DOI:
10.1039/D2NJ04668G
(Paper)
New J. Chem., 2022,
46, 23225-23238
Amidines from cyclic amines and nitriles in the presence of zinc(II): other nitriles in place of acetonitrile†
Received
21st September 2022
, Accepted 18th November 2022
First published on 18th November 2022
Abstract
[Zn(quin)2(H2O)] (quin− = an anionic form of quinoline-2-carboxylic acid, quinaldinate) was reacted with secondary cyclic amines (piperidine or pyrrolidine) and nitriles (propionitrile or benzonitrile) to form a series of products, including two types of amidine compound. Amidines result from the nucleophilic addition of amine to nitrile, promoted by zinc(II) species. Complexes of zinc(II) with intact amines were also obtained, with compositions of [Zn(quin)2(amine)] and trans-[Zn(quin)2(amine)2]. The nature of the products depended heavily upon the reaction conditions. Propionitrile reaction systems afforded only one zinc(II) amidine complex, [Zn(quin)2(pipepropioam)] (5) (pipepropioam = amidine, obtained from piperidine and propionitrile), and two amidinium salts, pipepropioamH[Zn(quin)3] (6) and pyropropioamH[Zn(quin)3] (7) (pipepropioamH+, pyropropioamH+ = protonated amidines). Benzonitrile reactions led to zinc(II) complexes with both benzoamidines, [Zn(quin)2(pipebenzoam)] (8) and [Zn(quin)2(pyrobenzoam)] (9) (pipebenzoam, pyrobenzoam = amidines from benzonitrile and amines, piperidine or pyrrolidine, respectively). Both benzoamidine complexes displayed polymorphism, with pairs of polymorphs being labeled 8a/8b or 9a/9b. [Zn(quin)3]− salts with protonated benzoamidines, pipebenzoamH[Zn(quin)3] (10) and pyrobenzoamH[Zn(quin)3] (11) were also isolated. X-Ray structure analysis has revealed that the [Zn(quin)3]− ions of 6, 7 and 11 featured zinc(II) in a five-coordinate environment. In the case of pyrrolidinobenzoamidine, a co-crystal, pyrobenzoamH[Zn(quin)3]·[Zn(quin)2(pyrobenzoam)] (12), which is composed of both the amidinium salt and the amidine complex, was obtained. Interestingly, the metal ion of [Zn(quin)3]− in 12 was in a six-coordinate environment.
1. Introduction
RC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR)NR2 (R = substituent), a structural fragment with a central carbon atom singly bonded to a nitrogen atom and doubly bonded to a second nitrogen atom, is known as carboxamidine or, more generally, as amidine.1 This nomenclature makes a reference to the acid obtained upon hydrolysis of the amidine.2 Amidines are among the strongest uncharged bases.3,4 On protonation at the sp2-hybridized nitrogen, the positive charge is delocalized onto both nitrogen atoms, ensuring that the C–N bonds in amidinium cations are of almost identical length. Owing to their basicity, amidines have found application in coordination chemistry.2,5 Due to the CN double bond and the preference to bind to metal ions via the imine nitrogen, two stereochemical configurations are possible for amidine complexes, commonly described as E and Z isomers.6,7 The E isomer is kinetically favored due to its reduced steric hindrance.8 The existence of only E isomer in a series of amidine rhenium complexes is attributed to the bulkiness of the amidine substituents.9
NR)NR2 (R = substituent), a structural fragment with a central carbon atom singly bonded to a nitrogen atom and doubly bonded to a second nitrogen atom, is known as carboxamidine or, more generally, as amidine.1 This nomenclature makes a reference to the acid obtained upon hydrolysis of the amidine.2 Amidines are among the strongest uncharged bases.3,4 On protonation at the sp2-hybridized nitrogen, the positive charge is delocalized onto both nitrogen atoms, ensuring that the C–N bonds in amidinium cations are of almost identical length. Owing to their basicity, amidines have found application in coordination chemistry.2,5 Due to the CN double bond and the preference to bind to metal ions via the imine nitrogen, two stereochemical configurations are possible for amidine complexes, commonly described as E and Z isomers.6,7 The E isomer is kinetically favored due to its reduced steric hindrance.8 The existence of only E isomer in a series of amidine rhenium complexes is attributed to the bulkiness of the amidine substituents.9
The literature describes several synthetic routes for the synthesis of amidines.10–13 The simplest starting materials, allowing 100% atom economy, are amines and nitriles. However, the electrophilicity of nitriles is not sufficient for a direct nucleophilic addition of amines.14 Activation of nitrile can be achieved through substitution with a strong electron-withdrawing group, such as CCl3.15 The activating group remains incorporated in the amidine product, and may occasionally also cause unwanted cross-reactivity. Other strategies for nitrile activation rely on the presence of Lewis acids such as AlCl3, ZnCl2 at high temperatures16,17 or highly acidic conditions. The latter refers to the multi-step Pinner method with a time-consuming reaction of nitrile with alcohol in the presence of substantial amounts of HCl or HBr to yield an imino ester salt. This intermediate is then reacted with a huge excess of amine.18 On the other hand, some metal ions have been shown to act as extremely strong activators of nitriles toward nucleophilic attack.14,19–29 Ir(III), Pt(II), Pt(IV), Pd(II), Rh(III) and Ru(II) are a few examples that display this type of behavior. In the majority of metal-mediated syntheses, nitrile is first coordinated to the metal ion,7,30,31 leading to a redistribution of the electron density in the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N moiety, making it more reactive toward nucleophiles. Typically, the amidine formed in situ remains coordinated to the metal ion. Reactions of this type using ammonia or primary and secondary amines are well-documented.6,9,28,29,32–37 By analogy, nitroanilines,38 diamines, triamines39 and some rather complex amines,23,40,41 (e.g., alkyldiamine-substituted 2,6-diaminopurines,24 1H-indazole,27) have also been reacted in this way. As well as acetonitrile, the other nitriles tested include propionitrile,38,39 benzonitrile,39,41,42 phenylacetonitrile39 and 2-cyanopyridine.23
N moiety, making it more reactive toward nucleophiles. Typically, the amidine formed in situ remains coordinated to the metal ion. Reactions of this type using ammonia or primary and secondary amines are well-documented.6,9,28,29,32–37 By analogy, nitroanilines,38 diamines, triamines39 and some rather complex amines,23,40,41 (e.g., alkyldiamine-substituted 2,6-diaminopurines,24 1H-indazole,27) have also been reacted in this way. As well as acetonitrile, the other nitriles tested include propionitrile,38,39 benzonitrile,39,41,42 phenylacetonitrile39 and 2-cyanopyridine.23
Recently, we have observed formation of amidine in the reaction of the secondary cyclic amines, piperidine or pyrrolidine, with zinc(II) quinaldinate (quinaldinate = an anionic form of quinoline-2-carboxylic acid) in acetonitrile.43 Surprisingly, the amines underwent reaction with the solvent, acetonitrile. Thus formed amidines, piperidinoacetamidine or pyrrolidinoacetamidine, were found either in a neutral form, coordinated to zinc(II) via imine nitrogen, or in a protonated form as amidinium cations in [Zn(quin)3]− salts. In the absence of zinc(II), no amidine could be detected. This result agrees with the known role of many zinc(II) compounds in catalysis.44
In order to test the general validity of our reaction system for the formation of amidines, we have expanded our study to two other nitriles, propionitrile and benzonitrile. As these molecules have only negligible difference in electrophilicity,45 their reactivities should be similar to that of acetonitrile. For comparison purposes, the same amines were used as in the previous study; piperidine (abbreviated as pipe) and pyrrolidine (pyro). Four reaction systems were screened, each consisting of zinc(II) quinaldinate, one of the two amines, and propionitrile or benzonitrile. As in our previous study, two types of amidine-containing product were obtained; amidine complexes and amidinium salts. Complexes of zinc(II) with intact amines were also obtained. All were analyzed and fully characterized by means of infrared (IR) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy and X-ray structure analysis of a single crystal. Crystal structures of the novel amidine compounds are presented in detail, whereas complexes with amines are briefly described in the ESI.† Structural formulas and abbreviations of amidines are given in Scheme 1. The abbreviation of each is derived from the names of the reacting amine and nitrile, with the addition of the ‘am’ suffix; e.g., pipepropioam represents piperidinopropioamidine, an amidine formed from piperidine and propionitrile. Protonated forms of the amidines included an additional H+; e.g., pipepropioam changes on protonation to pipepropioamH+.
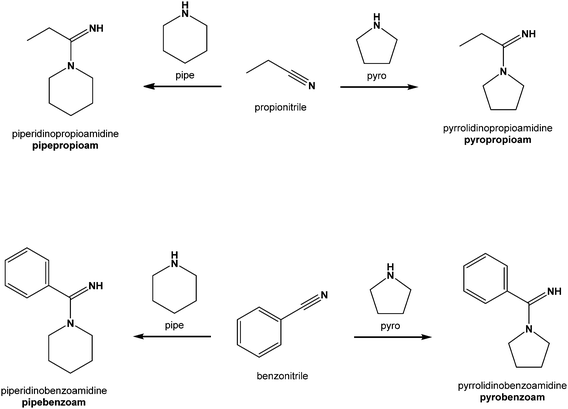 |
| | Scheme 1 Structural formulas and abbreviations (in bold) of amidines described in this study. | |
2. Experimental
2.1. General
Reagents were purchased from commercial sources and used as received, with the exception of propionitrile and benzonitrile which were dried over molecular sieves before use.46 The starting material, [Zn(quin)2(H2O)], was synthesized as previously described.471H NMR spectra were obtained on a Bruker Avance III 500 at 500 MHz or on a Bruker Avance NEO at 600 MHz, while 13C NMR spectra were obtained on a Bruker Avance NEO at 151 MHz. The solvent was (CD3)2SO with 0.03% tetramethylsilane (TMS). Chemical shifts were referenced to the central peak of the residual resonance for (CD3)2SO at 2.50 ppm (1H) or to the TMS standard at 0.00 ppm (13C).48 Chemical shifts (δ) are given in ppm and coupling constants (J) in Hz. Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and br = broad signal. Data were processed using the MestReNova program (version 11.0.4).49 The 13C NMR spectra were obtained for amidine compounds 6–11. In all, two resonances were observed in the 162–167 ppm spectral range. One signal may be attributed to the carboxylate carbon,47 whereas the other derives from the imine carbon in the amidine/amidinium cation. Similar values were reported for the imine carbon in acetamidine or acetamidinium chloride.28,50 High-resolution mass spectra (HRMS) were obtained on an Agilent 6224 Accurate Mass TOF LC Mass Spectrometer. IR spectra were recorded on a Bruker Alpha II FT-IR spectrophotometer with the attenuated total reflection (ATR) module between 4000 cm−1 and 400 cm−1. Elemental analyses on carbon, nitrogen and hydrogen were performed on a PerkinElmer 2400 II analyzer.
2.2. Preparation of [Zn(quin)2(pipe)]·CH3CH2CN (1·CH3CH2CN)
Procedure A. A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), propionitrile (7 mL) and piperidine (2 mL). The container was closed and inserted into a steel autoclave which was heated for 24 h at 105 °C. Afterward, the reaction mixture was allowed to cool slowly to room temperature. The reaction mixture was filtered, and the filtrate was stored at 4 °C. Needle-like crystals of [Zn(quin)2(pipe)]·CH3CH2CN (1·CH3CH2CN) were filtered off after a few days. Yield: 74 mg, 58%. Procedure B. [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), propionitrile (10 mL) and piperidine (1 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. White solid, 1·CH3CH2CN, was filtered off. Yield: 95 mg, 74%. IR (ATR, cm−1): 3126w, 3050w, 3013w, 2937m, 2900w, 2858w, 2240w, 1639vs, 1597s, 1568s, 1556s, 1509m, 1479w, 1461s, 1431m, 1368s, 1357vs, 1345vs, 1301m, 1279s, 1269s, 1211m, 1179s, 1151s, 1104m, 1086m, 1072w, 1044s, 1024m, 1011w, 964m, 943m, 897m, 875s, 855s, 802vs, 778vvs, 770vvs, 743s, 735s, 636s, 627m, 617m, 605s, 560m, 522s, 499s, 486w, 455w, 404s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.92 (2H, d, J = 8.6 Hz, quin−), 8.79 (2H, d, J = 8.4 Hz, quin−), 8.41 (2H, d, J = 8.4 Hz, quin−), 8.20 (2H, d, J = 8.0 Hz, quin−), 8.03–8.00 (2H, m, quin−), 7.84–7.81 (2H, m, quin−), 2.61–2.59 (4H, m, pipe), 2.46 (2H, q, J = 7.6 Hz, CH3CH2CN), 1.39–1.32 (6H, m, pipe), 1.14 (3H, t, J = 7.6 Hz, CH3CH2CN) ppm. ESI-HRMS: m/z calcd for [C25H24N3O4Zn]+ = [Zn(quin)2(pipe) + H]+: 494.1058, found: 494.1072. Elemental analysis calcd for C28H28N4O4Zn = [Zn(quin)2(pipe)]·CH3CH2CN (%): C, 61.15; H, 5.13; N, 10.19. Found (%): C, 61.00; H, 5.33; N, 10.34.
2.3. Preparation of [Zn(quin)2(pipe)2]·CH3CH2CN (2·CH3CH2CN)
A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), propionitrile (7 mL) and piperidine (1 mL). The container was closed and inserted into a steel autoclave which was heated for 24 h at 105 °C. Afterward, the reaction mixture was allowed to cool slowly to room temperature. The reaction mixture was filtered, and the filtrate was stored at 4 °C. After a few days, a mixture of crystals of [Zn(quin)2(pipe)2]·CH3CH2CN (2·CH3CH2CN) and [Zn(quin)2(pipe)]·CH3CH2CN (1·CH3CH2CN) was obtained as a one-time event. Note. The crystals of 2·CH3CH2CN are not stable outside of the mother liquor. An IR spectrum was run on the non-transparent crystals that were manually selected. IR (ATR, cm−1): 3221w, 3056w, 3043w, 2931m, 2854w, 2244w, 1634vs, 1597m, 1566s, 1509m, 1462s, 1434m, 1365vs, 1349s, 1272s, 1262s, 1219w, 1191m, 1177m, 1157m, 1114w, 1088m, 1072m, 1047m, 1023s, 1003s, 983w, 962w, 943m, 894m, 871s, 851s, 800vs, 778vvs, 747m, 661w, 635s, 598s, 555w, 523m, 502s, 456w, 438w, 403s.
2.4. Preparation of [Zn(quin)2(pipe)2]·2.5PhCN (2·2.5PhCN)
[Zn(quin)2(H2O)] (100 mg, 0.23 mmol), benzonitrile (10 mL) and piperidine (1 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. Afterward, the reaction mixture was filtered, and the filtrate was stored at 4 °C. After a few days, a small number of needle-like crystals of [Zn(quin)2(pipe)2]·2.5PhCN (2·2.5PhCN) was obtained as a one-time event. Note. The crystals of 2·2.5PhCN are not stable outside of the mother liquor. An IR spectrum was run on a dried sample. IR (ATR, cm−1): 3231w, 3057w, 2932m, 2855w, 2226m, 1639s, 1597s, 1566s, 1508m, 1490m, 1461s, 1447s, 1430m, 1365vs, 1288m, 1271m, 1218w, 1206w, 1190m, 1176s, 1154m, 1116w, 1088w, 1072s, 1048m, 1023s, 999s, 986m, 962w, 943m, 928w, 892s, 870s, 854s, 800vs, 777vvs, 758vvs, 688s, 630s, 599s, 547s, 521s, 503s, 455w, 427w, 404s.
2.5. Preparation of [Zn(quin)2(pyro)] (3)
A mixture of [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), propionitrile (10 mL) and pyrrolidine (0.5 mL) was heated in a round-bottom flask under reflux for 6 h. Afterward, the reaction mixture was allowed to cool slowly to room temperature. A white solid, [Zn(quin)2(pyro)] (3), was filtered off. Yield: 41 mg, 36%. IR (ATR, cm−1): 3171m, 3079w, 3036w, 2956w, 2885w, 1640vs, 1594m, 1567m, 1509m, 1458m, 1427w, 1369s, 1348s, 1299m, 1250m, 1214m, 1172s, 1152m, 1140s, 1116m, 1048m, 1020m, 951m, 896s, 873m, 854m, 797s, 771vvs, 729s, 637s, 628m, 603s, 556w, 522m, 492s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.90 (2H, d, J = 8.6 Hz, quin−), 8.79 (2H, d, J = 8.4 Hz, quin−), 8.41 (2H, d, J = 8.4 Hz, quin−), 8.20 (2H, d, J = 8.1 Hz, quin−), 8.03–8.01 (2H, m, quin−), 7.84–7.81 (2H, m, quin−), 2.62 (4H, t, J = 6.4 Hz, pyro), 1.50–1.45 (4H, m, pyro) ppm. ESI-HRMS: m/z calcd for [C24H22N3O4Zn]+ = [Zn(quin)2(pyro) + H]+: 480.0902, found: 480.0894. Elemental analysis calcd for C24H21N3O4Zn (%): C, 59.95; H, 4.40; N, 8.74. Found (%): C, 59.96; H, 4.42; N, 8.76.
2.6. Preparation of [Zn(quin)2(pyro)2] (4)
Synthesis of [Zn(quin)2(pyro)2] and its characterization (with the exception of X-ray structure analysis) has been reported previously.43 Single crystals were prepared in the course of this study. [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), propionitrile (5 mL), 2-propanol (5 mL) and pyrrolidine (1 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. Afterward, the reaction mixture was filtered. A glass vial with diethyl ether was carefully inserted into the filtrate. After a few days, a mixture of crystals of [Zn(quin)2(pyro)2] (4) and [Zn(quin)2(pyro)] (3) was obtained.
2.7. Preparation of [Zn(quin)2(pipepropioam)] (5)
[Zn(quin)2(H2O)] (100 mg, 0.23 mmol), propionitrile (5 mL), methanol (5 mL) and piperidine (1 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. Afterward, the reaction mixture was filtered and concentrated on a rotary evaporator. The concentrate was transferred to an Erlenmeyer flask. A glass vial with diethyl ether was carefully inserted into the flask. A crystalline solid, [Zn(quin)2(pipepropioam)] (5), was filtered off on the next day. Yield: 45 mg, 35%. Single crystals of 5, suitable for X-ray diffraction analysis, were not obtained. The identity of the solid was determined by IR, 1H NMR spectroscopy and mass spectrometry. IR (ATR, cm−1): 3405w, 3288m, 3059w, 3021w, 2932w, 2855w, 1640vs, 1582s, 1571s, 1509m, 1492m, 1461s, 1439m, 1342vs, 1281m, 1258m, 1218s, 1183s, 1145m, 1113m, 1081m, 1067w, 1040w, 1024m, 970m, 953w, 899s, 875m, 852m, 832m, 798vs, 771vvs, 740s, 715m, 693w, 638s, 629s, 602s, 560m, 522m, 495s, 405s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.96 (1H, br s, pipepropioam), 8.76 (2H, d, J = 8.4 Hz, quin−), 8.49 (1H, br s, pipepropioam), 8.32 (2H, d, J = 8.6 Hz, quin−), 8.25 (2H, d, J = 8.4 Hz, quin−), 8.19 (2H, d, J = 7.9 Hz, quin−), 7.99–7.96 (2H, m, quin−), 7.84–7.81 (2H, m, quin−), 3.58–3.53 (4H, m, pipepropioam), 2.59–2.57 (2H, m, pipepropioam, overlapped with the residual peak for (CD3)2SO), 1.68–1.60 (6H, m, pipepropioam), 1.16 (3H, t, J = 7.6 Hz, pipepropioam) ppm. Note. Due to the poor solubility of 5 in (CD3)2SO, trifluoroacetic acid (TFA) was added. TFA causes a dissociation of the complex. The signals in the spectrum thus belong to the protonated species and are located at different chemical shifts. A signal at 14.47 ppm belongs to TFA. ESI-HRMS: m/z calcd for [C28H29N4O4Zn]+ = [Zn(quin)2(pipepropioam) + H]+: 549.1480, found: 549.1482.
2.8. Preparation of pipepropioamH[Zn(quin)3] (6)
[Zn(quin)2(H2O)] (100 mg, 0.23 mmol), propionitrile (5 mL), methanol (5 mL) and piperidine (0.5 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. Afterward, the reaction mixture was filtered. A glass vial with diethyl ether was carefully inserted into the filtrate. After a few days, single crystals of pipepropioamH[Zn(quin)3] (6) were filtered off. Yield: 27 mg, 16%. IR (ATR, cm−1): 3007w, 2944w, 2885w, 2858w, 1703w, 1652m, 1623vs, 1558s, 1508m, 1461m, 1428w, 1367s, 1344vs, 1307m, 1267m, 1242w, 1222m, 1187w, 1175m, 1167m, 1150m, 1109m, 1087w, 1072w, 1022w, 981w, 952w, 892m, 853m, 805s, 797vs, 776vvs, 742s, 637s, 628s, 603m, 560w, 522m, 500m, 483w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.59 (3H, d, J = 8.6 Hz, quin−), 8.54 (3H, d, J = 8.4 Hz, quin−), 8.19 (3H, d, J = 8.4 Hz, quin−), 8.04 (3H, d, J = 8.0 Hz, quin−), 7.74–7.71 (3H, m, quin−), 7.67–7.64 (3H, m, quin−), 3.52–3.50 (4H, m, pipepropioamH+), 2.53 (2H, q, J = 7.6 Hz, pipepropioamH+), 1.59–1.57 (6H, m, pipepropioamH+), 1.11 (3H, t, J = 7.6 Hz, pipepropioamH+) ppm. 13C NMR ((CD3)2SO with 0.03% v/v TMS, 151 MHz): δ 166.51, 166.23, 152.33, 144.46, 138.83, 130.23, 128.58, 128.52, 127.69, 127.64, 120.29, 24.59, 22.77, 10.96 ppm. ESI-HRMS: m/z calcd for [C30H18N3O6Zn]− = [Zn(quin)3]−: 580.0487, found: 580.0479. Elemental analysis calcd for C38H35N5O6Zn (%): C, 63.12; H, 4.88; N, 9.69. Found (%): C, 62.53; H, 4.88; N, 9.72.
2.9. Preparation of pyropropioamH[Zn(quin)3] (7)
Procedure A. A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), propionitrile (6 mL), methanol (1 mL) and pyrrolidine (1 mL). The container was closed and inserted into a steel autoclave which was heated for 24 h at 105 °C. Afterward, the reaction mixture was allowed to cool slowly to room temperature. The reaction mixture was filtered and concentrated on a rotary evaporator. The concentrate was transferred to an Erlenmeyer flask and a glass vial with diethyl ether was carefully inserted. Crystalline solid, pyropropioamH[Zn(quin)3] (7), was filtered off after a few days. Yield: 86 mg, 52%. Procedure B. [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), propionitrile (5 mL), methanol (5 mL) and pyrrolidine (0.5 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. Afterward, the reaction mixture was filtered and concentrated on a rotary evaporator. The concentrate was transferred to an Erlenmeyer flask. A glass vial with diethyl ether was carefully inserted into the flask. Crystals of 7 were filtered off on the next day (32 mg). IR (ATR, cm−1): 3039w, 2972w, 2881w, 1694w, 1652m, 1615s, 1559s, 1509m, 1460m, 1427w, 1368s, 1352s, 1267m, 1214m, 1188m, 1175m, 1166m, 1149s, 1109m, 1082m, 1022w, 951w, 893m, 854m, 807s, 798s, 776vvs, 743s, 711m, 637s, 628s, 598s, 560m, 523s, 500s, 484m, 417m, 403s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.59 (3H, d, J = 8.6 Hz, quin−), 8.54 (3H, d, J = 8.5 Hz, quin−), 8.18 (3H, d, J = 8.5 Hz, quin−), 8.04 (3H, d, J = 8.1 Hz, quin−), 7.74–7.71 (3H, m, quin−), 7.67–7.64 (3H, m, quin−), 3.59 (2H, t, J = 6.5 Hz, pyropropioamH+), 3.35–3.32 (2H, m, pyropropioamH+, overlapped with the peak for H2O), 1.96–1.90 (4H, m, pyropropioamH+), 1.15 (3H, t, J = 7.6 Hz, pyropropioamH+) ppm. Note. The residual signal for ((CD3)2SO hides a signal for another two pyropropioamH+ protons. 13C NMR ((CD3)2SO with 0.03% v/v TMS, 151 MHz): δ 166.50, 165.60, 144.46, 138.81, 130.22, 128.58, 128.53, 127.68, 127.63, 120.29, 48.56, 48.15, 25.13, 24.69, 24.14, 9.92 ppm. ESI-HRMS: m/z calcd for [C30H18N3O6Zn]− = [Zn(quin)3]−: 580.0487, found: 580.0477. Elemental analysis calcd for C37H33N5O6Zn (%): C, 62.67; H, 4.69; N, 9.88. Found (%): C, 62.46; H, 5.02; N, 9.94.
2.10. Preparation of [Zn(quin)2(pipebenzoam)] (8)
Procedure A. A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), benzonitrile (7 mL) and piperidine (1 mL). The container was closed and inserted into a steel autoclave which was heated for 24 h at 105 °C. Afterward, the reaction mixture was allowed to cool slowly to room temperature. The reaction mixture was filtered. A glass vial with diethyl ether was carefully inserted into the filtrate. Crystals of [Zn(quin)2(pipebenzoam)] (8) were filtered off after a few days. Yield: 97 mg, 69%. Procedure B. [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), benzonitrile (5 mL), methanol (5 mL) and piperidine (1 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. Afterward, the reaction mixture was filtered and a glass vial with diethyl ether was carefully inserted into the filtrate. After a few days, colorless crystals of [Zn(quin)2(pipebenzoam)] were filtered off (44 mg). [Zn(quin)2(pipebenzoam)] crystallizes in two polymorphic modifications; triclinic (8a) and monoclinic (8b). IR (ATR, cm−1): 3205m, 3062w, 3019w, 2925w, 2853w, 1634vvs, 1583s, 1550s, 1510m, 1462s, 1446m, 1435m, 1357s, 1340s, 1286w, 1262m, 1247s, 1230m, 1216m, 1175m, 1157m, 1142m, 1116m, 1074w, 1025w, 1006w, 961w, 951w, 926w, 897m, 877s, 854s, 801s, 782vs, 772vs, 736s, 704s, 654w, 637s, 627s, 603s, 578w, 561w, 531m, 522m, 497s, 458w, 434w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.71 (2H, d, J = 8.4 Hz, quin−), 8.59–8.58 (2H, m, quin−), 8.24 (2H, d, J = 7.6 Hz, quin−), 8.19 (2H, d, J = 8.2 Hz, quin−), 8.05–8.02 (2H, m, quin−), 7.84–7.81 (2H, m, quin−), 7.30–6.93 (5H, br m, pipebenzoam), 3.16 (4H, br s, pipebenzoam), 1.51–1.50 (2H, m, pipebenzoam), 1.36 (4H, br s, pipebenzoam) ppm. 13C NMR ((CD3)2SO with 0.03% v/v TMS, 151 MHz): δ 167.40, 165.35, 151.18, 143.51, 140.07, 130.90, 129.39, 129.03, 128.14, 128.01, 127.94, 126.52, 119.95, 25.17, 23.54 ppm. ESI-HRMS: m/z calcd for [C32H29N4O4Zn]+ = [Zn(quin)2(pipebenzoam) + H]+: 597.1480, found: 597.1498. Elemental analysis calcd for C32H28N4O4Zn (%): C, 64.28; H, 4.72; N, 9.37. Found (%): C, 63.96; H, 4.62; N, 9.35.
2.11. Preparation of [Zn(quin)2(pyrobenzoam)] (9)
A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), benzonitrile (7 mL) and pyrrolidine (1 mL). The container was closed and inserted into a steel autoclave which was heated for 24 h at 105 °C. Afterward, the reaction mixture was allowed to cool slowly to room temperature. The reaction mixture was filtered. A glass vial with diethyl ether was carefully inserted into the filtrate. Single crystals of [Zn(quin)2(pyrobenzoam)] (9) grew after a few days. Yield: 79 mg, 58%. When [Zn(quin)2(pyrobenzoam)] was left in the mother liquor, the crystals of pyrobenzoamH[Zn(quin)3] (11) also formed. [Zn(quin)2(pyrobenzoam)] crystallizes in two polymorphic modifications; triclinic (9a) and monoclinic (9b). IR (ATR, cm−1): 3212m, 3057w, 2968w, 2944w, 2874w, 1639vs, 1584m, 1553s, 1510m, 1461s, 1371s, 1356s, 1344vs, 1324s, 1263m, 1252m, 1216m, 1177s, 1155m, 1141m, 1116w, 1077w, 1023w, 989w, 967w, 924w, 896m, 853s, 839m, 800vs, 781vs, 773vvs, 738s, 704s, 663m, 637s, 627m, 603s, 560w, 522m, 499s, 435w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.66 (2H, d, J = 8.4 Hz, quin−), 8.52 (2H, d, J = 8.1 Hz, quin−), 8.19–8.17 (4H, m, quin−), 8.03–8.00 (2H, m, quin−), 7.83–7.80 (2H, m, quin−), 7.14–6.68 (5H, br m, pyrobenzoam), 3.11 (4H, br s, pyrobenzoam), 1.75 (4H, br s, pyrobenzoam) ppm. 13C NMR ((CD3)2SO with 0.03% v/v TMS, 151 MHz): δ 165.76, 165.38, 151.14, 143.51, 139.80, 130.76, 129.36, 128.97, 128.02, 127.91, 127.53, 126.22, 119.87, 24.69 ppm. ESI-HRMS: m/z calcd for [C31H27N4O4Zn]+ = [Zn(quin)2(pyrobenzoam) + H]+: 583.1324, found: 583.1335. Elemental analysis calcd for C31H26N4O4Zn (%): C, 63.76; H, 4.49; N, 9.59. Found (%): C, 63.43; H, 4.28; N, 9.52.
2.12. Preparation of pipebenzoamH[Zn(quin)3] (10)
[Zn(quin)2(pipebenzoam)] (50 mg, 0.08 mmol), quinaldinic acid (29 mg, 0.17 mmol) and benzonitrile (10 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. White solid, pipebenzoamH[Zn(quin)3] (10), was filtered off. Yield: 48 mg, 74%. Single crystals of 10, suitable for X-ray diffraction analysis, could not be obtained. The true identity of the solid was determined by IR, 1H NMR spectroscopy and mass spectrometry. IR (ATR, cm−1): 3046w, 2943m, 2859w, 1699w, 1653s, 1630s, 1609s, 1563s, 1508m, 1467s, 1447m, 1432w, 1375vs, 1355s, 1344s, 1308m, 1259w, 1220m, 1174s, 1154m, 1113m, 1076w, 1023w, 992w, 964w, 924w, 895m, 858s, 809s, 800s, 785s, 773vvs, 709s, 697s, 639m, 631s, 599s, 581m, 549w, 524m, 500m, 481w, 455w, 420w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 9.23 (2H, br s, pipebenzoamH+), 8.58 (3H, d, J = 7.5 Hz, quin−), 8.53 (3H, d, J = 8.5 Hz, quin−), 8.18 (3H, d, J = 8.5 Hz, quin−), 8.04 (3H, d, J = 8.0 Hz, quin−), 7.74–7.71 (3H, m, quin−), 7.66–7.57 (3H + 5H, m, quin− + pipebenzoamH+), 3.48 (4H, s, pipebenzoamH+), 1.64 (6H, s, pipebenzoamH+) ppm. 13C NMR ((CD3)2SO with 0.03% v/v TMS, 151 MHz): δ 166.51, 163.24, 152.31, 144.49, 138.77, 133.25, 132.11, 131.79, 130.20, 129.59, 129.38, 129.05, 128.57, 127.77, 127.68, 127.64, 120.29, 24.96, 22.77 ppm. ESI-HRMS: m/z calcd for [C30H18N3O6Zn]− = [Zn(quin)3]−: 580.0487, found: 580.0494. Elemental analysis calcd for C42H35N5O6Zn (%): C, 65.42; H, 4.57; N, 9.08. Found (%): C, 65.25; H, 4.30; N, 8.93.
2.13. Preparation of pyrobenzoamH[Zn(quin)3] (11)
[Zn(quin)2(pyrobenzoam)] (50 mg, 0.09 mmol), quinaldinic acid (30 mg, 0.17 mmol) and benzonitrile (10 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. A white solid, pyrobenzoamH[Zn(quin)3] (11), was filtered off. Yield: 49 mg, 76%. IR (ATR, cm−1): 3233w, 3053w, 2961w, 2879w, 1679w, 1660s, 1626s, 1615s, 1595s, 1562s, 1509m, 1464m, 1448m, 1435m, 1386s, 1373s, 1346vs, 1303m, 1271w, 1260w, 1217m, 1162m, 1150m, 1130w, 1119w, 1109w, 1078w, 1033w, 1024w, 966w, 892m, 867w, 851m, 807s, 797s, 775vvs, 741m, 719m, 700s, 639m, 628s, 603s, 572w, 522m, 500s, 420w, 404m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 9.02 (2H, br s, pyrobenzoamH+), 8.58 (3H, d, J = 8.4 Hz, quin−), 8.53 (3H, d, J = 8.4 Hz, quin−), 8.18 (3H, d, J = 8.4 Hz, quin−), 8.04 (3H, d, J = 8.0 Hz, quin−), 7.74–7.71 (3H, m, quin−), 7.66–7.60 (3H + 5H, m, quin− + pyrobenzoamH+), 3.50–3.40 (4H, m, pyrobenzoamH+), 1.99–1.87 (4H, m, pyrobenzoamH+) ppm. 13C NMR ((CD3)2SO with 0.03% v/v TMS, 151 MHz): δ 166.50, 161.76, 152.32, 144.47, 138.80, 132.11, 131.79, 130.21, 130.01, 128.87, 128.58, 128.55, 127.68, 127.64, 127.55, 120.29, 51.40, 48.63, 24.96, 24.32 ppm. ESI-HRMS: m/z calcd for [C30H18N3O6Zn]− = [Zn(quin)3]−: 580.0487, found: 580.0485. Elemental analysis calcd for C41H33N5O6Zn (%): C, 65.04; H, 4.39; N, 9.25. Found (%): C, 64.75; H, 4.09; N, 9.34.
2.14. Preparation of pyrobenzoamH[Zn(quin)3]·[Zn(quin)2(pyrobenzoam)] (12)
[Zn(quin)2(H2O)] (100 mg, 0.23 mmol), benzonitrile (5 mL), methanol (5 mL) and pyrrolidine (0.5 mL) were added to a round-bottom flask. The contents were stirred at room temperature for 3 days. Afterward, the reaction mixture was filtered. A glass vial with diethyl ether was carefully inserted into the filtrate. After a few days, a small number of colorless crystals of pyrobenzoamH[Zn(quin)3]·[Zn(quin)2(pyrobenzoam)] (12) was obtained. IR (ATR, cm−1): 3229w, 3054w, 2977w, 2876w, 2814w, 1694w, 1623s, 1581s, 1558s, 1508m, 1460s, 1366vs, 1344s, 1302m, 1269m, 1256m, 1216m, 1179m, 1153m, 1113m, 1075w, 1028m, 965w, 927w, 896s, 856m, 801s, 772vvs, 734s, 706s, 696s, 668m, 636s, 602s, 571m, 522s, 498s, 484m, 434w.
2.15. X-Ray structure analysis
Single crystal X-ray diffraction data were collected on an Agilent SuperNova diffractometer with molybdenum (Mo-Kα, λ = 0.71073 Å) micro-focus sealed X-ray source at 150 K. Each crystal was placed on a glass fiber tip using silicone grease and then mounted on the goniometer head. CrysAlis PRO51 was used for data processing. The structures were solved with Olex2 software52 using ShelXT53 and refined using the least squares methods in ShelXL.54 Anisotropic displacement parameters were determined for all non-hydrogen atoms. All NH or NH2+ hydrogen atoms of amines, amidines or protonated amidines were located in the final stages of refinement from difference Fourier maps and refined with isotropic displacement parameters. The remaining hydrogen atoms were added in calculated positions. Several compounds crystallized with solvent molecules that tended to be disordered. The disorder of propionitrile in 2·CH3CH2CN and of pyrrolidine ligand in 3 were successfully modeled using PART −1 instruction, while that of benzonitrile in 2·2.5PhCN was resolved using PART instruction. The disorder of the solvent molecules in 9a and 12 could not be modeled. The contribution of the disordered solvent to the scattering factors was therefore accounted for using the solvent mask function. The Platon55 and Mercury56 programs were used for crystal structure analysis and preparation of figures. Crystallographic data for 6–12 are collected in Table 1, while crystallographic data for 1·CH3CH2CN–4 are collected in Table S1 (ESI†). All crystal structures were deposited to the CCDC and were assigned deposition numbers 2170736 (1·CH3CH2CN), 2170737 (2·CH3CH2CN), 2170738 (2·2.5PhCN), 2170739 (3), 2170740 (4), 2170741 (6), 2170742 (7), 2170743 (8a), 2170744 (8b), 2170745 (9a), 2170746 (9b), 2170747 (11), 2170748 (12).
Table 1 Crystallographic data for 6–12
|
|
6
|
7
|
8a
|
8b
|
| Empirical formula |
C38H35N5O6Zn |
C37H33N5O6Zn |
C32H28N4O4Zn |
C32H28N4O4Zn |
| Formula weight |
723.08 |
709.05 |
597.95 |
597.95 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
I2/a |
|
T [K] |
150.00(10) |
150.00(10) |
150.00(10) |
150.00(10) |
|
λ [Å] |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
|
a [Å] |
9.7450(2) |
9.6687(3) |
11.9685(3) |
16.8851(9) |
|
b [Å] |
12.8471(3) |
12.7174(4) |
14.7358(4) |
15.2488(6) |
|
c [Å] |
13.6923(2) |
13.5705(3) |
16.8453(3) |
21.6499(9) |
|
α [°] |
86.967(2) |
94.784(2) |
84.163(2) |
90 |
|
β [°] |
88.015(2) |
90.751(2) |
81.961(2) |
95.594(5) |
|
γ [°] |
77.889(2) |
103.734(2) |
69.405(2) |
90 |
|
V [Å3] |
1673.22(6) |
1614.35(8) |
2749.31(12) |
5547.8(4) |
|
Z
|
2 |
2 |
4 |
8 |
|
D
calc [g cm−3] |
1.435 |
1.459 |
1.445 |
1.432 |
|
μ [mm−1] |
0.790 |
0.818 |
0.939 |
0.931 |
| Collected reflections |
49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 630 630 |
29798 |
41024 |
15678 |
| Unique reflections |
9380 |
8862 |
14520 |
7240 |
| Observed reflections |
7758 |
7499 |
11730 |
4692 |
|
R
int
|
0.0349 |
0.0296 |
0.0297 |
0.0387 |
|
R
1 (I > 2σ(I)) |
0.0412 |
0.0406 |
0.0347 |
0.0442 |
| wR2 (all data) |
0.1117 |
0.1062 |
0.0815 |
0.0987 |
|
|
9a
|
9b
|
11
|
12
|
| Empirical formula |
C31H26N4O4Zn |
C31H26N4O4Zn |
C41H33N5O6Zn |
C72H59N9O10Zn2 |
| Formula weight |
583.93 |
583.93 |
757.09 |
1341.02 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P |
P21/c |
C2/c |
P |
|
T [K] |
150.00(10) |
150.00(10) |
150.00(10) |
150.00(10) |
|
λ [Å] |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
|
a [Å] |
10.1047(2) |
16.7335(5) |
40.104(2) |
9.8931(3) |
|
b [Å] |
15.6035(3) |
15.2906(4) |
9.6775(4) |
17.7112(6) |
|
c [Å] |
16.7005(3) |
21.2757(6) |
17.8447(9) |
19.4839(6) |
|
α [°] |
94.937(2) |
90 |
90 |
107.957(3) |
|
β [°] |
90.8110(10) |
95.203(3) |
98.273(5) |
98.357(3) |
|
γ [°] |
90.7760(10) |
90 |
90 |
93.583(3) |
|
V [Å3] |
2622.83(9) |
5421.3(3) |
6853.6(6) |
3192.21(18) |
|
Z
|
4 |
8 |
8 |
2 |
|
D
calc [g cm−3] |
1.479 |
1.431 |
1.467 |
1.395 |
|
μ [mm−1] |
0.982 |
0.950 |
0.776 |
0.820 |
| Collected reflections |
77591 |
32431 |
19516 |
32819 |
| Unique reflections |
14668 |
13877 |
9053 |
16589 |
| Observed reflections |
11968 |
10451 |
4896 |
10407 |
|
R
int
|
0.0338 |
0.0333 |
0.0812 |
0.0379 |
|
R
1 (I > 2σ(I)) |
0.0350 |
0.0370 |
0.0804 |
0.0584 |
| wR2 (all data) |
0.0905 |
0.0956 |
0.1772 |
0.1488 |
3. Results and discussion
3.1. Synthetic considerations
The aim of this study was to investigate generality of a three-component reaction system which consisted of zinc(II) quinaldinate, cyclic amine (piperidine or pyrrolidine) and nitrile in the formation of amidines. Acetonitrile, used in our previous study, was replaced with propionitrile or benzonitrile. When it comes to a direct reaction of nitrile with amine, both can be considered as ordinary unactivated nitriles. The differences in reactivity, if any, would thus reflect the different bulk of their substituents. The reaction systems were tested under mild and harsh reaction conditions. In line with the already recognized beneficial effect of alcohols over the amidine synthesis,14 the addition of methanol was assessed for each system. The products obtained from the four reaction systems are depicted in Schemes 2–5. Details of their preparation are given in the Experimental section. The products were of three types: (i) zinc(II) complexes with intact amines, [Zn(quin)2(amine)] and [Zn(quin)2(amine)2]; (ii) zinc(II) complexes with amidines, [Zn(quin)2(amidine)]; and (iii) amidinium salts of [Zn(quin)3]− ions.
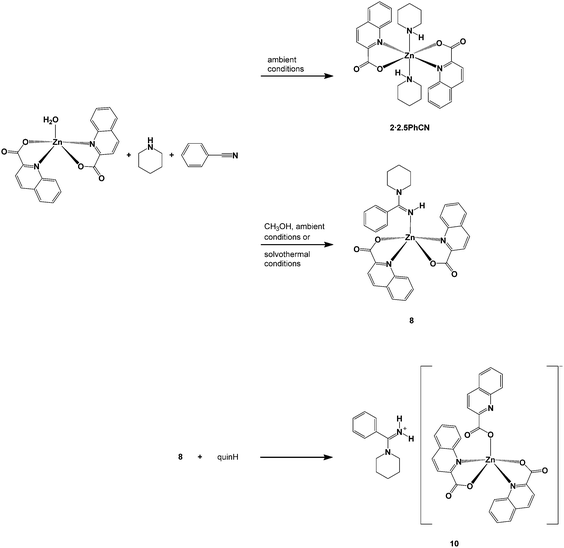 |
| | Scheme 2 Products of benzonitrile/piperidine reactions. | |
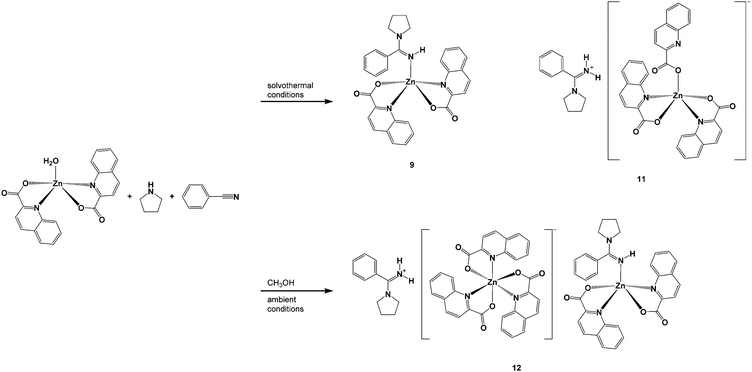 |
| | Scheme 3 Products of benzonitrile/pyrrolidine reactions. | |
 |
| | Scheme 4 Products of propionitrile/piperidine reactions. | |
 |
| | Scheme 5 Products of propionitrile/pyrrolidine reactions. | |
The reactions of both amines with benzonitrile under forcing conditions (i.e., heating for 24 h in an autoclave), produced the anticipated corresponding benzoamidines that were isolated as their zinc(II) complexes, [Zn(quin)2(pipebenzoam)] (8) and [Zn(quin)2(pyrobenzoam)] (9). From the pyrrolidine reaction system, an amidinium salt, pyrobenzoamH[Zn(quin)3] (11), was obtained on a prolonged standing of the reaction mixture. The pipebenzoamH+ analog could only be obtained by direct reaction of [Zn(quin)2(pipebenzoam)] with quinaldinic acid (Scheme 2). Similarly, the reaction of [Zn(quin)2(pyrobenzoam)] with quinaldinic acid serves as a reproducible and rational synthetic pathway for pyrobenzoamH[Zn(quin)3] (11):
| [Zn(quin)2(pyrobenzoam)] (9) + quinH → pyrobenzoamH[Zn(quin)3] (11) |
The presence of methanol was found to be a prerequisite for the formation of amidine complexes at ambient conditions. Interestingly, the combination of pyrrolidine, benzonitrile and methanol at ambient conditions produced pyrobenzoamH[Zn(quin)3]·[Zn(quin)2(pyrobenzoam)] (12).
In the case of propionitrile (Schemes 4 and 5), we were less successful at the isolation of amidine complexes. The piperidinopropioamidine complex, [Zn(quin)2(pipepropioam)] (5), was obtained in one system only; i.e., from the methanol-containing mixture under ambient conditions. Unfortunately, single crystals of 5 could not be obtained. Both amines afforded [Zn(quin)3]− salts with amidinium cations, pipepropioamH[Zn(quin)3] (6) and pyropropioamH[Zn(quin)3] (7). The pipepropioamH+ salt formed only under ambient conditions, whereas the pyropropioamH+ salt also formed under forcing conditions. It is noteworthy that the systems that yielded the amidinium salts 6 and 7 share methanol as a common ingredient. In its absence, complexes with intact amines formed. The monoamine piperidine compound, [Zn(quin)2(pipe)]·CH3CH2CN (1·CH3CH2CN), formed not only under mild reaction conditions, but also under forcing conditions. Similarly, the pyrrolidine system afforded the monoamine compound [Zn(quin)2(pyro)] (3) under reflux conditions.
Final comments pertain to the amidinium salts of the [Zn(quin)3]− ion, whose formation from systems, where no extraneous acid was added, is interesting in its own right. The source of protons has not been identified, although the outcome does meet the expectations. Of the two bases inherent to the system, amidine and quinaldinate, the one with the higher basicity has accepted the proton. Notably, the formation of the [Zn(quin)3]− ion implies the presence of zinc(II) species with less than two quinaldinates per metal. The isolation of both structural isomers of [Zn(quin)3]−, a five- and a six-coordinate structure, shows their incidence as a universal phenomenon. The latter is in accord with our previous results. Namely, the existence of the [Zn(quin)3]− structural isomers, conveniently denoted as 5-[Zn(quin)3]− or 6-[Zn(quin)3]−, was observed before. Density functional theory (DFT) calculations on the isolated isomers have shown 6-[Zn(quin)3]− to be more stable by 5.6 kcal mol−1 than the five-coordinate isomer, and their interconversion in the solution to be almost barrier-free. Weak intermolecular interactions in the solid state have been shown to stabilize both isomers and reduce the energy difference between them.43
3.2. Crystal structures
The piperidinobenzoamidine and pyrrolidinobenzoamidine zinc(II) complexes crystallize in two polymorphic forms; triclinic (labeled a) and monoclinic (labeled b). The crystal structures of [Zn(quin)2(pipebenzoam)] (8a and 8b) and [Zn(quin)2(pyrobenzoam)] (9a and 9b) are similar to each other. In all complex molecules, the zinc(II) ion is surrounded by two N,O-bidentate chelating quinaldinates and a monodentate amidine, bound via imine nitrogen. Oak Ridge Thermal-Ellipsoid Plot Program (ORTEP) drawings of complex molecules are shown in Fig. 1, and their relevant geometric parameters are given in Table 2. The N3O2 donors occupy vertices of a distorted polyhedron which cannot be described as either a square pyramid or a trigonal bipyramid. The corresponding τ values lie in the 0.40–0.58 interval.57 Bond lengths are comparable to those determined for the related piperidinoacetamidine and pyrrolidinoacetamidine zinc(II) complexes.43 The amidine-to-zinc(II) bond is almost 0.2 Å shorter than the bonds between zinc(II) and the quinaldinate nitrogen. Both benzoamidines complexes are E isomers. The explanation for this cannot lie in the steric hindrance, as both benzoamidine parts (the phenyl group and the amine residue) are bulky. It is noteworthy that the proximity and orientation of the benzoamidine phenyl group and the adjacent quinaldinate ligand in the E isomer enable an intramolecular π⋯π stacking interaction between them. A closer look at the structures of 8a/8b and 9a/9b reveals such an interaction in all cases except 9a. The interaction in 8a and a less favorable orientation of the benzoamidine phenyl group with respect to the closest quinaldinate in 9a can also be seen in Fig. 1. The CN imine bond is around 1.30 Å, whereas the C–N amine bonds span a 1.33–1.36 Å interval. Comparison with the bond lengths in non-coordinated amidines (1.28 and 1.38 Å,58 respectively) suggests a more delocalized nature in our compounds.
 |
| | Fig. 1 ORTEP diagrams of the [Zn(quin)2(pipebenzoam)] (8a) and [Zn(quin)2(pyrobenzoam)] (9a) complex molecules. The displacement ellipsoids are shown at the 50% probability level. | |
Table 2 Relevant geometric parameters [Å] of novel zinc(II) complexes with amidines
| Compound |
Amidine |
Zn–N (amidine) |
Zn–N (quin−) |
Zn–O (quin−) |
τ
57
|
|
Pertains to [Zn(quin)2(pyrobenzoam)].
|
|
8a
|
pipebenzoam |
2.0097(15) |
2.1732(14), 2.2183(14) |
1.9913(12), 1.9983(11) |
0.57 |
| 2.0117(14) |
2.1668(13), 2.1809(14) |
2.0054(13), 2.0129(12) |
0.57 |
|
8b
|
pipebenzoam |
2.001(2) |
2.1678(17), 2.1902(17) |
1.9934(16), 1.9955(16) |
0.57 |
|
9a
|
pyrobenzoam |
2.0138(14) |
2.1943(14), 2.1997(13) |
1.9866(11), 2.0116(11) |
0.40 |
| 1.9962(15) |
2.1751(14), 2.1836(13) |
2.0035(12), 2.0128(12) |
0.47 |
|
9b
|
pyrobenzoam |
2.0085(16) |
2.1703(15), 2.1801(15) |
1.9995(14), 2.0003(14) |
0.57 |
| 2.0052(16) |
2.1723(15), 2.1749(14) |
1.9982(14), 2.0053(13) |
0.58 |
|
12
|
pyrobenzoam |
1.991(3) |
2.164(3), 2.184(2) |
2.015(2), 2.029(2) |
0.44 |
The same type of hydrogen bonds (i.e., NH(amidine)⋯COO− interactions) may be observed in all cases. In the 8a/8b polymorphs, these hydrogen bonds link complex molecules into supramolecular chains. The pair differs only minutely in their intermolecular connectivity, and their densities are consequently almost the same. Although the densities of the 9a/9b polymorphs are also very similar, their differences are easier to perceive. 9b features the same supramolecular structure as the polymorphs 8a/8b, whereby the NH⋯COO− hydrogen bonds link [Zn(quin)2(pyrobenzoam)] molecules into infinite chains. In 9a, somewhat longer interactions link complex molecules into pairs (Fig. 2).
 |
| | Fig. 2 Connectivity patterns in the [Zn(quin)2(pyrobenzoam)] polymorphs: a pair of complex molecules in 9a (top) and a section of an infinite chain in 9b (bottom). | |
The solid-state structures of pipepropioamH[Zn(quin)3] (6), pyropropioamH[Zn(quin)3] (7) and pyrobenzoamH[Zn(quin)3] (11) are very similar and all consist of amidinium cations and the [Zn(quin)3]− complex anions. ORTEP diagrams of amidinium ions are shown in Fig. 3, with relevant geometric parameters given in Table 3. The difference between the CN imine and the C–N amine bonds can be used for assessing the extent of delocalization within the –NC–N– fragment.59 These differences in 6, 7 and 11 closely approach zero and thereby suggest fully delocalized systems. Their [Zn(quin)3]− counter-anions share a common structural feature in that the zinc(II) ion is penta-coordinated by the N2O3 donors of three quinaldinate ligands. Two quinaldinates are bound in the usual N,O-bidentate chelating manner, while a third is monodentate, bound via only one carboxylate oxygen atom (Fig. 4). Although the other carboxylate oxygen of this quinaldinate is in a favorable orientation for a bonding interaction, its distance to zinc(II) is too long, at over 2.8 Å. The τ values for the N2O3 coordination polyhedron are around 0.4 (Table 4).57
 |
| | Fig. 3 ORTEP diagrams of amidinium cations: pipepropioamH+ of 6, pyropropioamH+ of 7 and pyrobenzoamH+ of 11. The displacement ellipsoids are shown at the 50% probability level. | |
Table 3 Relevant geometric parameters [Å] of amidinium cations
| Compound |
C–NH2+ |
C–N(amine) |
C–C |
|
6
|
1.311(3) |
1.323(3) |
1.503(3) |
|
7
|
1.312(3) |
1.314(3) |
1.498(3) |
|
11
|
1.306(6) |
1.306(6) |
1.489(6) |
|
12
|
1.320(4) |
1.306(4) |
1.488(4) |
 |
| | Fig. 4 ORTEP diagram of the [Zn(quin)3]− ion in 7. The displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
Table 4 Relevant geometric parameters [Å] of [Zn(quin)3]− ionsa
| Compound |
Zn–O (quin−)b |
Zn–N (quin−) |
Zn–O (quin−) |
Zn⋯O |
τ
57
|
|
In 6, 7 and 11, the complex ions are five-coordinate. In 12, the complex ions are six-coordinate.
Monodentate quinaldinate.
|
|
6
|
1.9834(14) |
2.1618(16), 2.1680(16) |
2.0144(14), 2.0179(14) |
2.8812(1) |
0.42 |
|
7
|
1.9699(14) |
2.1592(16), 2.1727(16) |
2.0059(14), 2.0116(13) |
2.8387(1) |
0.39 |
|
11
|
1.983(3) |
2.132(4), 2.167(4) |
2.013(3), 2.025(3) |
2.9428(1) |
0.43 |
|
12
|
— |
2.211(3)–2.455(3) |
2.002(3)–2.055(2) |
— |
— |
The salts 6 and 7 share the same pattern of intermolecular connectivity which involves the nitrogen atom of the monodentate quinaldinate. In both, the NH2+ group of amidinium cations forms two hydrogen bonding interactions, one to the carboxylate oxygen and another to the quinaldinate nitrogen (Fig. 5). As a result, infinite supramolecular chains of alternating amidinium cations and [Zn(quin)3]− complex anions are formed. The chains are packed in a parallel fashion so that each is surrounded by six others. Among adjacent chains, π⋯π stacking interactions may be observed. Although pyrobenzoamH[Zn(quin)3] (11) features a markedly longer NH2+⋯N(quin−) contact (longer than the sum of the van der Waals radii),60 the connectivity motif is the same as in the other two salts.
 |
| | Fig. 5 Section of a supramolecular chain in pyropropioamH[Zn(quin)3] (7): the NH2+⋯COO− and the NH2+⋯N(quin−) contacts are 2.900(2) and 2.991(2) Å, respectively. | |
The solid-state structure of pyrobenzoamH[Zn(quin)3]·[Zn(quin)2(pyrobenzoam)] (12) consists of [Zn(quin)2(pyrobenzoam)] amidine complex molecules, [Zn(quin)3]− complex anions and pyrobenzoamH+ counter-cations in a 1:1:1 molar ratio. Because of the separate existence of both the neutral amidine complex (polymorphs 9a/9b) and the ionic part (salt 11), pyrobenzoamH[Zn(quin)3]·[Zn(quin)2(pyrobenzoam)] is referred to as a co-crystal. The geometry of the [Zn(quin)2(pyrobenzoam)] complex molecules does not differ significantly from those in the polymorphs 9a/9b. The same observation pertains to the pyrobenzoamH+ cations in relation to those in the structure of 11. A major difference may be observed in the geometry of the [Zn(quin)3]− complex anions, which are not the same as those in the structure of 11. The anions of 12 feature a six-coordinate zinc(II) surrounded by the N3O3 donors (Fig. 6). The three quinaldinate ligands are all bound in the usual N,O-bidentate chelating fashion. On the whole, the quinaldinate-to-zinc(II) bonds in the six-coordinate [Zn(quin)3]− ions are longer than those in the five-coordinate ions. The longest distance exceeds 2.4 Å and occurs between zinc(II) and quinaldinate nitrogen that is trans to carboxylate. An inspection of the connectivity patterns in 12 reveals that the amidine complex molecules are linked via NH⋯COO− hydrogen bonds into dimers, whereas the pyrobenzoamH+/[Zn(quin)3]− ions are linked via NH2+⋯COO− hydrogen bonds into infinite chains. The chains pack to form layers. The amidine complex molecules are accommodated in between these layers (Fig. S6 and S7, ESI†).
 |
| | Fig. 6 ORTEP diagram of the [Zn(quin)3]− ion in 12. The displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
3.3. IR spectroscopy
The spectra of all new compounds, as provided in the ESI,† are dominated by various quinaldinate vibrations. Among these, the νas(COO−) and νs(COO−) absorptions are the most intense bands. In the complexes with amidines, the νas(COO−) band may be found at ca. 1640 cm−1, whereas the νs(COO−) vibration usually appears as two distinct bands at 1360–1340 cm−1.
The presence of amidine in the compound may be seen by the characteristic bands in several spectral regions.61 It is to be noted that the two constituent parts of amidine, the amine and the nitrile residues, both leave a characteristic imprint over the spectrum. The amine part may be seen as the ν(C–H) vibrations of the methylene groups. In the case of benzoamidines, the nitrile residue, the phenyl moiety, adds significantly to the complexity of the spectra. Therefore, only selected bands were assigned (Table 5). A medium intense band at the highest frequency, at ca. 3200 cm−1, may be associated with the ν(N–H) vibration. An intense absorption at ca. 1550 cm−1 may be ascribed to the ν(CN) vibration. Similarly, the IR spectra of related quinaldinate zinc(II) complexes with piperidinoacetamidine and pyrrolidinoacetamidine displayed bands in the 1591–1562 cm−1 spectral region. The ν(CN) frequencies in our compounds are in agreement with the literature values of ca. 1550 cm−1.41,62
Table 5 Amidine signature bands [cm−1] in the spectra of novel zinc(II) complexes with amidinesa
| Compound |
ν(N–H) |
ν(CN) |
|
m = medium and s = strong.
|
| [Zn(quin)2(pipepropioam)] (5) |
3288 m |
1582 s |
| [Zn(quin)2(pipebenzoam)] (8) |
3205 m |
1550 s |
| [Zn(quin)2(pyrobenzoam)] (9) |
3212 m |
1553 s |
On amidine protonation, the spectra undergo dramatic changes. With no exceptions, the spectra of all amidinium salts feature a broad band at 3000 cm−1 that masks the ν(C–H) vibrations of methylene groups in amidinium cations. Additionally, several strong bands may be observed in the 1660–1560 cm−1 spectral region making an unambiguous assignation of the νas(COO−), ν(CN) and NH2+ deformation vibrations very difficult. In the spectra of the related [Zn(quin)3]− ionic compounds with protonated piperidinoacetamidine and pyrrolidinoacetamidine, a new band (as compared to parent amidine complexes) was observed at 1658 cm−1 and ascribed to the interaction between the ν(CN) and NH2+ deformation vibrations.43
The IR spectrum of co-crystal 12 combines features of both the coordinated amidine and of the amidinium cation. Its most relevant features are a weak absorption at 3229 cm−1, which may be associated with the ν(N–H) vibration of amidine, a very broad band at ca. 3000 cm−1, typical for amidinium salts, and several bands in the 1623–1558 cm−1 region.
3.4.
1H NMR spectroscopy
The 1H NMR spectra of the dimethyl sulfoxide (DMSO)-d6 solutions of the analyzed compounds confirm their compositions. The chemical shifts of quinaldinate depend upon the compound type. For complexes with amines, the signals appear within the 7.81–8.92 ppm interval, while the quinaldinate signals in complexes with amidines (8 and 9) are shifted slightly upfield; i.e., 7.80–8.71 ppm. A more pronounced shift upfield may be observed for the ionic compounds with the [Zn(quin)3]− ions; i.e., 7.65–8.59 ppm.
On the amidine formation and its coordination to zinc(II), signals of both parts comprising the amidine experience changes in chemical shifts as compared with the “free” components. The shapes of the signals change as well. In the spectra of the benzoamidine complexes, 8 and 9, the signals of the phenyl residue may be seen as one or two broad multiplets. The piperidine moiety of pipebenzoam in 8 has three signals, at 1.36, 1.50 and 3.16 ppm in a 4:2:4 integral ratio. On the other hand, the piperidine ligand of [Zn(quin)2(pipe)2]·2.5PhCN (2·2.5PhCN) is characterized by signals at 1.36, 1.43 and 2.62 ppm. As the comparison shows, the signal of the methylene group adjacent to the ring nitrogen in pipebenzoam is shifted downfield. The same observation pertains to the pyrrolidine signals in the pyrobenzoam complex 9 as compared with the amine complex 3. Contrary to expectations, a signal from the azomethine hydrogen was not seen in the spectra of amidine complexes 8 and 9. The spectra of zinc(II) complexes with piperidinoacetamidine or pyrrolidinoacetamidine displayed the corresponding resonance at 6.98 ppm.43
On protonation, the signals of the amine residue in the amidinium cations of the [Zn(quin)3]− salts (i.e., salts 6, 7, 10 and 11) move even further downfield, as exemplified by the piperidine signals at 1.64 and 3.48 ppm in the spectrum of the pipebenzoamH+ salt 10. Of the four salts with amidinium cations, the pipebenzoamH+ (compound 10) and pyrobenzoamH+ (11) ions displayed resonance at 9.23 or 9.02 ppm that belonged to the NH2+ protons, whereas no signals could be discerned in the spectra of the pipepropioamH+ and pyropropioamH+ salts 6 and 7, respectively.
4. Concluding remarks
In summary, our study clearly confirms amidine formation in the reaction systems of both propionitrile and benzonitrile. This finding is consistent with their similar reactivity. The propionitrile reactions favor amidinium salts as products, whereas the benzonitrile reactions may be characterized by the ready formation of amidine complexes. The benzoamidine ligands are bulkier, yet a π⋯π stacking interaction between the benzoamidine phenyl moiety and quinaldinate imparts additional stability to the [Zn(quin)2(benzoamidine)] complex molecule. The general validity of the above argument is challenged by pyrrolidinobenzoamidine which exists not only as a ligand in [Zn(quin)2(pyrobenzoam)] (9) or in a protonated state as a counter-cation in pyrobenzoamH[Zn(quin)3] (11), but as both in the co-crystal pyrobenzoamH[Zn(quin)3]·[Zn(quin)2(pyrobenzoam)] (12). This study again confirms the beneficial influence of methanol in amidine formation. The exact role of the zinc(II) species in the reaction of nitrile with amine remains a subject for future studies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the Junior Researcher Grant for N. P. and the Program Grant P1-0134 of the Slovenian Research Agency.
References
- IUPAC, compiled by: A. D. McNaught and A. Wilkinson, Compendium of Chemical Terminology: IUPAC Recommendations, Blackwell Scientific Publications; Oxford, UK, 2nd edn, 1997 Search PubMed.
- J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219–300 CrossRef CAS.
-
J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, Oxford, UK, 2nd edn, 2012 Search PubMed.
- M. Baidya and H. Mayr, Chem. Commun., 2008, 1792–1794 RSC.
- M. P. Coles, Dalton Trans., 2006, 985–1001 RSC.
- D. Cornacchia, R. Z. Pellicani, F. P. Intini, C. Pacifico and G. Natile, Inorg. Chem., 2009, 48, 10800–10810 CrossRef CAS PubMed.
- V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef CAS PubMed.
- U. Belluco, F. Benetollo, R. Bertani, G. Bombieri, R. A. Michelin, M. Mozzon, A. J. L. Pombeiro and F. C. Guedes da Silva, Inorg. Chim. Acta, 2002, 330, 229–239 CrossRef CAS.
- P. Abhayawardhana, P. A. Marzilli, T. Perera, F. R. Fronczek and L. G. Marzilli, Inorg. Chem., 2012, 51, 7271–7283 CrossRef CAS PubMed.
- R. L. Shriner and F. W. Neumann, Chem. Rev., 1944, 35, 351–425 CrossRef CAS.
- M. Anbazhagan, D. W. Boykin and C. E. Stephens, Tetrahedron Lett., 2002, 43, 9089–9092 CrossRef CAS.
- W.-X. Zhang, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2005, 127, 16788–16789 CrossRef CAS PubMed.
- A. A. Aly, S. Bräse and M. A.-M. Gomaa, ARKIVOC, 2018, 6, 85–138 Search PubMed.
- G. Rousselet, P. Capdevielle and M. Maumy, Tetrahedron Lett., 1993, 34, 6395–6398 CrossRef CAS.
- J. C. Grivas and A. Taurins, Can. J. Chem., 1961, 39, 761–764 CrossRef CAS.
- P. Oxley, M. W. Partridge and W. F. Short, J. Chem. Soc., 1947, 1110–1116 RSC.
- J. D. Bower and G. R. Ramage, J. Chem. Soc., 1957, 4506–4510 RSC.
-
Z. Wang, Comprehensive Organic Name Reactions and Reagents, Wiley, Hoboken, NJ, 2010 Search PubMed.
- J. H. Forsberg, V. T. Spaziano, T. M. Balasubramanian, G. K. Liu, S. A. Kinsley, C. A. Duckworth, J. J. Poteruca, P. S. Brown and J. L. Miller, J. Org. Chem., 1987, 52, 1017–1021 CrossRef CAS.
- J. Wang, F. Xu, T. Cai and Q. Shen, Org. Lett., 2008, 10, 445–448 CrossRef CAS PubMed.
- S. D. Veer, K. V. Katkar and K. G. Akamanchi, Tetrahedron Lett., 2016, 57, 4039–4043 CrossRef CAS.
- A. K. Jain, M. R. Gau, P. J. Carroll and K. I. Goldberg, Organometallics, 2022 DOI:10.1021/acs.organomet.2c00302.
-
S. Roy, S. Bhattacharya and S. Chattopadhyay, J. Coord. Chem., 2016, 69, 112–122 Search PubMed.
- M. A. Galindo, D. Amantia, A. Martinez-Martinez, W. Clegg, R. W. Harrington, V. Moreno Martinez and A. Houlton, Inorg. Chem., 2009, 48, 11085–11091 CrossRef CAS PubMed.
- N. Antón, M. Arroyo, P. Gómez-Iglesias, D. Miguel and F. Villafañe, J. Organomet. Chem., 2008, 693, 3074–3080 CrossRef.
- M. Arroyo, D. Miguel, F. Villafañe, S. Nieto, J. Pérez and L. Riera, Inorg. Chem., 2006, 45, 7018–7026 CrossRef CAS PubMed.
- E. Reisner, V. B. Arion, A. Rufińska, I. Chiorescu, W. F. Schmid and B. K. Keppler, Dalton Trans., 2005, 2355–2364 RSC.
- B. Longato, G. Bandoli, A. Mucci and L. Schenetti, Eur. J. Inorg. Chem., 2001, 3021–3029 CrossRef CAS.
- C. S. Chin, D. Chong, B. Lee, H. Jeong, G. Won, Y. Do and Y. J. Park, Organometallics, 2000, 19, 638–648 CrossRef CAS.
- R. A. Michelin, M. Mozzon and R. Bertani, Coord. Chem. Rev., 1996, 147, 299–338 CrossRef CAS.
-
A. J. L. Pombeiro and V. Y. Kukushkin, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, UK, 2004, vol. 1 Search PubMed.
- S. M. Sbovata, F. Bettio, C. Marzano, M. Mozzon, R. Bertani, F. Benetollo and R. A. Michelin, Inorg. Chim. Acta, 2008, 361, 3109–3116 CrossRef CAS.
- M. R. Tyan, N. A. Bokach, M.-J. Wang, M. Haukka, M. L. Kuznetsov and V. Y. Kukushkin, Dalton Trans., 2008, 5178–5188 RSC.
- C. Marzano, S. M. Sbovata, V. Gandin, R. A. Michelin, A. Venzo, R. Bertani and R. Seraglia, J. Inorg. Biochem., 2009, 103, 1113–1119 CrossRef CAS PubMed.
- T. Perera, F. R. Fronczek, P. A. Marzilli and L. G. Marzilli, Inorg. Chem., 2010, 49, 7035–7045 CrossRef CAS PubMed.
- C. Marzano, S. Mazzega Sbovata, V. Gandin, D. Colavito, E. Del Giudice, R. A. Michelin, A. Venzo, R. Seraglia, F. Benetollo, M. Schiavon and R. Bertani, J. Med. Chem., 2010, 53, 6210–6227 CrossRef CAS PubMed.
- S. M. Sbovata, R. A. Michelin, M. Mozzon, A. Venzo, R. Bertani and F. Benetollo, Inorg. Chim. Acta, 2010, 363, 487–494 CrossRef CAS.
- A. N. Chernyshev, N. A. Bokach, P. V. Gushchin, M. Haukka and V. Y. Kukushkin, Dalton Trans., 2012, 41, 12857–12864 RSC.
- N. A. Bokach, N. P. Konovalova, Y. Wang, Y. E. Moskalenko, A. V. Gribanov and V. Y. Kukushkin, Dalton Trans., 2010, 39, 4619–4623 RSC.
- T. Perera, P. A. Marzilli, F. R. Fronczek and L. G. Marzilli, Inorg. Chem., 2010, 49, 2123–2131 CrossRef CAS PubMed.
- S. Rajak, K. Chair, L. K. Rana, P. Kaur, T. Maris and A. Duong, Inorg. Chem., 2020, 59, 14910–14919 CrossRef CAS PubMed.
- C. Marzano, S. M. Sbovata, F. Bettio, R. A. Michelin, R. Seraglia, T. Kiss, A. Venzo and R. Bertani, JBIC, J. Biol. Inorg. Chem., 2007, 12, 477–493 CrossRef CAS PubMed.
- N. Podjed, B. Modec, M. M. Alcaide and J. López-Serrano, RSC Adv., 2020, 10, 18200–18221 RSC.
- S. Enthaler, ACS Catal., 2013, 3, 150–158 CrossRef CAS.
- R. M. Oballa, J.-F. Truchon, C. I. Bayly, N. Chauret, S. Day, S. Crane and C. Berthelette, Bioorg. Med. Chem. Lett., 2007, 17, 998–1002 CrossRef CAS PubMed.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
- N. Podjed, P. Stare, R. Cerc Korošec, M. M. Alcaide, J. López-Serrano and B. Modec, New J. Chem., 2020, 44, 387–400 RSC.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- M. R. Willcott, J. Am. Chem. Soc., 2009, 131, 13180 CrossRef CAS.
- SDBSWeb (National Institute of Advanced Industrial Science and Technology), https://sdbs.db.aist.go.jp, (accessed 25 October 2022).
- Agilent ( 2014). CrysAlis PRO, Agilent Technologies Ltd; Yarnton, Oxfordshire, England Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
-
G. Häfelinger and F. K. H. Kuske, in The Chemistry of Amidines and Imidates, ed. S. Patai and Z. Rappoport, Wiley, Chichester, UK, 1991, vol. 2 Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
-
W. H. Prichard, in The Chemistry of Amidines and Imidates, ed. S. Patai, Wiley, London, 1975, vol. 1 Search PubMed.
- L. E. Clougherty, J. A. Sousa and G. M. Wyman, J. Org. Chem., 1957, 22, 462 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for 1·CH3CH2CN–4 (Table S1), ORTEP drawings of 3 and 4 (Fig. S1 and S3), geometric parameters of 3 and 4 (Tables S2 and S3), intermolecular interactions (Fig. S2, S4–S7), hydrogen bonds (Table S4), IR assignments for 1·CH3CH2CN–4 (Table S5), IR spectra of 1·CH3CH2CN to 12 (Fig. S8–S19); NMR spectra of 1·CH3CH2CN to 11 (Fig. S20–S34). CCDC 2170736–2170748. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2nj04668g |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Barbara
Modec
and
Barbara
Modec










