initiate cancer only under specific conditions or certain mutant accumulations. The underlying molecular mechanism of carcinogenicity remains obscure.
5.2. Colibactin modulates the tumour microenvironment to promote cancer growth
The tumour microenvironment is the environment surrounding the tumour and can affect cancer development. Colibactin directly interacts with DNA, induces mutations, and promotes a pro-carcinogenic environment. Colibactin contributes to the colonisation capacity of E. coli94 and alters the microbial diversity in the gut.95 Microbial dysbiosis exerts a cancer-promoting effect.96 After 35 days of colonisation by pks+ E. coli, the gut microbiota substantially changes at the taxonomical and functional levels.89 This phenomenon may be due to the antibiotic activity of pks+ E. coli, which helps genotoxic bacteria to create their own niches.97 Once niches are created, the genotoxic activity of the bacteria helps the bacteria to expand their niches by targeting unrelated microbial taxa in the gut. pks+ E. coli can also impair the permeability of the host intestinal mucosa, leading to increased ROS and in turn altering the composition and function of the gut microbiota.95Furthermore, pks+ E. coli shows immune–modulation activity. In CRC patients, a decrease in CD3+ T-cells is correlated with the colonisation of colibactin-producing E. coli.87 In an APCMin/+ mouse model, pks+ E. coli 11G5 significantly increases gut inflammation and reduces tumour-infiltrating T lymphocytes (CD3+ and CD8+ T cells) as well as antitumour T cells in mesenteric lymph nodes.87 CD45+ T cells and neutrophil percentage decrease in tumour areas treated with E. coli 11G5. Moreover, pks+ E. coli infection shows resistance to PD-1 immunotherapy in an MC38 CRC model.87pks+ E. coli also impairs intestinal permeability in newborn rats and thus promotes the trans-epithelial passage of luminal antigens and hyperimmune response, thereby contributing to the development of immune-dysregulation diseases.98 These results indicate that colibactin-producing E. coli induces a procarcinogenic immune microenvironment and facilitates the development and progress of CRC.87
Additionally, pks+ E. coli alters the physiology of epithelial cells by inducing the apoptosis of epithelial cells, which is compensated by the abnormal proliferation of epithelial cells.98,99 Colibactin-induced senescence is accompanied by the secretion of HGF, which promotes the proliferation of mutant cells,83 which in turn facilitates the development of colon cancer cells.
6. Discussion and perspectives
6.1. Proposed functions of ClbQ and ClbL in the late-stage biosynthesis
Research of the mechanisms of ClbL and ClbQ in the final-step biosynthesis of precolibactin-1491 (24) is inadequate. Nevertheless, according to the research of Balskus and co-workers,16 ClbL likely participates in the offloading of intermediate-ClbI. During this offloading, the amino group of α-aminoketone in 25 possesses stronger nucleophilicity than the oxygen in H2O, thereby forming an amide bond. Additionally, in the study of Crawford, Herzon, and co-workers,15,30 ClbL plays a role similar to that of amide transferase. In the process of hydrolysing the amide bond, the amino group of α-aminoketone attacks intermediate-O-Ser-ClbL to form an amide bond. However, neither of these two proposed mechanisms is backed by sufficient evidence to prove the real function of ClbL. In our re-analysis of all the information to date, we made an interesting discovery about ClbL. In Müller and co-workers' research of precolibactin-546 (6),38 the knockout of clbL significantly decreased the production of 6, indicating that it may be produced by ClbL through the hydrolysis of the larger precolibactin. In contrast, knocking out the downstream clbJ increases the production of 6. These observations imply that 6 may originate not from the hydrolysis of the larger precolibactin, but instead from the ClbL-dependent offloading. A similar observation was reported by Balskus.16 Conversely, in Crawford's research,56 the S179A site-mutation S179A of ClbL did not affect the yield of 6. These contradictory observations may be due to the different methods used in their research (the Müller and Balskus laboratories used gene knockout, and the Crawford laboratory used site mutagenesis). Nevertheless, the two independent studies discussed above provide new insights into the function of ClbL, suggesting that it behaves like a TE to offload intermediate-ClbI (Fig. 16a).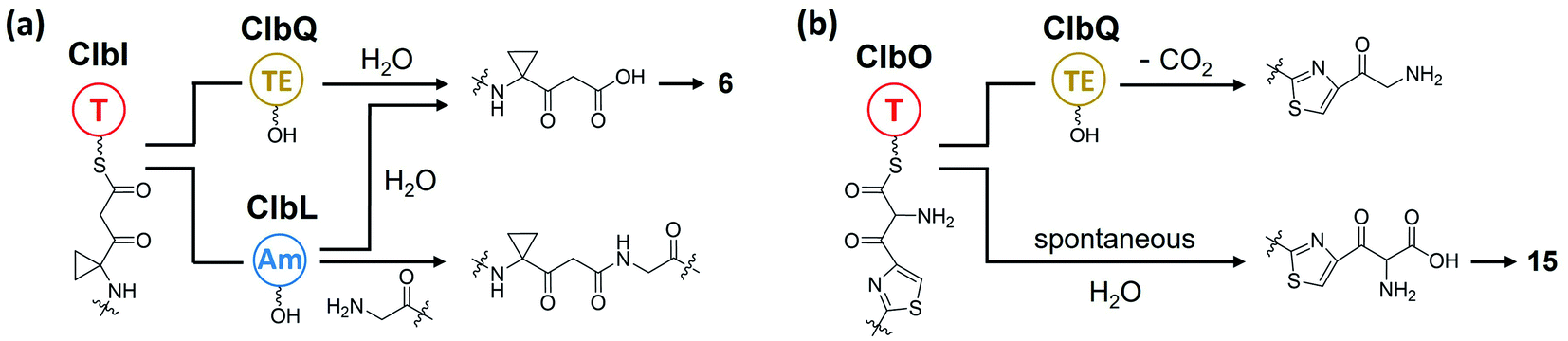 | ||
| Fig. 16 (a) Proposed biosynthesis of 6 and machinery of ClbL catalysis of amide-bond formation. ClbQ and ClbL offload intermediate-ClbI to generate 6; in ClbL-mediated offloading, the amino group of α-aminoketone is a stronger nucleophile than the oxygen in H2O, thereby forming an amide bond. (b) Proposed role of ClbQ in offloading, decarboxylation of intermediate-ClbO, and biosynthesis of 15. | ||
Regarding the role of ClbQ, the observation of 15 in the ΔclbQ mutant18 suggests that the release of the ClbO-bound intermediate may be ClbQ independent. Moreover, if ClbL does indeed catalyse amide-bond formation upon offloading of the intermediate of ClbI, then ClbQ cannot be involved in the offloading of ClbI- and ClbO-bound intermediates. Therefore, it is difficult to propose the role of ClbQ, which may participate in the decarboxylation of intermediate-ClbO (Fig. 16b), similarly to SgcE10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 and CalE7.101 It may also play a similar role such as being a ‘waiting room’68 to tether the intermediate-ClbO and search for ClbL.
100 and CalE7.101 It may also play a similar role such as being a ‘waiting room’68 to tether the intermediate-ClbO and search for ClbL.
6.2. Is clb a flexible or rigorous NRPS/PKS assembly line?
The clb NRPS/PKS assembly line appears to be highly flexible. The starting module, ClbN, prefers myristoyl-CoA (C14), but it can also prefer N-acylates and other substrates ranging from hexanoyl-CoA (C6) to lauryl-CoA (C12).13 This finding is consistent with identified precolibactins with varying chain lengths and patterns in the prodrug motif, ranging from decoacyl to palmitoyl,19,36 similar to xenocoumacin biosynthesis.33 The biosynthesis logic in ClbK and ClbO is also very complex, because AM can be incorporated or not into downstream products with different mechanisms.18,35 The offloading mechanism and the amidase ClbL are also promiscuous.16 In addition to the intrinsically flexible logic of clb, some components of clb also participate in the biosynthesis of other metabolites in E. coli, which further increases the diversity of clb-related metabolites. For instance, ClbA is involved in the biosynthesis of yersiniabactin,102 and AM-ACP-ClbE cooperates with fatty acid biosynthesis enzymes to generate γ-lactam derivatives.103 These results indicate that the clb NRPS/PKS assembly line tends to be flexible, similar to the thalassospiramide biosynthesis gene cluster,104,105 and can synthesise various metabolites in vivo.In contrast, clb shows extremely tight control over the synthesis of colibactin-770 (19)15,16 as the final product. The formation of 19 requires an intact intermediate and an incomplete intermediate of the assembly line. These intermediates are heterodimerically linked through amide-bond formation. Later, the heterodimer is exported from the cytoplasm to the periplasm and cleaves the prodrug motif. Through subsequent spontaneous intramolecular nucleophilic amino addition and Knoevenagel condensation, the genotoxic colibactin finally forms. In this procedure, any mistake abolishes the production.
In summary, clb possesses the paradoxical character of being either flexible or rigorous. It is rigorous in synthesising the final product colibactin-770 (19), which requires the precise designation of the synthesis route, especially for the two-step spontaneous reaction that forms the bioactive moiety. Meanwhile, its flexibility lies in the promiscuity of multiple enzymes and unpredictable offloading mechanism. These properties enable clb to specifically act on DNA DSBs via colibactin coupled with diverse bioactivities on other targets via shunt metabolites.
6.3. Is the genotoxic colibactin a single compound or a mixture?
At present, colibactin-770 (19)15,16 is viewed as being the long-sought genotoxic colibactin produced by pks+ E. coli, or at least its major component. However, owing to the promiscuous activities of multiple enzymes in the clb assembly line, we must reconsider whether there are other genotoxic shunt/final products and underlying genotoxic mechanisms that have not yet been characterised. For example, the macrocyclic colibactin-645 (18)18 exerts direct DNA DSB activity through copper-mediated oxidative cleavage. In addition, as we mentioned in the previous section, ClbA participates in biosynthesis of yersiniabactin,102 a siderophore responsible for mediating the copper density of E. coli.106 We cannot reach a conclusion as to whether yersiniabactin plays a role in the DNA DSBs caused by colibactin-645 (18) in vivo, but it does provide a new perspective on the role of clb metabolites in causing DNA DSBs. Furthermore, the research of Balskus and co-workers53 inspired us to make a more inclusive reconsideration. They observed that compound 39 (Fig. 17a), an ethyl-ester-modified colibactin possessing only one cyclopropane ring, induces DNA DSBs in HeLa cells at 20 μM. The mechanism of this induction is proposed to go through DNA alkylation. This observation suggests that shunt metabolites of clb containing a cyclopropane ring may also be a component of colibactin in inducing DSBs. In addition, we note that in their DNA adductomics analysis, only the m/z 540.1772 adduct, which is the left-hand fragment of colibactin-770 (19), is detected. Later, in their subsequent research,16 they detected the right-hand fragment of 19, the m/z 568.1721 adduct, by exposing pks+ E. coli to plasmid DNA. Despite the influences of the different DNA adductomic methods in their two studies, these observations still raise the question of whether the clb metabolites, specifically colibactins, which are produced by the same/different strains, are the same or not in different studies. For instance, we noted that in several studies, different strains of pks+ E. coli show different genotoxicities and cartridges of colibactin(s).17,24,107 When comparing the E. coli M1/5 strain with E. coli Nissle 1917 in terms of the induction of DNA DSBs and ICLs, E. coli Nissle 1917 causes a lower-level induction of DSBs and ICLs and fails to induce Wnt-independent organoids in healthy primary colon epithelial cells.24 Apparently, while it is known that the expression level of the clb cluster differs in different strains, the question of whether colibactin acts as a single compound or a mixture remains to be discussed.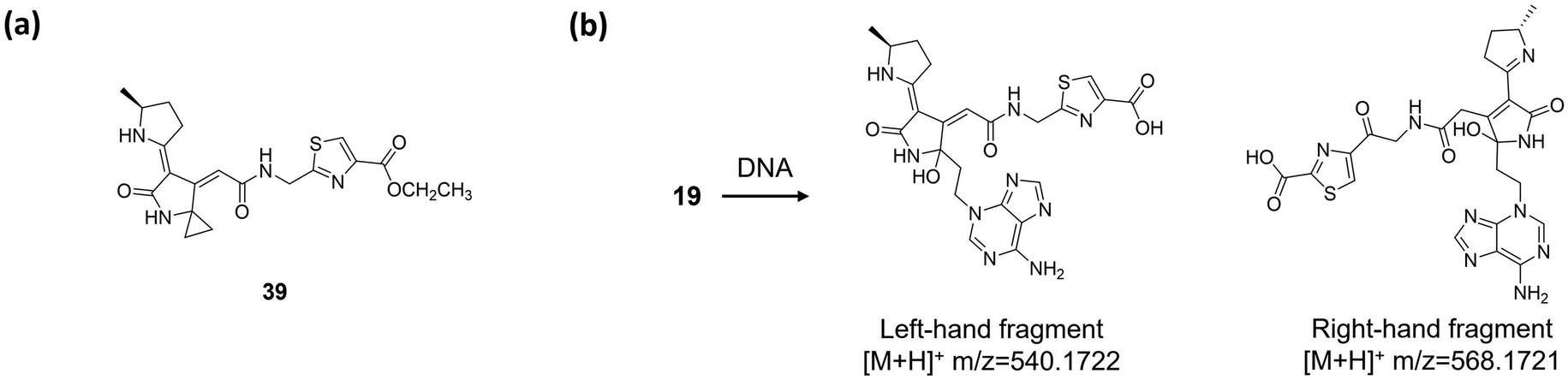 | ||
| Fig. 17 (a) Structure of compound 39. (b) DNA adductomics of colibactin (19) (spontaneous breakage of the C–C bond of the 1,2-diketone moiety generates the left-hand and right-hand fragments). | ||
In addition, considering the fact that the clb cluster produces various active products, we propose distinguishing between pks+ E. coli and colibactin. Most studies of colibactin utilise pks+ E. coli to assess the activities of colibactin in vivo and use the Δpks or ΔclbA/clbQ/clbP mutants as a negative control. The deletion of the clb cluster or the functional clb gene abolishes colibactin production and affects the production of other metabolites, such as lipopeptides and yersiniabactin. Thus, the observed differences in activities between pks+ E. coli and E. coli mutants in vivo may not be attributed solely to colibactin. To be more rigorous, the biological effects should be linked to the clb metabolites instead of colibactin.
6.4. Ecological role of clb in E. coli
In evolutionary terms, the commensal E. coli featuring clb gene clusters may aim to adapt to the ecological niche instead of killing the host. Colibactin encoded by clb confers E. coli with stronger colonisation capacity, and its antibacterial activity offers E. coli a competitive edge over other gut bacteria. Meanwhile, pks+ E. coli induces inflammation by releasing proinflammatory cytokines,87 which in turn promotes the growth of E. coli.89 The immunomodulation activity facilitates the escape of E. coli from immunity. clb is also involved in the synthesis of siderophores,102 which import iron and copper to E. coli as nutrients.106,108 Therefore, E. coli can thrive under iron- or copper-limited conditions. The self-resistant gene clbS inactivates colibactin and protects the bacterial DNA from nucleolytic degradation.47 At present, in our opinion, the ecological function of clb in E. coli remains a mystery because of the limited data. Therefore, future studies should further explore the real natural product of the clb pathway. More physiological studies of ClbM, the clb natural transporter, may provide some answers to these questions, considering the fact that the mutation of ClbM failed to abolish the genotoxic colibactin-induced DSBs. Moreover, all the alleged final clb natural products are highly complicated. It is difficult to imagine that the E. coli hosts consume large amounts of energy to synthesise these complex molecules without using them for specific physiological functions.6.5. Is ROS a possible trigger for the biosynthesis of colibactin?
To date, knowledge regarding the regulation of the clb cluster is limited. Nevertheless, several studies demonstrated that bacterial growth state and composition of the growth medium affect clb expression.109 Heat shock protein HtpG also regulates clb via a non-transcriptional pathway.110 As the sole transcriptional regulator in the clb cluster, ClbR, a LuxR family regulator, has been demonstrated to be the main transcriptional activator for the biosynthesis of colibactin.111 However, unlike the classical LuxR family proteins, ClbR lacks the N-terminal receiver (REC) domain (responsible for recognizing signal molecules), suggesting that ClbR is an autonomous effector domain regulator. Thus, the triggers for clbR expression and the relative regulation mechanism of clb remain uncharacterized. Several indirect studies on this topic warrant brief discussion, although the information is limited.It is a consensus that inflammation is associated with cancer.112,113 In inflammatory disorders, ROS production is a typical event in most cases.114 Thus, the frequent observation of a higher amount ROS in enteritis and CRC patients compared with healthy populations114 indicates the potential role of ROS in the development of CRC caused by pks+ E. coli. These results lead us to consider the possibility of ROS as a trigger for colibactin biosynthesis. The ROS-initiated oxidative-stress response regulator OxyR,115 which belongs to LysR transcriptional factor family, has also been reported as a positive regulator in E. coli.116 The activation of biosynthesis-related genes also regulates the secondary metabolites of Streptomyces avermitilis117 and Streptomyces coelicolor.118 According to the known mechanism of OxyR regulation,119 ROS initiates the conformational change of OxyR from the reduced –SH state to the –S–S state; the oxidised OxyR–S–S binds to the upstream DNA sequences adjacent to the target gene,120 which further recruits RNA polymerase to promoter121 and subsequently initiates the expression. The sequences in the noncoding region similar to the conserved motifs120 binding to OxyR have also been found in the upstream of clbR. Thus, further validation of the possibility that OxyR acts as a regulator of clb is imperative in future studies.
6.6. Significance and therapeutic potential
Colibactin-producing E. coli exhibits diverse biological activities, including genotoxicity intermediated by colibactin, antibacterial activities to facilitate colonisation, decreasing the number of immune cells to achieve immune escape, analgesic activity for the survival of pks+ E. coli, alteration of intestinal-tract physiology via inflammation-inducing activity to generate more ROS, and causing CRC in humans. Recent studies have made substantial progress in illustrating the structure and carcinogenicity of colibactin and the distinct mutational signatures that could act as potential diagnostic markers of CRC. Given that short-term infection with colibactin induces DNA damage in healthy colon epithelial cells without the mutational signature,24 the colibactin–DNA adduct (instead of the signature) could also serve as a biomarker for early diagnosis.122 To retard the development and progress of CRC, diverse strategies should be developed to abolish or impede colibactin biosynthesis. The following approaches could be considered: (i) the knockout of key genes responsible for colibactin biosynthesis, such as clbP, clbA, and clbR; (ii) the addition of a Cu(II) chelator such as EDTA to deprive colibactin of Cu(II), thereby at least partially reducing its action; (iii) the introduction of the colibactin-resistant gene clbS to the host to transform colibactin into other adducts, thereby reducing the genotoxicity of colibactin; (iv) the introduction of a dCas9 element that targets the key genes for colibactin biosynthesis to abolish the expression of these genes, thereby eliminating colibactin production; and (v) the development of an inhibitor of the regulators of clbR, HtpG heat-shock protein, and other uncharacterised regulators to impede the production of colibactin. Furthermore, from another perspective, the utilisation of pks+ E. coli such as Nissle 1917 and the clb gene cluster to induce the apoptosis of tumour cells should also be considered. For instance, the attenuated Salmonella enterica strain, which can selectively target tumour tissues, could be an ideal host for the expression of the clb gene cluster to produce colibactin in cancer, induce tumour-cell apoptosis and death, and ultimately impede CRC development and progress. Additionally, colibactin-645 (18) causes DNA damage in various eukaryotic cells at the nanomolar level18 and the cytotoxicity of precolibactin-886 (14) indicates 18's potential cytotoxicity. Thus, this compound could provide a new scaffold template for anticancer drug development.7. Author contributions
P. Y. Qian conceptualised the project in response to an invitation from editors of NPR. P. Y. Qian and Z. R. Li drafted and submitted the synopsis. J. W. Tang, X. Liu and W. Ye drafted the manuscript with input from Z. R. Li. (J. W. Tang drafted the chemistry and biosynthesis related part; X. Liu drafted the bioactivity related part). All authors contributed to the revision of the manuscript.8. Conflicts of interest
All authors in this manuscript declare no conflict of interest.9. Acknowledgements
This research was funded by the National Key R & D Program of China (2018YFA0903200), the Hong Kong Branch of Southern Marine Science and Engineering Guangdong Laboratory (Guangdong) (MSEGL20SC01), and a CRF grant from HKSAR government (C6026-19G-A).10. References
- M. Hanus, D. Parada-Venegas, G. Landskron, A. M. Wielandt, C. Hurtado, K. Alvarez, M. A. Hermoso, F. López-Köstner and M. De la Fuente, Front. Immunol., 2021, 12, 612826 CrossRef CAS PubMed.
- J. C. Clemente, L. K. Ursell, L. W. Parfrey and R. Knight, Cell, 2012, 148, 1258–1270 CrossRef CAS PubMed.
- M. Bonnet, E. Buc, P. Sauvanet, C. Darcha, D. Dubois, B. Pereira, P. Dechelotte, R. Bonnet, D. Pezet and A. Darfeuille-Michaud, Clin. Cancer Res., 2014, 20, 859–867 CrossRef PubMed.
- A. Shariati, S. Razavi, E. Ghaznavi-Rad, B. Jahanbin, A. Akbari, S. Norzaee and D. Darban-Sarokhalil, Infect. Agents Cancer, 2021, 16, 41 CrossRef CAS PubMed.
- J. Butt, M. Jenab, J. Werner, V. Fedirko, E. Weiderpass, C. C. Dahm, A. Tjønneland, A. Olsen, M. C. Boutron-Ruault, J. A. Rothwell, G. Severi, R. Kaaks, R. Turzanski-Fortner, K. Aleksandrova, M. Schulze, D. Palli, V. Pala, S. Panico, R. Tumino, C. Sacerdote, B. Bueno-de-Mesquita, C. H. Van Gils, I. T. Gram, M. Lukic, N. Sala, M. J. Sánchez Pérez, E. Ardanaz, M. D. Chirlaque, R. Palmquist, T. Löwenmark, R. C. Travis, A. Heath, A. J. Cross, H. Freisling, S. Zouiouich, E. Aglago, T. Waterboer and D. J. Hughes, Gut Microbes, 2021, 13, 1–14 CrossRef PubMed.
- Y. Cheng, Z. Ling and L. Li, Front. Immunol., 2020, 11, 615056 CrossRef CAS PubMed.
- J. P. Nougayrède, S. Homburg, F. Taieb, M. Boury, E. Brzuszkiewicz, G. Gottschalk, C. Buchrieser, J. Hacker, U. Dobrindt and E. Oswald, Science, 2006, 313, 848–851 CrossRef PubMed.
- G. Cuevas-Ramos, C. R. Petit, I. Marcq, M. Boury, E. Oswald and J. P. Nougayrède, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11537–11541 CrossRef CAS PubMed.
- T. Secher, A. Samba-Louaka, E. Oswald and J. P. Nougayrède, PLoS One, 2013, 8, e77157 CrossRef CAS PubMed.
- I. Marcq, P. Martin, D. Payros, G. Cuevas-Ramos, M. Boury, C. Watrin, J. P. Nougayrède, M. Olier and E. Oswald, J. Infect. Dis., 2014, 210, 285–294 CrossRef CAS PubMed.
- A. Cougnoux, L. Gibold, F. Robin, D. Dubois, N. Pradel, A. Darfeuille-Michaud, G. Dalmasso, J. Delmas and R. Bonnet, J. Mol. Biol., 2012, 424, 203–214 CrossRef CAS PubMed.
- D. Dubois, O. Baron, A. Cougnoux, J. Delmas, N. Pradel, M. Boury, B. Bouchon, M. A. Bringer, J. P. Nougayrède, E. Oswald and R. Bonnet, J. Biol. Chem., 2011, 286, 35562–35570 CrossRef CAS PubMed.
- C. A. Brotherton and E. P. Balskus, J. Am. Chem. Soc., 2013, 135, 3359–3362 CrossRef CAS PubMed.
- X. Bian, J. Fu, A. Plaza, J. Herrmann, D. Pistorius, A. F. Stewart, Y. Zhang and R. Müller, ChemBioChem, 2013, 14, 1194–1197 CrossRef CAS PubMed.
- M. Xue, C. S. Kim, A. R. Healy, K. M. Wernke, Z. Wang, M. C. Frischling, E. E. Shine, W. Wang, S. B. Herzon and J. M. Crawford, Science, 2019, 365, eaax2685 CrossRef CAS PubMed.
- Y. Jiang, A. Stornetta, P. W. Villalta, M. R. Wilson, P. D. Boudreau, L. Zha, S. Balbo and E. P. Balskus, J. Am. Chem. Soc., 2019, 141, 11489–11496 CrossRef CAS PubMed.
- T. Zhou, Y. Hirayama, Y. Tsunematsu, N. Suzuki, S. Tanaka, N. Uchiyama, Y. Goda, Y. Yoshikawa, Y. Iwashita, M. Sato, N. Miyoshi, M. Mutoh, H. Ishikawa, H. Sugimura, K. Wakabayashi and K. Watanabe, J. Am. Chem. Soc., 2021, 143, 5526–5533 CrossRef CAS PubMed.
- Z. R. Li, J. Li, W. Cai, J. Y. H. Lai, S. M. K. McKinnie, W. P. Zhang, B. S. Moore, W. Zhang and P. Y. Qian, Nat. Chem., 2019, 11, 880–889 CrossRef CAS PubMed.
- M. I. Vizcaino, P. Engel, E. Trautman and J. M. Crawford, J. Am. Chem. Soc., 2014, 136, 9244–9247 CrossRef CAS PubMed.
- T. Fais, J. Delmas, N. Barnich, R. Bonnet and G. Dalmasso, Toxins, 2018, 10, 151 CrossRef PubMed.
- E. Buc, D. Dubois, P. Sauvanet, J. Raisch, J. Delmas, A. Darfeuille-Michaud, D. Pezet and R. Bonnet, PLoS One, 2013, 8, e56964 CrossRef CAS PubMed.
- J. C. Arthur, E. Perez-Chanona, M. Mühlbauer, S. Tomkovich, J. M. Uronis, T.-J. Fan, B. J. Campbell, T. Abujamel, B. Dogan, A. B. Rogers, J. M. Rhodes, A. Stintzi, K. W. Simpson, J. J. Hansen, T. O. Keku, A. A. Fodor and C. Jobin, Science, 2012, 338, 120–123 CrossRef CAS PubMed.
- L. Salesse, C. Lucas, M. H. T. Hoang, P. Sauvanet, A. Rezard, P. Rosenstiel, C. Damon-Soubeyrand, N. Barnich, C. Godfraind, G. Dalmasso and H. T. T. Nguyen, Cancers, 2021, 13, 2060 CrossRef CAS PubMed.
- A. Iftekhar, H. Berger, N. Bouznad, J. Heuberger, F. Boccellato, U. Dobrindt, H. Hermeking, M. Sigal and T. F. Meyer, Nat. Commun., 2021, 12, 1003 CrossRef CAS PubMed.
- C. Pleguezuelos-Manzano, J. Puschhof, A. Rosendahl Huber, A. van Hoeck, H. M. Wood, J. Nomburg, C. Gurjao, F. Manders, G. Dalmasso, P. B. Stege, F. L. Paganelli, M. H. Geurts, J. Beumer, T. Mizutani, Y. Miao, R. van der Linden, S. van der Elst, K. C. Garcia, J. Top, R. J. L. Willems, M. Giannakis, R. Bonnet, P. Quirke, M. Meyerson, E. Cuppen, R. van Boxtel and H. Clevers, Nature, 2020, 580, 269–273 CrossRef CAS PubMed.
- P. J. Dziubańska-Kusibab, H. Berger, F. Battistini, B. A. M. Bouwman, A. Iftekhar, R. Katainen, T. Cajuso, N. Crosetto, M. Orozco, L. A. Aaltonen and T. F. Meyer, Nat. Med., 2020, 26, 1063–1069 CrossRef PubMed.
- H. B. Bode, Angew. Chem., Int. Ed., 2015, 54, 10408–10411 CrossRef CAS PubMed.
- E. P. Balskus, Nat. Prod. Rep., 2015, 32, 1534–1540 RSC.
- P. C. Williams, K. M. Wernke, A. Tirla and S. B. Herzon, Nat. Prod. Rep., 2020, 37, 1532–1548 RSC.
- K. M. Wernke, M. Xue, A. Tirla, C. S. Kim, J. M. Crawford and S. B. Herzon, Bioorg. Med. Chem. Lett., 2020, 30, 127280 CrossRef CAS PubMed.
- D. Reimer and H. B. Bode, Nat. Prod. Rep., 2014, 31, 154–159 RSC.
- Y. Li, Z. Li, K. Yamanaka, Y. Xu, W. Zhang, H. Vlamakis, R. Kolter, B. S. Moore and P. Y. Qian, Sci. Rep., 2015, 5, 9383 CrossRef CAS PubMed.
- D. Reimer, K. M. Pos, M. Thines, P. Grun and H. B. Bode, Nat. Chem. Biol., 2011, 7, 888–890 CrossRef CAS PubMed.
- B. M. Kevany, D. A. Rasko and M. G. Thomas, Appl. Environ. Microbiol., 2009, 75, 1144–1155 CrossRef CAS PubMed.
- Z. R. Li, J. Li, J. P. Gu, J. Y. Lai, B. M. Duggan, W. P. Zhang, Z. L. Li, Y. X. Li, R. B. Tong, Y. Xu, D. H. Lin, B. S. Moore and P. Y. Qian, Nat. Chem. Biol., 2016, 12, 773–775 CrossRef CAS PubMed.
- M. I. Vizcaino and J. M. Crawford, Nat. Chem., 2015, 7, 411–417 CrossRef CAS PubMed.
- C. A. Brotherton, M. Wilson, G. Byrd and E. P. Balskus, Org. Lett., 2015, 17, 1545–1548 CrossRef CAS PubMed.
- X. Bian, A. Plaza, Y. Zhang and R. Müller, Chem. Sci., 2015, 6, 3154–3160 RSC.
- L. Zha, Y. Jiang, M. T. Henke, M. R. Wilson, J. X. Wang, N. L. Kelleher and E. P. Balskus, Nat. Chem. Biol., 2017, 13, 1063–1065 CrossRef CAS PubMed.
- A. R. Healy, M. I. Vizcaino, J. M. Crawford and S. B. Herzon, J. Am. Chem. Soc., 2016, 138, 5426–5432 CrossRef CAS PubMed.
- Z. R. Li, Y. Li, J. Y. Lai, J. Tang, B. Wang, L. Lu, G. Zhu, X. Wu, Y. Xu and P. Y. Qian, ChemBioChem, 2015, 16, 1715–1719 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- T. L. Schneider, B. Shen and C. T. Walsh, Biochemistry, 2003, 42, 9722–9730 CrossRef CAS PubMed.
- L. Zha, M. R. Wilson, C. A. Brotherton and E. P. Balskus, ACS Chem. Biol., 2016, 11, 1287–1295 CrossRef CAS PubMed.
- A. O. Brachmann, C. Garcie, V. Wu, P. Martin, R. Ueoka, E. Oswald and J. Piel, Chem. Commun., 2015, 51, 13138–13141 RSC.
- P. Tripathi and S. D. Bruner, Biochemistry, 2021, 60, 1619–1625 CrossRef CAS PubMed.
- K. Molan, Z. Podlesek, V. Hodnik, M. Butala, E. Oswald and D. Žgur Bertok, DNA Repair, 2019, 79, 50–54 CrossRef CAS PubMed.
- P. Tripathi, E. E. Shine, A. R. Healy, C. S. Kim, S. B. Herzon, S. D. Bruner and J. M. Crawford, J. Am. Chem. Soc., 2017, 139, 17719–17722 CrossRef CAS PubMed.
- N. Bossuet-Greif, D. Dubois, C. Petit, S. Tronnet, P. Martin, R. Bonnet, E. Oswald and J. P. Nougayrède, Mol. Microbiol., 2016, 99, 897–908 CrossRef CAS PubMed.
- A. R. Healy, H. Nikolayevskiy, J. R. Patel, J. M. Crawford and S. B. Herzon, J. Am. Chem. Soc., 2016, 138, 15563–15570 CrossRef CAS PubMed.
- N. Bossuet-Greif, J. Vignard, F. Taieb, G. Mirey, D. Dubois, C. Petit, E. Oswald and J.-P. Nougayrède, mBio, 2018, 9, e02393-17 CrossRef PubMed.
- M. Xue, E. Shine, W. Wang, J. M. Crawford and S. B. Herzon, Biochemistry, 2018, 57, 6391–6394 CrossRef CAS PubMed.
- M. R. Wilson, Y. Jiang, P. W. Villalta, A. Stornetta, P. D. Boudreau, A. Carra, C. A. Brennan, E. Chun, L. Ngo, L. D. Samson, B. P. Engelward, W. S. Garrett, S. Balbo and E. P. Balskus, Science, 2019, 363, eaar7785 CrossRef CAS PubMed.
- Y. Hirayama, Y. Tsunematsu, Y. Yoshikawa, R. Tamafune, N. Matsuzaki, Y. Iwashita, I. Ohnishi, F. Tanioka, M. Sato, N. Miyoshi, M. Mutoh, H. Ishikawa, H. Sugimura, K. Wakabayashi and K. Watanabe, Org. Lett., 2019, 21, 4490–4494 CrossRef CAS PubMed.
- A. R. Healy, K. M. Wernke, C. S. Kim, N. R. Lees, J. M. Crawford and S. B. Herzon, Nat. Chem., 2019, 11, 890–898 CrossRef CAS PubMed.
- E. P. Trautman, A. R. Healy, E. E. Shine, S. B. Herzon and J. M. Crawford, J. Am. Chem. Soc., 2017, 139, 4195–4201 CrossRef CAS PubMed.
- E. J. Helfrich and J. Piel, Nat. Prod. Rep., 2016, 33, 231–316 RSC.
- J. Piel, Nat. Prod. Rep., 2010, 27, 996–1047 RSC.
- P. Engel, M. I. Vizcaino and J. M. Crawford, Appl. Environ. Microbiol., 2015, 81, 1502–1512 CrossRef PubMed.
- Y. A. Chan, M. T. Boyne II, A. M. Podevels, A. K. Klimowicz, J. Handelsman, N. L. Kelleher and M. G. Thomas, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14349–14354 CrossRef CAS PubMed.
- T. C. Holmes, A. E. May, K. Zaleta-Rivera, J. G. Ruby, P. Skewes-Cox, M. A. Fischbach, J. L. DeRisi, M. Iwatsuki, S. Omura and C. Khosla, J. Am. Chem. Soc., 2012, 134, 17797–17806 CrossRef CAS PubMed.
- H. B. Park and J. M. Crawford, J. Nat. Prod., 2015, 78, 1437–1441 CrossRef CAS PubMed.
- K. L. Wang, H. Li and J. R. Ecker, Plant Cell, 2002, 14, S131–S151 CrossRef CAS PubMed.
- T. Robbins, J. Kapilivsky, D. E. Cane and C. Khosla, Biochemistry, 2016, 55, 4476–4484 CrossRef CAS PubMed.
- C. T. Calderone, W. E. Kowtoniuk, N. L. Kelleher, C. T. Walsh and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8977–8982 CrossRef CAS PubMed.
- V. Simunovic and R. Müller, ChemBioChem, 2007, 8, 1273–1280 CrossRef CAS PubMed.
- M. E. Horsman, T. P. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255–278 RSC.
- H. D. Luo, M. Y. Jin, H. Wu and H. Jiang, Protein Pept. Lett., 2016, 23, 1032–1037 CrossRef CAS PubMed.
- D. Schwarzer, H. D. Mootz, U. Linne and M. A. Marahiel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14083–14088 CrossRef CAS PubMed.
- M. L. Heathcote, J. Staunton and P. F. Leadlay, Chem. Biol., 2001, 8, 207–220 CrossRef CAS PubMed.
- N. S. Guntaka, A. R. Healy, J. M. Crawford, S. B. Herzon and S. D. Bruner, ACS Chem. Biol., 2017, 12, 2598–2608 CrossRef CAS PubMed.
- A. L. Valina, D. Mazumder-Shivakumar and T. C. Bruice, Biochemistry, 2004, 43, 15657–15672 CrossRef CAS PubMed.
- S. Shin, T. H. Lee, N. C. Ha, H. M. Koo, S. Y. Kim, H. S. Lee, Y. S. Kim and B. H. Oh, EMBO J., 2002, 21, 2509–2516 CrossRef CAS PubMed.
- A. ThiéRy, M. Maestracci, A. Arnaud and P. Galzy, Microbiology, 1986, 132, 2205–2208 Search PubMed.
- A. Krah, R. G. Huber, U. Zachariae and P. J. Bond, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183137 CrossRef PubMed.
- J. J. Mousa, R. C. Newsome, Y. Yang, C. Jobin and S. D. Bruner, Biochem. Biophys. Res. Commun., 2017, 482, 1233–1239 CrossRef CAS PubMed.
- J. J. Mousa, Y. Yang, S. Tomkovich, A. Shima, R. C. Newsome, P. Tripathi, E. Oswald, S. D. Bruner and C. Jobin, Nat. Microbiol., 2016, 1, 15009 CrossRef CAS PubMed.
- M. Xue, K. M. Wernke and S. B. Herzon, Biochemistry, 2020, 59, 892–900 CrossRef CAS PubMed.
- J. Gagnière, V. Bonnin, A.-S. Jarrousse, E. Cardamone, A. Agus, N. Uhrhammer, P. Sauvanet, P. Déchelotte, N. Barnich, R. Bonnet, D. Pezet and M. Bonnet, Clin. Sci., 2017, 131, 471–485 CrossRef PubMed.
- M. Pitié and G. Pratviel, Chem. Rev., 2010, 110, 1018–1059 CrossRef PubMed.
- J. Stubbe and J. W. Kozarich, Chem. Rev., 1987, 87, 1107–1136 CrossRef CAS.
- A. Cougnoux, G. Dalmasso, R. Martinez, E. Buc, J. Delmas, L. Gibold, P. Sauvanet, C. Darcha, P. Déchelotte, M. Bonnet, D. Pezet, H. Wodrich, A. Darfeuille-Michaud and R. Bonnet, Gut, 2014, 63, 1932–1942 CrossRef CAS PubMed.
- T. Faïs, A. Cougnoux, G. Dalmasso, F. Laurent, J. Delmas and R. Bonnet, Antimicrob. Agents Chemother., 2016, 60, 6986–6988 CrossRef PubMed.
- J. C. Arthur, R. Z. Gharaibeh, M. Mühlbauer, E. Perez-Chanona, J. M. Uronis, J. McCafferty, A. A. Fodor and C. Jobin, Nat. Commun., 2014, 5, 4724 CrossRef CAS PubMed.
- S. Tomkovich, Y. Yang, K. Winglee, J. Gauthier, M. Mühlbauer, X. Sun, M. Mohamadzadeh, X. Liu, P. Martin, G. P. Wang, E. Oswald, A. A. Fodor and C. Jobin, Clin. Cancer Res., 2017, 77, 2620–2632 CAS.
- A. Lopès, E. Billard, A. H. Casse, R. Villéger, J. Veziant, G. Roche, G. Carrier, P. Sauvanet, A. Briat, F. Pagès, S. Naimi, D. Pezet, N. Barnich, B. Dumas and M. Bonnet, Int. J. Cancer, 2020, 146, 3147–3159 CrossRef PubMed.
- I. Demirel, A. Persson, A. Brauner, E. Särndahl, R. Kruse and K. Persson, Sci. Rep., 2020, 10, 21837 CrossRef CAS PubMed.
- I. Demirel, A. Persson, A. Brauner, E. Särndahl, R. Kruse and K. Persson, Front. Cell. Infect. Microbiol., 2018, 8, 81 CrossRef PubMed.
- T. Pérez-Berezo, J. Pujo, P. Martin, P. Le Faouder, J. M. Galano, A. Guy, C. Knauf, J. C. Tabet, S. Tronnet, F. Barreau, M. Heuillet, G. Dietrich, J. Bertrand-Michel, T. Durand, E. Oswald and N. Cenac, Nat. Commun., 2017, 8, 1314 CrossRef PubMed.
- G. Dalmasso, A. Cougnoux, J. Delmas, A. Darfeuille-Michaud and R. Bonnet, Gut Microbes, 2014, 5, 675–680 CrossRef PubMed.
- C. M. Dejea, P. Fathi, J. M. Craig, A. Boleij, R. Taddese, A. L. Geis, X. Wu, C. E. DeStefano Shields, E. M. Hechenbleikner, D. L. Huso, R. A. Anders, F. M. Giardiello, E. C. Wick, H. Wang, S. Wu, D. M. Pardoll, F. Housseau and C. L. Sears, Science, 2018, 359, 592–597 CrossRef CAS PubMed.
- A. J. Rowan, H. Lamlum, M. Ilyas, J. Wheeler, J. Straub, A. Papadopoulou, D. Bicknell, W. F. Bodmer and I. P. M. Tomlinson, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3352 CrossRef CAS PubMed.
- A. J. McCarthy, P. Martin, E. Cloup, R. A. Stabler, E. Oswald and P. W. Taylor, Infect. Immun., 2015, 83, 3704–3711 CrossRef CAS PubMed.
- S. Tronnet, P. Floch, L. Lucarelli, D. Gaillard, P. Martin, M. Serino and E. Oswald, mSphere, 2020, 5, e00589–20 CrossRef CAS PubMed.
- A. M. Sheflin, A. K. Whitney and T. L. Weir, Curr. Oncol. Rep., 2014, 16, 406 CrossRef PubMed.
- M. A. Bauer, K. Kainz, D. Carmona-Gutierrez and F. Madeo, Microb. Cell, 2018, 5, 215–219 CrossRef PubMed.
- T. Secher, D. Payros, C. Brehin, M. Boury, C. Watrin, M. Gillet, I. Bernard-Cadenat, S. Menard, V. Theodorou, A. Saoudi, M. Olier and E. Oswald, Infect. Immun., 2015, 83, 2420–2429 CrossRef CAS PubMed.
- D. Payros, T. Secher, M. Boury, C. Brehin, S. Ménard, C. Salvador-Cartier, G. Cuevas-Ramos, C. Watrin, I. Marcq, J. P. Nougayrède, D. Dubois, A. Bedu, F. Garnier, O. Clermont, E. Denamur, P. Plaisancié, V. Theodorou, J. Fioramonti, M. Olier and E. Oswald, Gut Microbes, 2014, 5, 313–325 CrossRef PubMed.
- T. Annaval, J. D. Rudolf, C. Y. Chang, J. R. Lohman, Y. Kim, L. Bigelow, R. Jedrzejczak, G. Babnigg, A. Joachimiak, G. N. Phillips Jr and B. Shen, ACS Omega, 2017, 2, 5159–5169 CrossRef CAS PubMed.
- M. Kotaka, R. Kong, I. Qureshi, Q. S. Ho, H. Sun, C. W. Liew, L. P. Goh, P. Cheung, Y. Mu, J. Lescar and Z. X. Liang, J. Biol. Chem., 2009, 284, 15739–15749 CrossRef CAS PubMed.
- P. Martin, I. Marcq, G. Magistro, M. Penary, C. Garcie, D. Payros, M. Boury, M. Olier, J. P. Nougayrède, M. Audebert, C. Chalut, S. Schubert and E. Oswald, PLoS Pathog., 2013, 9, e1003437 CrossRef CAS PubMed.
- C. S. Kim, T. Turocy, G. Moon, E. E. Shine and J. M. Crawford, Org. Lett., 2021, 23, 6895–6899 CrossRef CAS PubMed.
- D. C. Oh, W. K. Strangman, C. A. Kauffman, P. R. Jensen and W. Fenical, Org. Lett., 2007, 9, 1525–1528 CrossRef CAS PubMed.
- A. C. Ross, Y. Xu, L. Lu, R. D. Kersten, Z. Z. Shao, A. M. Al-Suwailem, P. C. Dorrestein, P. Y. Qian and B. S. Moore, J. Am. Chem. Soc., 2013, 135, 1155–1162 CrossRef CAS PubMed.
- E. I. Koh, A. E. Robinson, N. Bandara, B. E. Rogers and J. P. Henderson, Nat. Chem. Biol., 2017, 13, 1016–1021 CrossRef CAS PubMed.
- F. Auvray, A. Perrat, Y. Arimizu, C. V. Chagneau, N. Bossuet-Greif, C. Massip, H. Brugère, J. P. Nougayrède, T. Hayashi, P. Branchu, Y. Ogura and E. Oswald, Microb. Genomes, 2021, 7, 000579 CAS.
- E. I. Koh, C. S. Hung, K. S. Parker, J. R. Crowley, D. E. Giblin and J. P. Henderson, Metallomics, 2015, 7, 1011–1022 CrossRef CAS PubMed.
- S. Tronnet, C. Garcie, N. Rehm, U. Dobrindt, E. Oswald and P. Martin, Infect. Immun., 2016, 84, 3358–3368 CrossRef CAS PubMed.
- C. Garcie, S. Tronnet, A. Garénaux, A. J. McCarthy, A. O. Brachmann, M. Pénary, S. Houle, J. P. Nougayrède, J. Piel, P. W. Taylor, C. M. Dozois, P. Genevaux, E. Oswald and P. Martin, J. Infect. Dis., 2016, 214, 916–924 CrossRef CAS PubMed.
- A. Walenstein, N. Rehm, M. Brinkmann, M. Selle, N. Bossuet-Greif, D. Sauer, B. Bunk, C. Sproer, H. T. Wami, S. Homburg, R. V. Bunau, S. Konig, J. P. Nougayrède, J. Overmann, E. Oswald, R. Muller and U. Dobrindt, mSphere, 2020, 5, e00591–20 Search PubMed.
- Y. Yang, R. Z. Gharaibeh, R. C. Newsome and C. Jobin, Nat. Cancer, 2020, 1, 723–734 CrossRef CAS PubMed.
- J. K. Dyson and M. D. Rutter, World J. Gastroenterol., 2012, 18, 3839–3848 CrossRef PubMed.
- G. Aviello and U. G. Knaus, Br. J. Pharmacol., 2017, 174, 1704–1718 CrossRef CAS PubMed.
- X. Liu, M. Sun, Y. Cheng, R. Yang, Y. Wen, Z. Chen and J. Li, Microbiology, 2016, 162, 707–716 CrossRef CAS PubMed.
- M. F. Christman, G. Storz and B. N. Ames, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 3484–3488 CrossRef CAS PubMed.
- M. Sun, L. Mengya, W. Ying, Y. Song, J. Li and Z. Chen, Front. Microbiol., 2018, 9, 1398 CrossRef PubMed.
- J. S. Hahn, S. Y. Oh and J. H. Roe, J. Bacteriol., 2002, 184, 5214–5222 CrossRef CAS PubMed.
- M. Zheng, F. Aslund and G. Storz, Science, 1998, 279, 1718–1721 CrossRef CAS PubMed.
- M. Zheng, X. Wang, D. Bernard, K. A. Lewis, T. D. Schneider and G. Storz, J. Bacteriol., 2001, 183, 4571–4579 CrossRef CAS PubMed.
- I. Kullik, M. B. Toledano, L. A. Tartaglia and G. Storz, J. Bacteriol., 1995, 177, 1275–1284 CrossRef CAS PubMed.
- K. J. Murray, E. S. Carlson, A. Stornetta, E. P. Balskus, P. W. Villalta and S. Balbo, Anal. Chem., 2021, 93, 5754–5762 CrossRef CAS PubMed.
Footnote |
| † J. W. Tang & X. Liu: equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |