
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards an understanding of the biological activity of naphthylisoquinoline alkaloids: DNA-binding properties of dioncophyllines A, B, and C†
Denisa
Soost
 a,
Gerhard
Bringmann
b and
Heiko
Ihmels
*a
a,
Gerhard
Bringmann
b and
Heiko
Ihmels
*a
aDepartment of Chemistry – Biology, University of Siegen, Center of Micro- and Nanochemistry and (Bio-)Technology (Cμ), Adolf-Reichwein-Str. 2, 57068 Siegen, Germany. E-mail: ihmels@chemie.uni-siegen.de
bInstitute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: bringman@chemie.uni-würzburg.de
First published on 10th October 2022
Abstract
To gain further insight into the mechanism of the promising cytotoxic and antitumoral actvities of naphthylisoquinoline (NIQ) alkaloids, the DNA-binding properties of three representative NIQ alkaloids, dioncophyllines A (1), B (2), and C (3), were, for the first time, investigated by means of spectroscopic studies. Thus, photometric and fluorimetric titrations as well as CD- and LD-spectroscopic analyses revealed an association of dioncophyllines A (1) and B (2) with duplex DNA and with abasic site-containing DNA (apurinic/apyrimidinic DNA:AP-DNA) with moderate affinity (Kb = 1–4 × 104 M−1). Notably, exemplary studies with dioncophylline A (1) also provided first evidence of a selectivity of this ligand towards AT-rich DNA sequences and abasic sites. Presumably, NIQs 1 and 2 bind to regular DNA in a half-intercalation mode, whereas they insert into the abasic site in AP-DNA. In contrast, dioncophylline C (3) does not bind to DNA, presumably because the 5,1′-connection of the biaryl unit in this NIQ leads to a sterically demanding structure that does not fit well into the DNA-binding sites. In summary, the results show that the DNA-binding properties of NIQ alkaloids should be considered in the assessment of their biological activities.
Introduction
Natural products constitute a huge, inexhaustible source of compounds with in part unique structures and promising bioactivities, making them candidates for drug discovery and development.1–7 In this context, the naphthylisoquinoline (NIQ) alkaloids8–11 figure as a unique class of natural products with regard to their structures, their biosynthetic origin, and their biological activities. They are obtained from two plant families exclusively, Ancistrocladaceae and Dioncophyllaceae, which are native to the tropical rainforests of Africa and Asia.8–12 These lianas are used in traditional medicine, for example for the treatment of malaria.13 Depending on their individual structure, several NIQs show promising activities against various pathogens causing tropical infectious diseases.10 For example, dioncophylline A (1) (Fig. 1) has pronounced activity against liver forms of the rodent malaria parasite Plasmodium berghei, while dioncophylline B (2)14 and dioncophylline C (3) (Fig. 1) show strong in vitro and in vivo activity against Plasmodium falciparum15,16 and P. berghei.17 Moreover, a high anti-babesial potential of dioncophylline A (1) and C (3) against Babesia canis has been demonstrated.18 To add to that, the dimeric derivative michellamine B is highly active against HIV,19 and the likewise dimeric jozimine A2 exhibits strong antiplasmodial activity.20 Along with these remarkable biological activities, it has been discovered that apparently NIQ-containing extracts of some Ancistrocladus species exhibit promising anticancer activities,21–25 which has recently prompted an investigation of the antitumoral activity of selected NIQs.18,26–30 It was demonstrated that some of these NIQs have significant cytotoxic effects against selected cancer cell lines.28,29,31 Thus, dioncophylline A (1) and dioncophylline C (3) are highly active against the breast cancer cell lines MCF-7 (1: IC50 = 1.6 μM, 3: IC50 = 2.1 μM) and MDA-MB-231 (1: IC50 = 0.9 μM, 3: IC50 = 1.8 μM),26 and dioncophylline A (1) has also a high and selective antitumor activity against multiple myeloma cells (INA-6) and leukemia cell lines.27 Furthermore, some NIQs exert a strong and selective cytotoxicity against human pancreatic cancer cells (PANC-1).29,31Overall, the recent studies on the pronounced bioactivities of NIQ derivatives point to a high potential of this class of natural products to provide lead structures of antitumor agents. For the further development and application of NIQ-based antitumoral substrates, however, the mode of action of the bioactive NIQs has to be assessed, because the knowledge of the mechanism of tumor growth inhibition may enable target-oriented variation and adjustment of the NIQ scaffold. In this context, it may be of importance that many chemotherapeutic drugs operate as DNA binders in tumor cells,32–34 and thus affect the biological activity of the DNA.35–37 For example, enzyme inhibition may be caused by occupation of the enzyme-binding sites with the drug or by significant changes of the DNA structure upon association of a ligand. In fact, some NIQ derivatives exhibit characteristic features of DNA-binding ligands, specifically an extended aromatic π system and amino and hydroxy functionalities that may establish attractive interactions in the DNA-binding sites.32–36 Moreover, these compounds possess a biaryl structure that may have the propensity to adapt to the helical structure of the nucleic acid and that has been shown also to cause preferential binding to abasic site-containing DNA, that is, DNA with an apyrimidinic or apurinic site, commonly referred to as AP-DNA.38,39 Surprisingly, the DNA-binding properties of NIQs have not been studied so far. In order to fill this knowledge gap and to clarify whether the cytotoxic activity of NIQs may involve DNA-binding processes, we investigated – in a case study – the interactions of the NIQ alkaloids dioncophyllines A (1), B (2), and C (3) with duplex DNA and AP-DNA (Fig. 1).
 | ||
| Fig. 1 Structures of the naturally occurring naphthylisoquinoline alkaloids dioncophylline A (1), dioncophylline B (2), and dioncophylline C (3). | ||
Results and discussion
Association with regular duplex DNA
Firstly, the interactions of the NIQ alkaloids 1, 2, and 3 with calf thymus (ct) DNA, as a representative duplex DNA form, were investigated by photometric and fluorimetric titrations in aqueous buffer solution (Fig. 2). Unfortunately, the absorption maxima of the NIQ derivatives 1–3 at λ < 300 nm overlapped with that of the DNA, so that only the changes of the weaker long-wavelength absorption bands of 1–3 at λ > 300 nm could be appropriately assessed during the titration. Notably, from the three NIQ derivatives under investigation, dioncophylline C (3) did not show significant interactions with the DNA, as clearly indicated by an essentially constant absorption of the alkaloid in the presence of increasing amounts of ct DNA (Fig. 2C1). The photometric DNA titrations of NIQs 1 and 2, on the contrary, revealed significant changes of the ligand absorption. Thus, the addition of ct DNA to 1 induced a continuous decrease of the absorption, with only a slight shift of the absorption maxima at 318 nm or 333 nm, along with the formation of a broad red-shifted band (Fig. 2A1). The maximum of the latter could not be determined because of overlapping bands. Overall, these developments of the spectrum on DNA addition, that is, hypochromicity and red shift of the absorption band, are characteristic features of DNA-binding ligands.40 Furthermore, an isosbestic point was formed at 335–336 nm during the titration (Fig. 2A1, red arrow), which implies that compound 1 interacts with the DNA in only one major binding mode. In the case of dioncophylline B (2), the addition of ct DNA also induced a continuous decrease of the absorption and a small red shift (Δλ = 3 nm) of the two absorption maxima at 337 nm and 323 nm (Fig. 2B1). Notably, no isosbestic points were formed during the titration, suggesting that ligand 2 interacts with ct DNA in more than one dominant binding mode. | ||
| Fig. 2 Photometric (1) and fluorimetric (2) titration of 1 (A), 2 (B), and 3 (C) with ct DNA (c1 = 40 μM; c2 = 40 μM, c3 = 40 μM) in BPE buffer (cNa+ = 16 mM, pH = 7.0). The black arrows indicate the development of the absorption or emission bands during the titration, the red arrow indicates an isosbestic point. Inset (1): Plot of the absorption at λ = 333 nm or λ = 337 nm versus cDNA. Inset (2): Plot of the emission at λ = 355 nm or λ = 365 nm versus ligand–DNA ratio LDR. | ||
The resulting binding isotherms obtained from the photometric DNA titrations of NIQs 1 and 2 were employed to determine the binding constants Kb.41 This analysis revealed moderate binding affinities of Kb = 2.1 × 104 M−1 (1) and Kb = 2.5 × 104 M−1 (2) (Table 1 and cf. ESI,† Fig. S3), which are comparable to those reported for well-known DNA-binding alkaloids.42
| Ligand | K ct/104 M−1 | %Hb | K AT-DNA/104 M−1 | K TX /104 M−1 | K CX /104 M−1 |
|---|---|---|---|---|---|
| a Binding constant (in nucleic bases) determined from fitting the Scatchard plots from photometric titrations to the theoretical model (ref. 41). b Percent hypochromicity, calculated from the absorbance at the long-wavelength maxima. c Not determined. d No interaction with the corresponding DNA. | |||||
| 1 | 2.1 | 12 | 0.9 | 4.2 | 1.9 |
| 2 | 2.5 | 40 | n.d.c | 4.4 | n.d.c |
| 3 | 3 | n.d.c | n.d.c | n.d.c | |
The binding properties of dioncophylline A (1) with the synthetic polynucleotides (poly[dA-dT]–poly[dA-dT]) and (poly[dG-dC]–poly[dG-dC]) were also examined exemplarily. Hence, the absorption of compound 1 changed during the addition of poly(dA-dT)–poly(dA-dT), showing essentially the same spectroscopic features as the photometric titration with ct DNA (cf. ESI,† Fig. S1A1). The analysis of the resulting binding isotherm revealed a binding constant of Kb = 8.7 × 103 M−1 (Table 1). The addition of poly(dG-dC)–poly(dG-dC) to NIQ 1, by contrast, did not lead to a change of the absorption spectrum (cf. ESI,† Fig. S1A2), which indicated that NIQ 1 has no significant interactions with GC-rich DNA sequences.
In aqueous solution, compounds 1, 2, and 3 showed a weak emission (Φfl = 0.15) with band maxima at 355 nm (1), 364 nm (2) and 357 nm (3). For compounds 1 and 2, the fluorescence intensity of the emission bands decreased steadily upon addition of ct DNA, without a significant shift of the emission maximum (Fig. 2A2 and B2). This observation confirms the association of the ligands 1 and 2 with ct DNA because the fluorescence quenching is a commonly observed effect of DNA-bound ligands as it is caused by a photoinduced electron transfer (PET) from the excited ligand to the DNA base.40 In contrast, the addition of ct DNA to NIQ 3 did not lead to a change of the emission spectrum (Fig. 2C2), which was consistent with the results of the photometric titration and also showed the lack of interaction between this NIQ and DNA. In the case of ligand 1, fluorescence quenching was also observed upon addition of poly(dA-dT)–poly(dAdT), whereas no significant changes occurred in the presence of (poly[dG-dC]–poly[dG-dC]) (cf. ESI,† Fig. S1A1 and A2), which was in agreement with the results of the photometric titrations with these polynucleotides (see above). Furthermore, this observation excluded a close association between 1 and (poly[dG-dC]–poly[dG-dC]), even in a loose backbone association, because guanine has the highest propensity of the DNA bases to be oxidized in a PET process and thus operates as an efficient fluorescence quencher.
In order to assess the binding mode of the NIQs 1 and 2 with DNA in more detail, circular dichroism (CD) and linear dichroism (LD) studies of the DNA–ligand complexes were performed (Fig. 3).43,44 Since 1 and 2 are chiral compounds, they already display intrinsic CD spectra in phosphate buffer (Fig. 3B1 and B2). Unfortunately, their short-wavelength CD absorption overlaps strongly with the one of DNA. Hence, the CD spectrum of 1 showed a negative CD signal at 300 nm and two positive ones at 280 nm and 230 nm, whereas compound 2 exhibited a positive CD signal at 280 nm and a negative one at 244 nm.45,46 The characteristic CD signals of B-DNA with maxima at 280 nm (−) and 250 nm (+) were observed,43 while in the presence of the ligands 1 and 2 the shifts and intensities of the bands in this absorption region changed significantly. However, because of the strong overlap of the CD signals of DNA with the ones of the chiral ligands at wavelength < 300 nm, these changes cannot be conclusively interpreted or assigned to a particular CD-active entity. Therefore, only the CD signals at wavelength > 300 nm, where solely the ligands absorb, were used for the analysis of the ligand–DNA complexes. Thus, with increasing concentration of ligand 1, a negative CD signal occurred at about 300 nm, along with an increase of intensity of both the positive and negative DNA signals (Fig. 3B1). In the presence of DNA, the NIQ derivative 2 showed only a very weak positive CD signal in the absorption range of the ligand (300–350 nm), whereas the intensity of the positive CD signal in the DNA absorption range increased and the negative one increased with increasing ligand–DNA ratio (LDR). Most notably, the comparison of the CD spectra of the free and the DNA-bound ligand 1 in a difference spectrum (cf. ESI,† Fig. S4) clearly revealed that even in the presence of DNA, only the intrinsic CD signal of 1 was detected. This means that even if an induced CD (ICD) developed because of the electronic coupling between transition dipole moments of the ligand and the DNA bases, it is superposed by the stronger intrinsic CD band of the ligand. At the same time, the maintenance of the CD pattern of the bound ligand indicated that the latter did not change its biaryl geometry, specifically the biaryl torsion angle, upon association with DNA, because it has been shown that the CD signature of this compound changes substantially with varying torsion angle.45,47
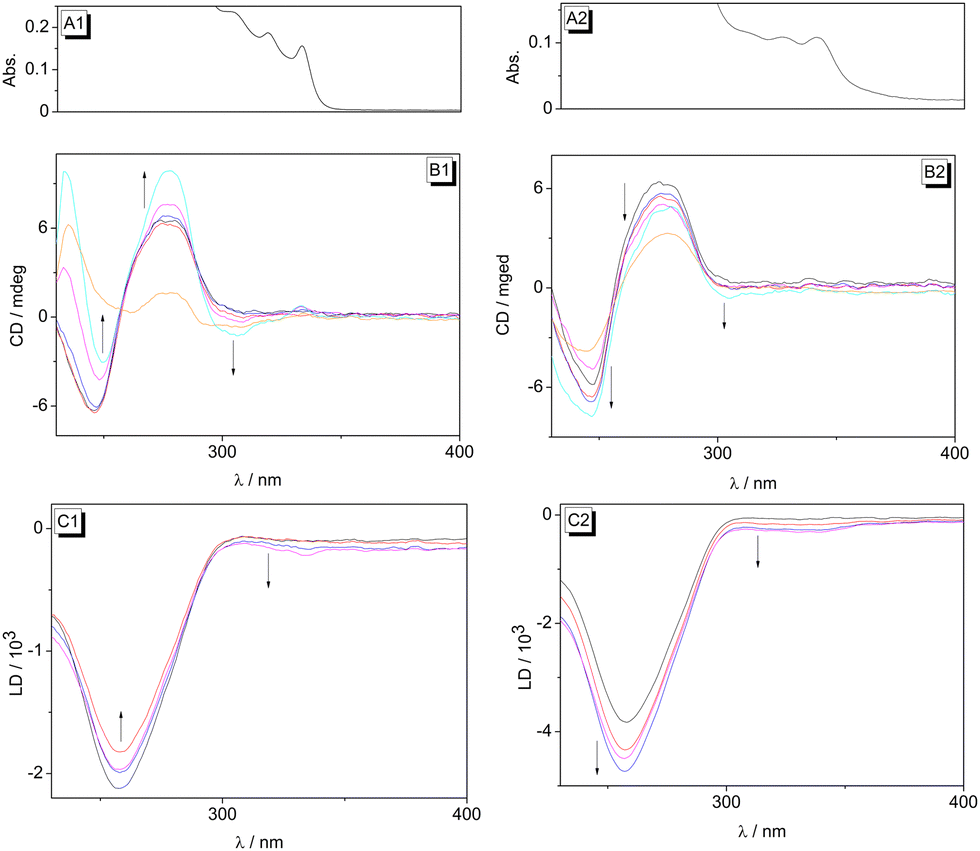 | ||
| Fig. 3 CD spectra of dioncophylline A (1) (B1) and dioncophylline B (2) (B2) in the presence of ct DNA (c = 20 μM) in BPE buffer (cNa+ = 16 mM, pH = 7.0) at LDR = 0.00 (black), 0.05 (red), 0.20 (blue), 1.00 (magenta), 2.00 (cyan); orange: ligand in BPE buffer (c = 20 μM). LD spectra of 1 (C1) and 2 (C2) in the presence of ct DNA (c = 20 μM) in BPE buffer (cNa+ = 16 mM, pH = 7.0) at LDR = 0.00 (black), 1.00 (red), 1.50 (blue), 2.50 (magenta). For better assignment of the CD and LD bands of the DNA-bound ligands at >300 nm, the absorption spectra of 1 (A1) and 2 (A2) in the presence of ct DNA (LDR = 1.0) are shown on top of the panel (x-axis with same scale as B and C). | ||
The LD spectra were obtained in a rotating cuvette to align the DNA as well as its bound ligand along the hydrodynamic field.44 Notably, for both ligands 1 and 2 a small, but significant negative LD signal was observed at 300–350 nm in the presence of DNA, with increasing intensity of the LD bands at higher LDR (Fig. 3C1 and C2). As the DNA did not give an LD signal in this absorption range, these bands can be assigned unambiguously to the DNA-bound ligands 1 and 2, and indicate that the corresponding chromophore, most likely the naphthalene unit, is oriented in an intercalative binding mode coplanar relative to the DNA bases.43,44
To sum up, the spectroscopic studies of the interactions of NIQs 1–3 with ct DNA provided evidence that dioncophylline A (1) and dioncophylline B (2) bind to duplex DNA with moderate affinity. Taking into account the particular biaryl structure of these ligands, along with the LD-spectroscopic analysis of their relative orientation within the host DNA, it may be deduced that the naphthalene unit intercalates at least partly between two base pairs whereas the isoquinoline unit is accommodated in the groove, thus resembling the half-intercalation binding mode that has been suggested for cyanine-type ligands.47,48 This proposed binding mode is in agreement with the observation that the ligand 1 binds to AT-rich DNA sequences and not to GC-rich regions, because in the latter a significant steric repulsion occurs between the groove-bound portion of the ligand and the amino substituent of the guanine, which protrudes in the minor groove. To add to that, AT-rich sequences usually have a slightly wider groove than GC regions, and thus provide more space for sterically demanding ligands. It should be noted that there is no obvious difference between the DNA-binding features of the NIQ 1, with its configurationally stable 7,1′-coupled biaryl axis, and the ones of the configurationally unstable, thus rapidly rotating, 7,6′-coupled derivative 2. This observation indicates that, at least in direct comparison of these NIQs, the higher structural flexibility of the biaryl fragment in NIQ 2, and thus its ability to form a more planarized conformation, does not lead to a more efficient binding to the DNA. Somewhat surprisingly, however, the 5,1′-coupled alkaloid dioncophylline C (3) does not bind to ct DNA at all. Obviously, the 5,1′-connection of the two aryl units in this compound leads to a structure that does not fit well into the DNA-binding sites because of serious steric repulsion.
Association with AP-DNA
It has recently been demonstrated that DNA ligands with a biaryl structure may exhibit even higher affinity to AP-DNA (Fig. 4 and Table 2 in Experimental) than to regularly-paired DNA.38 Therefore the interactions of NIQs 1 and 2 were also investigated with the commonly employed AP-DNA sequence TX (11-mer) (Fig. 5) and, for comparison, with its regularly paired analogue TA (cf. ESI,† Fig. S2). As a general trend, the photometric titration of AP-DNA TX to the ligands 1 and 2 led to the same characteristic development of absorption bands that were also observed upon binding to ct DNA (Fig. 5A1 and A3). Nevertheless, the hypochromicity as well as the formation of a red-shifted absorption band with increasing DNA concentration was more pronounced with the AP-DNA TX. Moreover, the analysis of the resulting binding isotherms gave slightly larger binding constants, Kb = 4.2 × 104 M−1 (1) and Kb = 4.4 × 104 M−1 (2), as compared to the ones with ct DNA (Table 1). To further examine whether the DNA base opposite to the abasic site has an influence on the binding properties to the AP-DNA, photometric titrations of 1 were performed exemplarily with another apurinic DNA, CX (Fig. 5A2). The general course of the titration is similar as with the AP-DNA TX (Fig. 5A1); however, the binding constant with CX is smaller (Kb = 1.9 × 104 M−1). | ||
| Fig. 4 Structures of the regular DNA TA and of the AP-DNA TX and CX. | ||
 | ||
| Fig. 5 Photometric (A) and fluorimetric (B) titration of ligand 1 with TX (1) (1: c1 = 30 μM), CX (2) (1: c1 = 30 μM) and ligand 2 with TX (3) (2: c2 = 40 μM) in ODN buffer (cNa+ = 38.1 mM, pH = 7.0). The black arrows indicate the development of the emission bands during the titration, the red arrows indicate isosbestic points. Inset (A): Plot of the absorption at λ = 333 nm or λ = 337 nm versus cDNA. Inset (B): Plot of the emission at λ = 357 nm or λ = 364 nm versus LDR. | ||
The fluorimetric titrations of NIQs 1 and 2 with AP-DNA TX also confirmed the ligand-DNA complex formation, as the emission intensity of the ligands was strongly quenched in the course of the titration (Fig. 5B1 and B3). In the case of dioncophylline A (1), the AP-DNA CX caused a slightly less efficient quenching than TX (Fig. 5B2), even at saturation, which is consistent with the photometric titrations (see above). Additional evidence of the association of the NIQ 1 with TX was obtained with a complementary fluorescent indicator displacement (FID) experiment (cf. ESI,† Fig. S5). It was demonstrated that the known AP-DNA ligand berberine49 is displaced from its binding site upon addition of NIQ 1.
Most notably, the photometric and fluorimetric titrations of the regularly paired DNA TA to compounds 1 and 2 did not result in changes of the absorption and emission spectra, which showed unambiguously that the NIQs do not bind to this particular duplex-DNA sequence. Presumably, the association of NIQs 1 and 2 with TA is hampered by the steric clash with the guanine residues in the grooves (see above), which cannot be avoided in this short DNA sequence other than in the much longer ct DNA. It should be emphasized also that the association of 1 and 2 with the corresponding AP-DNA TX is in good agreement with the observation that sterically congested ligands are able to differentiate between regular and abasic site-containing DNA.38 Specifically, the insertion of the bulky ligand into the abasic site is still energetically tolerated, i.e., it is still slightly exergonic, because – other than during intercalation – it is not accompanied by the energetically unfavorable DNA-unwinding and stiffening.
Conclusions
The initial intention of this work was the investigation of the DNA-binding properties of the NIQ alkaloids 1–3 in order to clarify whether the cytotoxic activity of these NIQs may involve DNA-binding processes. To sum up, the spectroscopic studies of the interactions of the NIQs 1–3 with ct DNA provided sufficient evidence that the alkaloids 1 and 2 bind to duplex DNA with moderate affinity, presumably in a half-intercalation binding mode,47,48 with an intercalating naphthalene unit and the isoquinoline unit accommodated in the groove. In addition, it was demonstrated that the NIQs 1 and 2 also bind to AP-DNA with even slightly higher affinity. This observation confirms the recent proposal that ligands with a biaryl structure exhibit higher affinities to AP-DNA than to the regularly-paired DNA.38 Overall these results clearly reveal – for the first time – the propensity of NIQs to bind to nucleic acids. More importantly, the exemplary studies with dioncophylline A (1) also provide first evidence of a selectivity of this ligand towards AT-rich regions and abasic sites. These properties may be a relevant feature that contributes to the biological activity of NIQs, specifically to their antitumor activity. We are confident that further studies along these lines will give more insight into the mode of action of these promising bioactive alkaloids.Experimental
Equipment
The absorption spectra were measured in Hellma quartz glass cuvettes 110-QS (d = 10 mm) with a Varian Cary 100 Bio absorption spectrometer. The emission spectra were recorded with a Varian Cary Eclipse fluorescence spectrometer in Hellma quartz glass cuvettes 114F-QS and 115F-QS (d = 10 mm). The CD and LD spectra were recorded with a Chirascan spectrometer (Applied Photophysics). For LD experiments the spectrometer was equipped with a High Shear Couette Cell Accessory. The samples were oriented in a rotating cuvette with a shear gradient of 1200 s−1. All measurements were recorded at a temperature of T = 20 °C as adjusted with a thermostat. The sample solutions for the DNA experiments were mixed with a reaction vessel shaker Top-Mix 11118 (Fisher Bioblock Scientific). E-Pure® water was obtained with an ultrapure water system D4632-33 (Wilhelm Werner GmbH, Leverkusen, Germany); filters: D0835, D0803, and D5027 (2×). The pH values were measured with the pH measuring device QpH 70 (Merck).Materials
Samples of dioncophylline A (1), dioncophylline B (2), and dioncophylline C (3) were obtained through isolation and/or total synthesis as described earlier.13,15,50–54Calf thymus DNA (ct DNA, type I; highly polymerized sodium salt; ε = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 824 cm−1 M−1) and the synthetic poly(dA-dT)–poly(dA-dT) (ε = 13200 cm−1 M−1) were purchased from Sigma-Aldrich (St. Louis, USA) and used without further purification. The synthetic poly(dG-dC)–poly(dG-dC) (ε = 16800 cm−1 M−1) was purchased from InvivoGen (San Diego, USA) and used without further purification. Oligonucleotides (HPLC purified, quality control by MS: MALDI-TOF) d(GCG-TGT-GTG-CG), d(CGC-ACA-CAC-GC), d(GCG-TGT-GTG-CG), d(GCG-TGC-GTG-CG), d(CGC-ACX-CAC-GC), X = tetrahydrofuran spacer (Fig. 4 and Table 2) were purchased from Biomers.net GmbH (Ulm, Germany) and used without further purification. The concentration of ct DNA is given in basepairs (bp) and for the AP-DNA in oligonucleotide.
824 cm−1 M−1) and the synthetic poly(dA-dT)–poly(dA-dT) (ε = 13200 cm−1 M−1) were purchased from Sigma-Aldrich (St. Louis, USA) and used without further purification. The synthetic poly(dG-dC)–poly(dG-dC) (ε = 16800 cm−1 M−1) was purchased from InvivoGen (San Diego, USA) and used without further purification. Oligonucleotides (HPLC purified, quality control by MS: MALDI-TOF) d(GCG-TGT-GTG-CG), d(CGC-ACA-CAC-GC), d(GCG-TGT-GTG-CG), d(GCG-TGC-GTG-CG), d(CGC-ACX-CAC-GC), X = tetrahydrofuran spacer (Fig. 4 and Table 2) were purchased from Biomers.net GmbH (Ulm, Germany) and used without further purification. The concentration of ct DNA is given in basepairs (bp) and for the AP-DNA in oligonucleotide.
Methods
The photometric and fluorimetric titrations with DNA,55,56 and the CD- and LD-spectroscopic analyses were performed according to reported protocols.57 Binding constants were determined from photometric titrations. The binding constants of the ligands with DNA were determined by fitting the binding isotherms to the established theoretical model according to the independent-site model.46Author contributions
Conceptualization, G. B. and H. I.; formal analysis, D. S.; funding acquisition: G. B. and D. S.; investigation, D. S.; writing – original draft, D. S.; writing – review and editing, H. I. and G. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the University of Siegen. D. S. is grateful to the House of Young Talents (University of Siegen) for a PhD fellowship. We thank Ms Jennifer Hermann and Mrs Sandra Uebach (University of Siegen) for technical assistance and Dr Doris Feineis (University of Würzburg) for valuable discussions. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, Br 699/14-2).References
- M. J. Balunas and A. D. Kinghorn, Life Sci., 2005, 78, 431–441 CrossRef CAS PubMed.
- I. Alami Merrouni and M. Elachouri, J. Ethnopharmacol., 2021, 266, 113–435 CrossRef PubMed.
- L. Pan, H. Chai and A. D. Kinghorn, Phytochem. Lett., 2010, 3, 1–8 CrossRef CAS PubMed.
- G. M. Cragg and D. J. Newman, J. Ethnopharmacol., 2005, 100, 72–79 CrossRef CAS PubMed.
- S. Mondal, S. Bandyopadhyay, M. K. Ghosh, S. Mukhopadhyay, S. Roy and C. Mandal, Anti-Cancer Agents Med. Chem., 2012, 12, 49–75 CrossRef CAS PubMed.
- D. J. Newman, G. M. Cragg and K. M. Snader, J. Nat. Prod., 2003, 66, 1022–1037 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- G. Bringmann and D. Feineis, Asian Ancistrocladus Lianas as Creative Producers of Naphthylisoquinoline Alkaloids, in Progress in the Chemistry of Organic Natural Products, ed. A. D. Kinghorn, H. Falk, S. Gibbons, Y. Asakawa, J.-K. Liu and V. M. Dirsch, Springer, Cham, Heidelberg, New York, Dordrecht, London, 2022, vol. 119 DOI:10.1007/978-3-031-10457-2_1.
- N. Tajuddeen, D. Feineis, H. Ihmels and G. Bringmann, Acc. Chem. Res., 2022, 55, 2370–2383 CrossRef CAS PubMed.
- B. K. Lombe, D. Feineis and G. Bringmann, Nat. Prod. Rep., 2019, 36, 1513–1545 RSC.
- N. Tajuddeen and G. Bringmann, Nat. Prod. Rep., 2021, 38, 2154–2186 RSC.
- B. K. Lombe, D. Feineis, V. Mudogo, M. Kaiser and G. Bringmann, J. Nat. Prod., 2021, 84, 1335–1344 CrossRef CAS PubMed.
- G. Bringmann, M. Rübenacker, J. R. Jansen, D. Scheutzow. and L. Aké Assi, Tetrahedron Lett., 1990, 31, 639–642 CrossRef CAS.
- G. François, G. Timperman, T. Steenackers, L. Aké Assi, J. Holenz and G. Bringmann, Parasitol. Res., 1997, 83, 673–679 CrossRef PubMed.
- G. Bringmann, J. Holenz, R. Weirich, M. Rübenacker, C. Funke, M. R. Boyd, R. J. Gulakowski and G. François, Tetrahedron, 1998, 54, 497–512 CrossRef CAS.
- G. François, G. Bringmann, J. Phillipson, L. Aké Assi, C. Dochez, M. Rübenacker, C. Schneider, M. Wéry, D. C. Warhurst and G. C. Kirby, Phytochemistry, 1994, 35, 1461–1464 CrossRef.
- G. François, G. Timperman, W. Eling, L. Aké Assi, J. Holenz and G. Bringmann, Antimicrob. Agents Chemother., 1997, 41, 2533–2539 CrossRef PubMed.
- G. Bringmann, S. Fayez, W. Shamburger, D. Feineis, S. Winiarczyk, R. Janecki and Ł. Adaszek, Vet. Parasitol., 2020, 283, 109177 CrossRef CAS PubMed.
- M. R. Boyd, Y. F. Hallock, J. H. Cardellina II, K. P. Manfredi, J. W. Blunt, J. B. McMahon, R. W. Buckheit, G. Bringmann, M. Schäffer and G. M. Cragg, J. Med. Chem., 1994, 37, 1740–1745 CrossRef CAS PubMed.
- G. Bringmann, G. Zhang, T. Büttner, G. Bauckmann, T. Kupfer, H. Braunschweig, R. Brun and V. Mudogo, Chem. – Eur. J., 2013, 19, 916–923 CrossRef CAS PubMed.
- G. Bringmann, R. Seupel, D. Feineis, G. Zhang, M. Xu, J. Wu, M. Kaiser, R. Brun, E.-J. Seo and T. Efferth, Fitoterapia, 2016, 115, 1–8 CrossRef CAS PubMed.
- G. Bringmann, R. Seupel, D. Feineis, M. Xu, G. Zhang, M. Kaiser, R. Brun, E.-J. Seo and T. Efferth, Fitoterapia, 2017, 121, 76–85 CrossRef CAS PubMed.
- S. Fayez, D. Feineis, V. Mudogo, E.-J. Seo, T. Efferth and G. Bringmann, Fitoterapia, 2018, 129, 114–125 CrossRef CAS PubMed.
- C. Jiang, Z.-L. Li, P. Gong, S.-L. Kang, M.-S. Liu, Y.-H. Pei, Y.-K. Jing and H.-M. Hua, Fitoterapia, 2013, 91, 305–312 CrossRef CAS PubMed.
- S. Fayez, D. Feineis, L. Aké Assi, E.-J. Seo, T. Efferth and G. Bringmann, RSC Adv., 2019, 9, 15738–15748 RSC.
- P. P. Kushwaha, A. K. Singh, K. S. Prajapati, M. Shuaib, S. Fayez, G. Bringmann and S. Kumar, Toxicol. Appl. Pharmacol., 2020, 409, 115297 CrossRef CAS PubMed.
- J. Li, R. Seupel, D. Feineis, V. Mudogo, M. Kaiser, R. Brun, D. Brünnert, M. Chatterjee, E.-J. Seo, T. Efferth and G. Bringmann, J. Nat. Prod., 2017, 80, 443–458 CrossRef CAS PubMed.
- B. K. Lombe, D. Feineis, V. Mudogo, R. Brun, S. Awale and G. Bringmann, RSC Adv., 2018, 8, 5243–5254 RSC.
- S. M. Kavatsurwa, B. K. Lombe, D. Feineis, D. F. Dibwe, V. Maharaj, S. Awale and G. Bringmann, Fitoterapia, 2018, 130, 6–16 CrossRef CAS PubMed.
- D. T. Tshitenge, D. Feineis, V. Mudogo, M. Kaiser, R. Brun, E.-J. Seo, T. Efferth and G. Bringmann, J. Nat. Prod., 2018, 81, 918–933 CrossRef CAS PubMed.
- S. Awale, D. F. Dibwe, C. Balachandran, S. Fayez, D. Feineis, B. K. Lombe and G. Bringmann, J. Nat. Prod., 2018, 81, 2282–2291 CrossRef CAS PubMed.
- S. Bhaduri, N. Ranjan and D. P. Arya, Beilstein J. Org. Chem., 2018, 14, 1051–1086 CrossRef CAS PubMed.
- A. Rescifina, C. Zagni, M. G. Varrica, V. Pistarà and A. Corsaro, Eur. J. Med. Chem., 2014, 74, 95–115 CrossRef CAS PubMed.
- M. S. Nafie, K. Arafa, N. K. Sedky, A. A. Alakhdar and R. K. Arafa, Chem.-Biol. Interact., 2020, 324, 109087 CrossRef CAS PubMed.
- G. Padroni, J. M. Withers, A. Taladriz-Sender, L. F. Reichenbach, J. A. Parkinson and G. A. Burley, J. Am. Chem. Soc., 2019, 141, 9555–9563 CrossRef CAS PubMed.
- S. Crisci, F. Amitrano, M. Saggese, T. Muto, S. Sarno, S. Mele, P. Vitale, G. Ronga, M. Berretta and R. Di Francia, Medicina, 2019, 55, 414 CrossRef PubMed.
- C. Chen, X. Li, H. Zhao, M. Liu, J. Du, J. Zhang, X. Yang, X. Hou and H. Fang, J. Med. Chem., 2022, 65, 3667–3683 CrossRef CAS PubMed.
- K. Benner, A. Bergen, H. Ihmels and P. M. Pithan, Chem. – Eur. J., 2014, 20, 9883–9887 CrossRef CAS PubMed.
- A. Granzhan and H. Ihmels, Synlett, 2016, 1775–1793 CAS.
- M. M. Aleksić and V. Kapetanović, Acta Chim. Slov., 2014, 61, 555–573 Search PubMed.
- F. H. Stootman, D. M. Fisher, A. Rodger and J. R. Aldrich- Wright, Analyst, 2006, 131, 1145–1151 RSC.
- A. Basu and G. S. Kumar, Biochim. Biophys. Acta, 2018, 1862, 1995–2016 CrossRef CAS PubMed.
- T. Šmidlehner, I. Piantanida and G. Pescitelli, Beilstein J. Org. Chem., 2018, 14, 84–105 CrossRef PubMed.
- B. Nordén, A. Rodger and T. Dafforn, Linear Dichroism and Circular Dichroism. A Textbook on Polarized-Light Spectroscopy, RSC Publishing, Cambridge, 2010 Search PubMed.
- G. Bringmann, K. Maksimenka, T. Bruhn, M. Reichert, T. Harada and R. Kuroda, Tetrahedron, 2009, 65, 5720–5728 CrossRef CAS.
- G. Bringmann, J. Mühlbacher, C. Repges and J. Fleischhauer, J. Comput. Chem., 2001, 22, 1273–1278 CrossRef CAS.
- S. M. Yarmoluk, V. B. Kovalska and M. Y. Losytskyy, Biotech. Histochem., 2008, 83, 131–145 CrossRef CAS PubMed.
- S. M. Yarmoluk, S. S. Lukashov, T. Y. Ogul’Chansky, M. Y. Losytskyy and O. S. Kornyushyna, Biopolymers, 2001, 62, 219–227 CrossRef CAS PubMed.
- F. Wu, Y. Shao, K. Ma, Q. Cui, G. Liu and S. Xu, Org. Biomol. Chem., 2012, 10, 3300–3307 RSC.
- G. Bringmann, J. R. Jansen, H. Reuscher, M. Rübenacker, K. Peters and H. G. von Schnering, Tetrahedron Lett., 1990, 31, 643–646 CrossRef CAS.
- G. Bringmann, M. Rübenacker, T. Geuder and L. Aké Assi, Phytochemistry, 1991, 30, 3845–3847 CrossRef CAS.
- G. Bringmann, C. Günther, P. Henschel, K. Peters and E.-M. Peters, Tetrahedron, 2001, 57, 1253–1259 CrossRef CAS.
- G. Bringmann and C. Günther, Synlett, 1999, 216–218 CrossRef CAS.
- G. Bringmann, M. Rübenacker, R. Weirich and L. Aké Assi, Phytochemistry, 1992, 31, 4019–4024 CrossRef CAS.
- K. Benner, A. Granzhan, H. Ihmels and G. Viola, Eur. J. Org. Chem., 2007, 4721–4730 CrossRef CAS.
- K. Benner, H. Ihmels, S. Kölsch and P. M. Pithan, Org. Biomol. Chem., 2014, 12, 1725–1734 RSC.
- H. Ihmels, K. Faulhaber, C. Sturm, G. Bringmann, K. Messer, N. Gabellini, D. Vedaldi and G. Viola, Photochem. Photobiol., 2001, 74, 505–511 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectra; photometric and fluorimetric DNA titrations, CD- and LD-spectroscopic investigations, fluorescent indicator displacement (FID) assay. See DOI: https://doi.org/10.1039/d2nj04081f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |