
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRibosome-independent peptide biosynthesis: the challenge of a unifying nomenclature
Maria
Dell
 a,
Kyle L.
Dunbar
a and
Christian
Hertweck
*ab
a,
Kyle L.
Dunbar
a and
Christian
Hertweck
*ab
aDepartment of Biomolecular Chemistry, Leibniz Institute for Natural Product Research and Infection Biology – Hans Knöll Institute (HKI), Beutenbergstr. 11a, 07745, Jena, Germany. E-mail: christian.hertweck@leibniz-hki.de
bFaculty of Biological Sciences, Friedrich Schiller University Jena, 07743, Jena, Germany
First published on 29th September 2021
Abstract
The first machineries for non-ribosomal peptide (NRP) biosynthesis were uncovered over 50 years ago, and the dissection of these megasynthetases set the stage for the nomenclature system that has been used ever since. Although the number of exceptions to the canonical biosynthetic pathways has surged in the intervening years, the NRP synthetase (NRPS) classification system has remained relatively unchanged. This has led to the exclusion of many biosynthetic pathways whose biosynthetic machineries violate the classical rules for NRP assembly, and ultimately to a rupture in the field of NRP biosynthesis. In an attempt to unify the classification of NRP pathways and to facilitate the communication within the research field, we propose a revised framework for grouping ribosome-independent peptide biosynthetic pathways based on recognizable commonalities in their biosynthetic logic. Importantly, the framework can be further refined as needed.
 Maria Dell | Maria Dell studied chemistry at Frankfurt University and spent a semester abroad at Bangor University during her master's degree. Her master thesis was focused on structural studies on fatty acid synthase using single-molecule FRET measurements under the supervision of M. Grininger and M. Heilemann. She is currently a PhD candidate in Natural Product Chemistry at the Leibniz Institute for Natural Product Research and Infection Biology (HKI) under the guidance of Christian Hertweck. Her research focuses on the biosynthesis of natural products. |
 Kyle L. Dunbar | Kyle Dunbar obtained his B.Sc. in Chemical Biology from the University of California, Berkeley. In 2014, he received his PhD in Chemistry from University of Illinois at Urbana-Champaign under the supervision of Douglas Mitchell. His PhD work focused on the biosynthesis of azoline heterocycles in ribosomal natural products. Following a short postdoctoral position at the University of Illinois at Urbana-Champaign, Kyle joined the group of Christian Hertweck at the Leibniz Institute for Natural Product Research and Infection Biology as an Alexander von Humboldt postdoctoral fellow. |
 Christian Hertweck | Christian Hertweck is Head of Department Biomolecular Chemistry at the Leibniz Institute for Natural Product Research and Infection Biology (HKI) and holds a Chair at the Friedrich Schiller University Jena. After his PhD studies at University of Bonn and MPI for Chemical Ecology (W. Boland), he has been Feodor Lynen postdoctoral fellow at the University of Washington, Seattle (H. G. Floss, B. S. Moore). His research focuses on microbial natural products, their biosynthesis, and their role in microbial interactions. He is elected member of the National Academy of Sciences (Leopoldina) and recipient of the Leibniz Award. |
1 Ribosome-independent peptide biosynthesis
The first example of a ribosome-independent peptide biosynthetic machinery was reported in the 1960s.1 From then on, these systems were studied intensively on the examples of gramicidin S and tyrocidine and were logically called non-ribosomal peptide (NRP) synthetases (NRPSs).2 While this term seemed to be ideal for describing alternative biosynthetic pathways to peptides, classification problems have been plaguing the field from the beginning.3–5 NRPs became synonymous with the so-called thiotemplate-directed products of (multi-)modular megasynthetases that are minimally composed of a condensation (C) domain, an adenylation (A) domain, and a carrier protein (CP, often also referred to as T domain). However, not all peptides that are assembled in ribosome-independent pathways are termed NRPs, excluding important examples such as peptidoglycan6 and glutathione.7 Numerous other peptides whose biosynthesis involves neither ribosomes nor NRPSs are classified by function (e.g. siderophores8) rather than by biosynthetic origin. At the turn of the century, adjustments to the nomenclature were introduced to accommodate NRPSs that lack the typical (multi-)modular architecture and involve free-standing components (type II NRPSs,4 by analogy to type II fatty acid synthases, for example). However, given the above-mentioned shortcomings, we still lack a unifying concept comprising the NRP field as a whole.In the past two decades, this nomenclature problem has only worsened as the genomic revolution has propelled the discovery of atypical NRP biosynthetic pathways that lack the key features of canonical NRPSs. Researchers have resorted to the inclusion of a string of qualifiers describing their NRP assembly lines to fit the current nomenclature. For example, when we discovered an atypical pathway for the biosynthesis of the antibiotic closthioamide, we were unsatisfied with needing to describe it as an NRPS-independent, yet thiotemplated NRP biosynthetic pathway.9 Such awkward descriptions are impractical in laboratory vernacular and hinder classification of systems that do not fit into the existing nomenclature. In studying closthioamide biosynthesis, we also found that there are even more unusual biosynthetic gene clusters20 that would not have been detected by conventional bioinformatic searches with the software that is available.10 This is primarily because these tools are trained on well-studied biosynthetic pathways.10 A reclassification of ribosome-independent biosynthetic machineries, when implemented in the current genome mining software and databases, could lead to the discovery of more atypical NRP assembly pathways and therefore provide a broader view into the biosynthetic landscape of this prominent natural product class.
2 A possible binning system for ribosome-independent peptide assembly systems
There are many ways to approach the challenge of developing a nomenclature for enzymes involved in ribosome-independent peptide biosynthesis. Obvious possibilities are to subdivide the ribosome-independent biosynthetic machineries based on the phylogeny, structural biology, or biochemistry of the amide bond-forming enzyme. While structural data are not readily available for each enzyme, a tentative fold could be modelled based on the protein sequence. The amino acid sequence can also be used to place the enzyme in an evolutionary context by performing a phylogenetic analysis. However, both of these features are based on protein sequence, and in the case of very low sequence similarity to other enzymes in the same protein family, these analyses can be a hurdle. Furthermore, it is not viable to establish phylogenies of unrelated enzyme families.To provide a more accessible option, we chose to use the protein family of the amide bond-forming enzyme and its underlying biochemistry as qualifiers for each classification. We found criteria for reclassification by examining the groups ‘NRPs’ and ‘others’ in MiBiG11 for possible peptides that are biosynthesised independent from the ribosome and do not fit the naming convention of NRPSs.
We propose to divide the field into five groups. A decision tree in Scheme 1 provides a visualised approach to the reclassification criteria for peptide biosynthetic machineries.
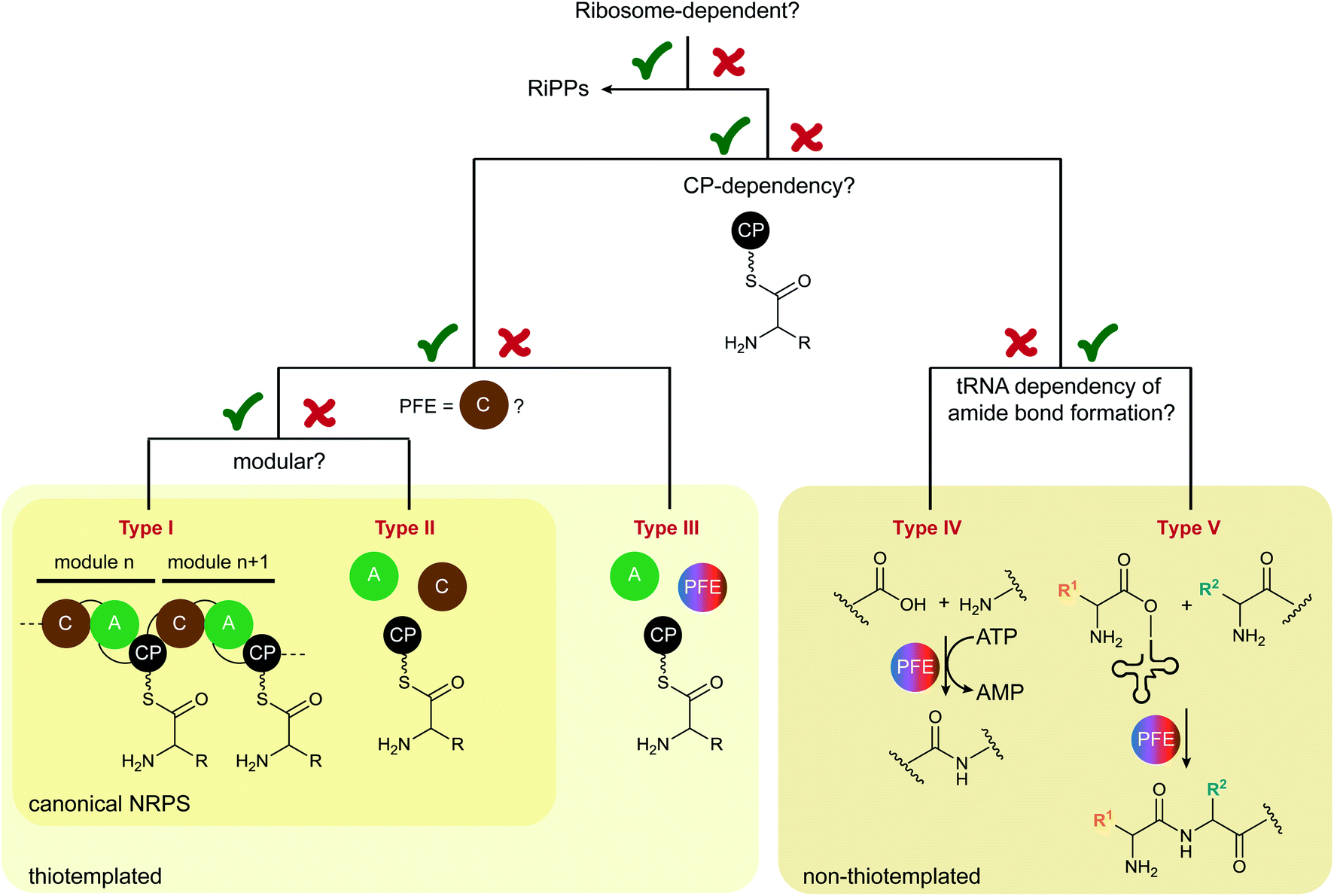 | ||
| Scheme 1 Decision tree to place ribosome-independent peptide biosynthetic machineries into different types. PFE = peptide bond-forming enzyme. | ||
The initial division point was, of course, whether or not the peptide is biosynthesised by the ribosome. Here, the biosynthetic pathways for ribosomally synthesised and posttranslationally modified peptides (RiPPs)12 and ribosome-independent pathways are distinguished. The second criterion was then the CP dependence of amide bond formation, that is, thiotemplated or not thiotemplated. Since this is one of the properties of NRPSs as currently defined,13 here the peptide field would have been divided into NRPSs and other enzymes that are termed “ribosome-independent”, but not “non-ribosomal” peptide synth(et)ases.14 Next, to base this expanded nomenclature on the established one that is aligned with the nomenclature for fatty acid (FA) and polyketide synthases (PKSs), we decide between modular and non-modular/freestanding, according to the definitions for type I and type II NRPSs. Another important decision is if C domains are required for amide bond formation. Biosynthetic machineries that do not use thiotemplated substrates are then placed into two other groups (IV and V) depending on the tRNA dependence of the amide bond-forming step.
Iterative NRPSs3,5 are considered a subclass, which can easily be denoted with a qualifier. They will therefore not have an own section in this nomenclature. These five types/groups of ribosome-independent peptide biosynthesis are described below with illustrative examples.
2.1 Thiotemplated, modular peptide synthetases: type I
Type I NRPSs are the prototypical linear megasynthetases that are historically associated with NRP biosynthesis. They are comprised of several connected enzymes, which are organized into modules. These modules can be initiation, extension and termination modules minimally comprised of a C domain, an A domain, and a CP (or T domain).13 The A domain catalyses the ATP-dependent activation and loading of amino acids onto the phosphopantetheine prosthetic group of the holo-CP. The C domain then catalyses peptide bond formation between two CP-tethered amino acids. The manner of biosynthesis is commonly likened to an assembly line.5 Examples of typical NRPS type I systems are the well-characterised assembly lines for surfactin and isopenicillin N (Fig. 1A).3 For comprehensive overviews on type I NRPSs see the review articles by the groups of Marahiel,15 Walsh,16 Süssmuth,5 and van Lanen,13 among others.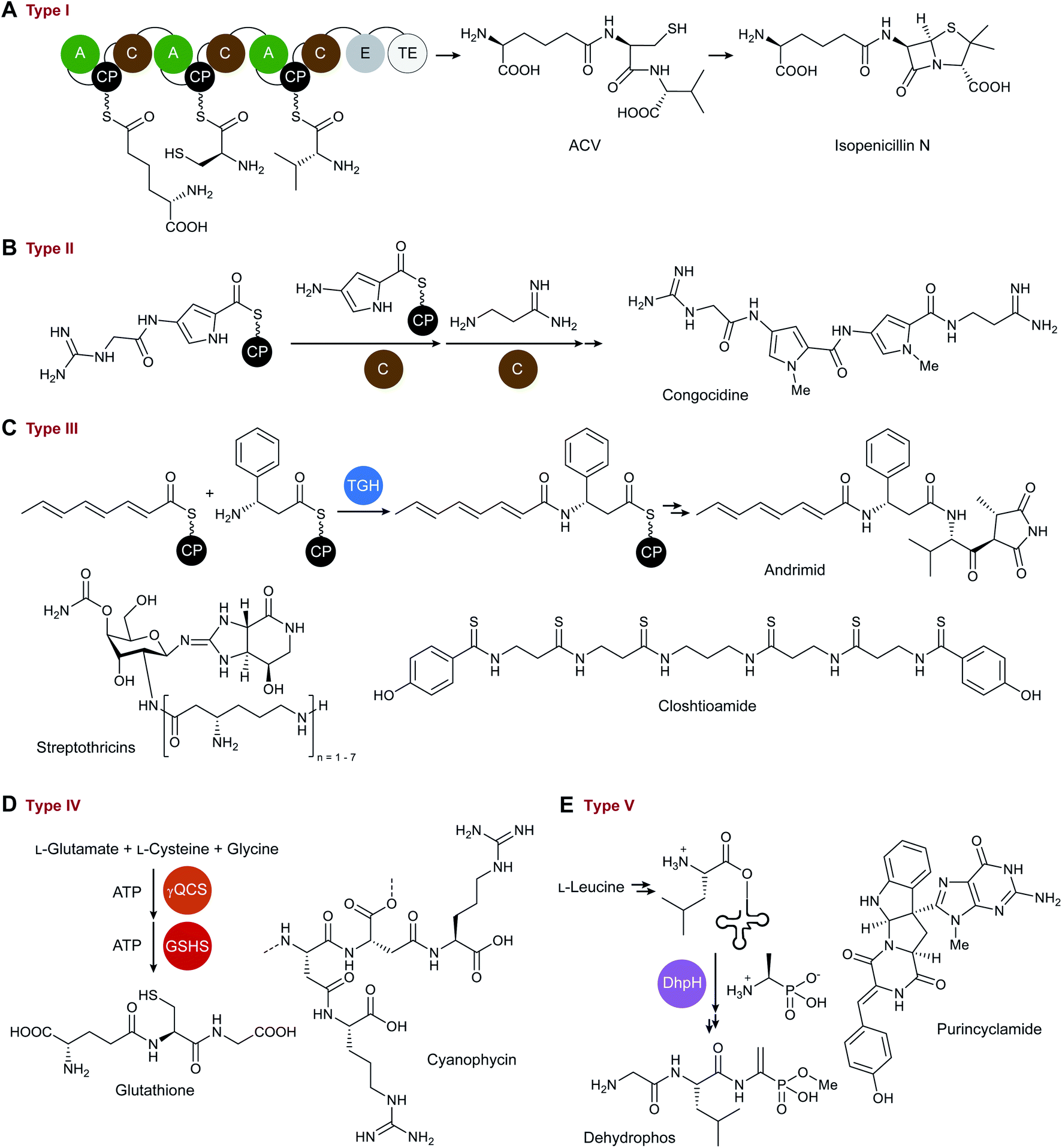 | ||
| Fig. 1 Examples for different types of ribosome-independent peptide biosynthesis. (A) The δ-aminoadipyl-cysteinyl-D-valine (ACV) synthetase is a typical type I NRPS. (B) Congocidine biosynthesis is an example for a type II NRPS in which free-standing C domains catalyse peptide bond formation. (C) Andrimid biosynthesis serves as an example for a type III NRPS, since it is CP-mediated, but the formation of the peptide bond is catalysed by a transglutaminase homolog (TGH). Two other examples for type III systems are the biosynthetic pathways for streptothricins and for closthioamide. (D) Type IV includes glutathione biosynthesis. γQCS = γ-glutamate-cysteine synthetase and GSHS = glutathione synthetase. Cyanophycin biosynthesis is another example for a ribosome-independent peptide assembly fitting to this class. (E) Group V includes dehydrophos biosynthesis where leucine is connected to tRNA for peptide bond formation with phosphonoalanine catalysed by the C-terminus of the peptidyltransferase DhpH. Purincyclamide also derives from a biosynthetic pathway of this type. | ||
This group also includes monomodular NRPSs with C domains as amide-bond forming catalysts. Examples for monomodular NRPSs can be found as part of the biosynthetic pathway to the conidiophore pigment17 as well as the route to hancockiamides.18 It should be noted, however, that not all products of NRPSs are peptides, since the A/C domains may also generate C–O and C–C bonds.19
2.2 Thiotemplated, freestanding peptide synthetases: type II
Various peptide synthetases deviate from the modular architecture of the type I NRPSs by employing catalytic units that are not covalently linked to each other and are thus termed type II NRPSs. Building on the previous definition of a type II NRPS by Shen and coworkers,4 we propose that the defining features of these machineries are (a) the CP-mediated substrate presentation, (b) the use of A domains for CP loading, and (c) the lack of the multi-modular architecture of a type I NRPS. In many biosynthetic pathways, however, only a portion of the biosynthetic enzymes are encoded as standalone proteins. Although a pure type II NRPS would require that all biosynthetic enzymes were standalone proteins, such examples are exceedingly rare,20,21 and type II systems predominantly occur as hybrids with type I NRPS or PKS assembly lines. The standalone portion of the biosynthetic enzymes are most commonly A domains, CPs or A-CP didomains.22 It would make sense to expand this definition and to include standalone C domains as well. However, initiation modules usually consist of an A-CP didomain and are therefore not considered type II NRPSs.23As mentioned above, most of the type II NRPSs occur together with type I systems or polyketide synthases. The prototypical example of such a type I–type II hybrid NRPS is the congocidine synthetase. The biosynthesis of congocidine, a pyrrolamide-containing NRP, involves two free-standing C domains that connect thioester-bound aminoacyl building blocks (Fig. 1B).24 Another rare example of an NRPS with standalone C (di-)domains was found in the biosynthesis of acinetobactin, a catecholate siderophore produced by Acinetobacter baumannii. Unusually, as described in congocidine biosynthesis,24 the amide bond-forming C domain also catalyses the offloading of the peptidic product.25
The siderophore enterobactin is also biosynthesised by a type I–type II hybrid NRPS.3 The enterobactin biosynthetic pathway involves the first reported type II NRPS, although it was not assigned as such at the time of its discovery. The type II NRPS component comprises a free-standing A domain and a CP-isochorismatase didomain and catalyses the formation of the substrate for the following type I NRPS assembly line. The standalone A-domain was first thought to be an AMP ligase only, but it was later discovered that it is also responsible for the prior loading of 2,3-dihydroxybenzoic acid onto a CP.22 Iterative action of the type I NRPS leads to the formation of the mature siderophore.3 More information and examples can be found in the detailed review article of Burkart and colleagues.22
Considering that type I and type II NRPSs are biochemically indistinguishable and occur almost exclusively as hybrids, one may question the separation of type I and type II NRPS and instead use qualifiers such as (multi-)modular, standalone, or in trans. Even though the type I/II subdivision is based on the systematics of fatty acid and polyketide synthases – and is therefore easy to remember – one may argue that the analogy to the FAS and PKS fields has shortcomings from an enzymatic point of view. While the latter belong to a protein family in which all enzymes catalyse the formation of C–C bonds and thus chain extensions in the same fashion,26,27 the amide bond-forming enzymes in NRPSs can belong to quite distinct families. In fact, numerous NRPS assembly lines have been identified that do not use C domains for peptide or amide bond formation, constituting the next group of ribosome-independent peptide synthetases.
2.3 Thiotemplated, freestanding, non-canonical peptide synthetases: type III
Thiotemplate systems that do not use typical C domains for amide bond formation but employ proteases or ATP-dependent enzymes28 clearly deviate from the classical NRPS assembly lines and constitute another type of NRPSs, here tentatively named type III NRPS. Examples of NRPs constructed by such unusual peptide synthetases are andrimid,21 closthioamide,20 and petrobactin.8 Although andrimid contains two amide bonds, no C domain is involved in its biosynthetic pathway. Instead, two members of the transglutaminase (TG) protein family are responsible for amide bond formation (Fig. 1C).21 A TG also participates in the biosynthesis of closthioamide (Fig. 1C),20 a thioamidated antibiotic isolated from the obligate anaerobe Ruminiclostridium cellulolyticum. Elucidation of the closthioamide biosynthetic pathway revealed a novel CP-dependent pathway for peptide backbone assembly using enzymes from three different protein families: TG, ATP-grasp, and BtrH (PF14399; an acyl CP aminoglycoside acyl-transferase).9,20,28 The amide bond-forming enzyme AsbE in petrobactin biosynthesis also belongs to the BtrH protein family,8 and catalyses the transfer of a CP-tethered 2,3-diydro benzoic acid-moiety to spermidine. Further processing of the resultant product by an ATP-dependent, CP-independent amide synthetase affords the mature siderophore.8 In principle, various alternative biocatalysts for amide bond formation are conceivable that would fit into this ‘type III NRPS’ group. There are numerous protein families known to catalyse this reaction,29 however, they are yet to be described in the context of NRP biosynthesis.Interestingly, there are also NRPSs where standalone A domains catalyse amide bond formation, as in the biosynthetic pathways to streptothricin (Fig. 1C)30 and vicenistatin.31 Although these synthetases contain A, C and CP domains, they clearly differ from canonical type I and type II NRPSs and would be binned into the type III group according to the decision tree (Scheme 1). By analogy to these non-modular NRPSs, it is conceivable that modular synthetases have evolved that utilize domains other than C domains for amide bond formation.
Although thiotemplate systems are typically regarded as the main avenue to non-ribosomal peptides, there are numerous ribosome-independent pathways that do not depend on CP-tethered substrates.
2.4 Thiotemplate- and tRNA-independent peptide synthetases: type IV
Several non-ribosomal peptides are produced by enzymes that require neither CP- or tRNA-mediated substrate presentation nor C domains for the formation of the amide bonds. Historically, the products of such pathways were not referred to as NRPs. In the early 2000s, the characterization of multiple siderophore biosynthetic pathways led to the development of an alternative nomenclature.32 Such systems were referred to as NRPS-independent siderophore (NIS) synthetases.8 While this classification effectively separates NIS pathways from the canonical type I and type II NRPS systems, it excludes pathways to NRPs that do not function as siderophores but that are formed similarly to NISs. One important example for such an NRP is glutathione. The biosynthesis of this tripeptide in Escherichia coli was elucidated in 1953![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and involves the ATP-dependent coupling of cysteine, glutamate and glycine by two amide synthetases (Fig. 1D).7 Although the enzymes responsible for peptide bond formation in glutathione biosynthesis are not related to those commonly described for NIS (IucA/IucC protein family),32 there is a shared biosynthetic logic (ATP-dependent, not CP-mediated) with aerobactin or staphyloferrin B8 biosynthesis, for example. In these cases, the amide bond is formed using the carboxylic group of citrate and a free amine of a specific substrate.
33 and involves the ATP-dependent coupling of cysteine, glutamate and glycine by two amide synthetases (Fig. 1D).7 Although the enzymes responsible for peptide bond formation in glutathione biosynthesis are not related to those commonly described for NIS (IucA/IucC protein family),32 there is a shared biosynthetic logic (ATP-dependent, not CP-mediated) with aerobactin or staphyloferrin B8 biosynthesis, for example. In these cases, the amide bond is formed using the carboxylic group of citrate and a free amine of a specific substrate.
In the last decades, the NRPS and NIS fields have continued to evolve in parallel, making it more challenging to do justice to both, when it comes to describing a novel biosynthetic pathway. It would be desirable to introduce a basic language for both worlds. More examples following the scheme of this group (IV) are the peptide synthetases for L-theanine,29 cyanophycin (Fig. 1D),34 peptidoglycan, and the 2,3-diaminobutyrate moiety in friulimicin biosynthesis,35 representing the protein families of synthetases,29 ligases, and lyases, respectively.35
Although one may object that this group would become quite large and perhaps too heterogenous, comprising such diverse enzyme families, it would nonetheless be advantageous to use this bin as a means to integrate these ribosome-independent peptide synthetases.
2.5 Non-thiotemplated, tRNA-dependent peptide synthases: type V
Finally, there is a group of ribosome-independent peptide synthetases that shares the mode of substrate presentation with the ribosome as both employ aminoacyl-tRNA. As such, the amino acid building blocks are already provided in the activated form, and the amide bond-forming step does not require additional ATP. Such ribosome-independent, yet aminoacyl-tRNA-utilizing peptide bond-forming enzymes play key roles in biosynthetic pathways to various bacterial cyclodipeptides or diketopiperazines36 that were previously termed ‘NRPS-independent’. For example, a cyclodipeptide synthase (CDPS) merges tRNA-tethered tryptophan and tyrosine to form the cyclodipeptide scaffold of purincyclamide (Fig. 1E).37 In a similar fashion, the ‘trojan horse’ antibiotic dehydrophos is biosynthesised.38 Phosphonated L-alanine and tRNA-tethered L-leucine are connected by the amide bond-forming C-terminal region of DhpH (Fig. 1E). This enzyme belongs to the FemX peptidyltransferase protein family and shows a mode of action similar to that of the ribosome.38 The utilization of tRNA-bound substrates may suggest that these could represent ancestral pathways to non-ribosomal peptides. As such, tRNA-dependent CDPS and peptidyltransferases should be included to the superfamily of ribosome-independent peptide synthetases as a new type V.3 Conclusions
It is obvious that the field of ribosome-independent peptide biosynthesis is highly diverse and unstructured. The aim of this Viewpoint article is to stimulate a discussion that would ideally lead to a unifying nomenclature for ribosome-independent peptide synthetases. Such a classification system is needed to provide a basic language in the field of natural product research. One can envision that a classification could also support the discovery of additional non-canonical NRPSs when implemented in genome mining tools and databases.To make this heterogeneous field a bit clearer, we propose here a decision tree (Scheme 1) as a binning system for ribosome-independent peptide biosynthesis.
If other atypical NRPSs emerge in the future that do not meet the criteria described here, it should be easy to extend, e.g., by applying subclasses to the main types and groups. Very soon, this may already be necessary for synthetic NRPSs.39 Such systems are currently on the rise due to the increased availability of synthetic biology methods and they could be included, for example, as type IIS or type IIIS, etc.39
There will always be different opinions about the structure of such a nomenclature, and there is probably no perfect solution that will satisfy everyone. For a critical reflection on this issue, it is crucial to clarify whether it is still useful to base an updated nomenclature on the PKS/FAS types, i.e. whether it still makes sense to discriminate between modular and standalone systems. Another important aspect to address is whether only typical modular megasynthetases should be referred to as NRPSs or if this generic term should also encompass all other ribosome-independent peptide bond-forming enzyme systems. In case it was used as an umbrella for all ribosome-independent peptide synthetases, what property (phylogeny, structure, cofactors, substrates, etc.) of the peptide bond-forming unit in the machinery should form the basis for this binning system?
What if there was an even better way to unify the diverse pathways to non-ribosomal peptides? Perhaps it would be necessary to replace the term ‘non-ribosomal peptide synthetases' with a term that does not require negation – but can truly describe the systems as they are. A term such as ribosome-independent peptide synthetase would meet this criterion. Even so, what abbreviation would be used? RIPS would be too confusing because of its similarity to RiPPs.
It will be necessary to discuss all these critical issues mentioned above with a combined effort. A future task could be to write a community review article with a more comprehensive literature survey, like the one in the field of RiPPs.12 Certainly, the existing information on unusual NRPS can then be used for the refinement of the available bioinformatic tools. At the same time, it will encourage scientist to take a closer look at their study systems. Revised systematics could drive the discovery of additional non-canonical NRPS systems, helping to overcome the limitations of genome mining, whereby only natural products similar to those already known can be predicted.
4 Author contributions
All authors created the nomenclature concept. M. D. reviewed the NRP literature, wrote the original draft and visualised the ideas in figures. K. L. D. reviewed, discussed, and edited the draft. C. H. reviewed, discussed, and edited the draft.5 Conflicts of interest
There are no conflicts to declare.6 Acknowledgments
We thank Sarah P. Niehs, Evelyn M. Molloy, Florian Bredy and Finn Gude for fruitful discussions on various biosynthetic pathways and where to place them in this proposed nomenclature. We are very grateful for all the comments and critical revision of this manuscript by members of the NRPS community. Namely these are: David F. Ackerley, Brian O. Bachmann, Kai Blin, Helge B. Bode, Gregory L. Challis, Max Cryle, Elke Dittmann, Tadashi Eguchi, Yang Hai, Donald Hilvert, Edward Kalkreuter, Hajo Kries, Marnix Medema, Henning D. Mootz, Jörn Piel, Simon Shaw, Ben Shen, Martin Schmeing, Pierre Stallforth, Roderich D. Süssmuth and his team, Tilmann Weber, and Nadine Ziemert. We are grateful for the financial support (Leibniz Award to C. H. and Humboldt Research Fellowship for Postdoctoral Researchers to K. L. D).7 Notes and references
- B. Mach, E. Reich and E. L. Tatum, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 175 CrossRef CAS PubMed.
- F. Lipmann, Acc. Chem. Res., 1973, 6, 361 CrossRef CAS.
- H. D. Mootz, D. Schwarzer and M. A. Marahiel, ChemBioChem, 2002, 3, 490 CrossRef CAS PubMed.
- L. Du and B. Shen, Chem. Biol., 1999, 6, 507 CrossRef CAS.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770 CrossRef.
- H. Barreteau, A. Kovac, A. Boniface, M. Sova, S. Gobec and D. Blanot, FEMS Microbiol. Rev., 2008, 32, 168 CrossRef CAS.
- M. E. Anderson, Chem.-Biol. Interact., 1998, 111–112, 1 CrossRef CAS.
- C. S. Carroll and M. M. Moore, Crit. Rev. Biochem. Mol. Biol., 2018, 53, 356 CrossRef CAS PubMed.
- K. L. Dunbar, H. Büttner, E. M. Molloy, M. Dell, J. Kumpfmuller and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 14080 CrossRef CAS PubMed.
- K. Blin, H. U. Kim, M. H. Medema and T. Weber, Briefings Bioinf., 2019, 20, 1103 CrossRef CAS.
- S. A. Kautsar, K. Blin, S. Shaw, J. C. Navarro-Munoz, B. R. Terlouw, J. J. J. van der Hooft, J. A. van Santen, V. Tracanna, H. G. Suarez Duran, V. Pascal Andreu, N. Selem-Mojica, M. Alanjary, S. L. Robinson, G. Lund, S. C. Epstein, A. C. Sisto, L. K. Charkoudian, J. Collemare, R. G. Linington, T. Weber and M. H. Medema, Nucleic Acids Res., 2020, 48, D454 Search PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108 RSC.
- M. McErlean, J. Overbay and S. Van Lanen, J. Ind. Microbiol. Biotechnol., 2019, 46, 493 CrossRef CAS PubMed.
- T. W. Giessen and M. A. Marahiel, FEBS Lett., 2012, 586, 2065 CrossRef CAS PubMed.
- R. Finking and M. A. Marahiel, Annu. Rev. Microbiol., 2004, 58, 453 CrossRef CAS PubMed.
- C. T. Walsh, Nat. Prod. Rep., 2016, 33, 127 RSC.
- Y. Hai, M. Jenner and Y. Tang, J. Am. Chem. Soc., 2019, 141, 16222 CrossRef CAS.
- H. Li, A. E. Lacey, S. Shu, J. A. Kalaitzis, D. Vuong, A. Crombie, J. Hu, C. L. M. Gilchrist, E. Lacey, A. M. Piggott and Y. H. Chooi, Org. Biomol. Chem., 2021, 19, 587 RSC.
- S. Dekimpe and J. Masschelein, Nat. Prod. Rep., 2021 10.1039/d0np00098a.
- K. L. Dunbar, M. Dell, F. Gude and C. Hertweck, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8850 CrossRef CAS PubMed.
- P. D. Fortin, C. T. Walsh and N. A. Magarvey, Nature, 2007, 448, 824 CrossRef CAS PubMed.
- M. J. Jaremko, T. D. Davis, J. C. Corpuz and M. D. Burkart, Nat. Prod. Rep., 2020, 37, 355 RSC.
- A. S. Brown, M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Prod. Rep., 2018, 35, 1210 RSC.
- A. H. Al-Mestarihi, A. Garzan, J. M. Kim and S. Garneau-Tsodikova, ChemBioChem, 2015, 16, 1307 CrossRef CAS PubMed.
- A. M. Gulick, Nat. Prod. Rep., 2017, 34, 981 RSC.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688 CrossRef CAS PubMed.
- Y. Chen, E. E. Kelly, R. P. Masluk, C. L. Nelson, D. C. Cantu and P. J. Reilly, Protein Sci., 2011, 20, 1659 CrossRef CAS PubMed.
- A. Goswami and S. G. Van Lanen, Mol. BioSyst., 2015, 11, 338 RSC.
- J. Pitzer and K. Steiner, J. Biotechnol., 2016, 235, 32 CrossRef CAS.
- C. Maruyama, J. Toyoda, Y. Kato, M. Izumikawa, M. Takagi, K. Shin-ya, H. Katano, T. Utagawa and Y. Hamano, Nat. Chem. Biol., 2012, 8, 791 CrossRef CAS.
- A. Miyanaga, F. Kudo and T. Eguchi, Curr. Opin. Chem. Biol., 2016, 35, 58 CrossRef CAS.
- G. L. Challis, ChemBioChem, 2005, 6, 601 CrossRef CAS PubMed.
- P. J. Samuels, Biochem. J., 1953, 55, 441 CrossRef CAS.
- K. Ziegler, A. Diener, C. Herpin, R. Richter, R. Deutzmann and W. Lockau, Eur. J. Biochem., 1998, 254, 154 CrossRef CAS PubMed.
- L. M. Iyer, S. Abhiman, A. Maxwell Burroughs and L. Aravind, Mol. BioSyst., 2009, 5, 1636 RSC.
- P. Belin, M. Moutiez, S. Lautru, J. Seguin, J. L. Pernodet and M. Gondry, Nat. Prod. Rep., 2012, 29, 961 RSC.
- J. Shi, X. Xu, E. J. Zhao, B. Zhang, W. Li, Y. Zhao, R. H. Jiao, R. X. Tan and H. M. Ge, Org. Lett., 2019, 21, 6825 CrossRef CAS PubMed.
- E. C. Ulrich, D. J. Bougioukou and W. A. van der Donk, ACS Chem. Biol., 2018, 13, 537 CrossRef CAS PubMed.
- K. A. J. Bozhueyuek, J. Watzel, N. Abbood and H. B. Bode, Angew. Chem., Int. Ed., 2021, 60, 17531 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |