
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrosion inhibition and disinfection of central heating and cooling water systems using in situ generated hydrogen peroxide†
Ye
Cao
a,
Yue
Xu
a,
Qi
Li
a,
Ruth-Sarah
Rose
 b,
Isaac
Abrahams
a,
Christopher R.
Jones
a and
Tippu S.
Sheriff
*a
b,
Isaac
Abrahams
a,
Christopher R.
Jones
a and
Tippu S.
Sheriff
*a
aDepartment of Chemistry, School of Physical and Chemical Sciences, Queen Mary University of London, London, E1 4NS, UK. E-mail: t.s.sheriff@qmul.ac.uk
bSchool of Biological and Behavioural Sciences, Queen Mary University of London, London, E1 4NS, UK
First published on 30th August 2022
Abstract
MnCl2·4H2O and Tiron (disodium 4,5-dihydroxy-1,3-benzenedisulfonate) rapidly remove dioxygen (O2) from aqueous solution at a rate of ∼20 mg L−1 min−1 with turnover frequencies (TOFs) of up to 440![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 in the pH range 7.50–11.0 and at 20–50 °C using hydroxylamine (NH2OH) as reducing substrate. These solutions remain deoxygenated for several hours despite being open to the atmosphere. During this time there is a steady rise in the concentration of in situ generated hydrogen peroxide (H2O2), reaching ∼12 mM after 17 h. The order of selectivity for selected 1st row transition metals was found to be: Mn(II) ≫ Co(II)∼Cu(II)∼Fe(II). No deuterium isotope effect was observed, which suggests that an electron transfer is the rate determining step. A mechanism is proposed that involves two 1-electron transfers from bound NH2OH to bound O2 to produce H2O2 concomitant with two proton transfers from catecholate oxygen atoms. This system can act as an anti-corrosion formulation as the catalytic reduction of O2 results in the removal of O2 from open aqueous solutions and the in situ generated H2O2 can be used as a biocide e.g. to kill L. pneumophila. Batch experiments were carried out to confirm the feasibility of this system to simultaneously inhibit corrosion and also potentially disinfect central heating and cooling water systems.
000 h−1 in the pH range 7.50–11.0 and at 20–50 °C using hydroxylamine (NH2OH) as reducing substrate. These solutions remain deoxygenated for several hours despite being open to the atmosphere. During this time there is a steady rise in the concentration of in situ generated hydrogen peroxide (H2O2), reaching ∼12 mM after 17 h. The order of selectivity for selected 1st row transition metals was found to be: Mn(II) ≫ Co(II)∼Cu(II)∼Fe(II). No deuterium isotope effect was observed, which suggests that an electron transfer is the rate determining step. A mechanism is proposed that involves two 1-electron transfers from bound NH2OH to bound O2 to produce H2O2 concomitant with two proton transfers from catecholate oxygen atoms. This system can act as an anti-corrosion formulation as the catalytic reduction of O2 results in the removal of O2 from open aqueous solutions and the in situ generated H2O2 can be used as a biocide e.g. to kill L. pneumophila. Batch experiments were carried out to confirm the feasibility of this system to simultaneously inhibit corrosion and also potentially disinfect central heating and cooling water systems.
Introduction
Cast iron has been traditionally used as a pipe material for water systems. However, cast iron is susceptible to corrosion in oxidising environments, resulting in environmental pollution, economic loss and safety incidents.1 Stainless steel is more resistant to corrosion due to its oxide protective surface,2 but corrosion still occurs in stainless steel pipes3 due to weak spots because of welding zones, poor coating or the coating not being thick enough thus exposing the metal to corrosion.4 There is a worldwide focus in the field of anti-corrosion technologies for water systems.5,6 Conventional anti-corrosion technologies include organic coatings,7 electrochemical protection,8 and corrosion inhibitors.9 The occurrence of corrosion scales in pipes has been ascribed to the interaction between pipe material and water with dissolved impurities, especially dissolved O2.10 Bacteria can grow on the surface of pipes with the development of biofilms which promote corrosion.11 Therefore, the removal of dissolved O2 provides a method to prevent corrosion and the build-up of sludge and scale. However, the removal of dissolved O2 in water systems is difficult, since the dissolved O2 is replenished while the piping system is in operation. The membrane-based dissolved O2 removal technique is a device made with hollow fibre membranes and enable effective removal of dissolved O2 by gas absorption and stripping.12 However, in spite of the known strengths of the membrane-based technique, its industrial applications are still rare because it suffers from high cost and low stability.13 Another technique for dissolved O2 removal is by dosing scavengers. Early dissolved O2 scavengers were based on polyphenols,14 but these were subsequently replaced by amine-based reducing substrates such as hydrazine (N2H4) and N,N-diethylhydroxylamine (DEHA, Et2NOH).15 The combination of DEHA with hydroquinone (H2Q) is an effective means of removing dissolved O2 under basic conditions (pH 10–11) since H2Q acts a catalyst to initiate a reaction with atmospheric triplet dioxygen (3O2).16 Aubry compared the ability of DEHA to reduce O2 in the presence of various polyphenols and quinones as alternatives to H2Q at pH 10.5 and found little variations in the rates,15 but with resorcinol (and derivatives of it) as co-catalysts with H2Q at pH 10.1, they reported synergistic effects in the depletion of dissolved O2.17,18 They also investigated other alkylhydroxylamines to remove dissolved O2 using H2Q, gallic acid and aminophenols as catalysts at pH 10.5.19 However, there has been limited use of these scavengers because of the environmental hazards associated with their toxicity.20Electron-deficient catechols such as disodium 4,5-dihydroxy-1,3-benzenedisulfonate (Tiron, Fig. 1), in the presence of Mn(II) form a catalytic system for the reduction of O2 to hydrogen peroxide (H2O2) in the presence of NH2OH (or N2H4) as a reducing substrate under ambient conditions and at pH values of ∼8.0 (eqn (1)).21
 | (1) |
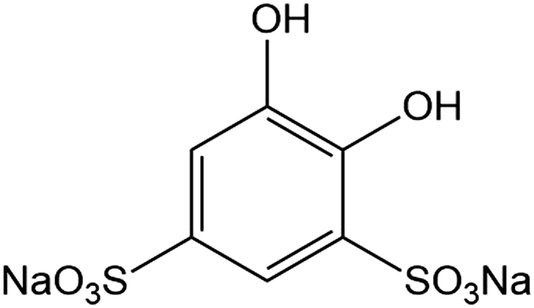 | ||
| Fig. 1 The structure of Tiron. | ||
Confined environments which are unhygienic, humid and gloomy such as central heating and cooling systems, are an ideal breeding ground for bacteria. In residential and public buildings, air may go through the central heating and cooling systems and may be exposed to bacteria and therefore induce the deterioration of indoor air quality by diffusion.22 Many studies have shown that indoor air pollution can cause health problems and microbial pollution was identified as a main source of responsibility.23
This work evaluates the efficacy of a formulation containing [Tiron] = 1.50 mM, [NH2OH] = 500 mM, [MnCl2·4H2O] = 50.0 μM in reducing and controlling the dissolved O2 concentration in aqueous solution. It investigates the influence of temperature and other factors on the efficiency of O2 removal, and suggests a mechanism for the in situ generation of H2O2. It also evaluates the inhibition of bacterial growth and corrosion on iron-nickel nails by visible inspection and using XPS and XRD for surface studies.
Experimental section
Materials and instruments
Manganese(II) chloride tetrahydrate (AnalaR, 99.5%, BDH), 1,2-dihydroxybenzene-3,5-disulfonate, disodium salt, monohydrate (Tiron, Sigma-Aldrich), N-2-hydroxyethylpiperazine-N′-3-propane-sulfonic acid (EPPS, Sigma-Aldrich), sodium hydrogen carbonate (AnalaR, Sigma-Aldrich), potassium dihydrogen phosphate (AnalaR, BDH), sodium hydroxide pellets (semiconductor grade, 99%, Sigma-Aldrich), disodium hydrogen phosphate (AnalaR, BDH), sodium chloride (AnalaR, BDH), ammonium chloride (AnalaR, BDH), hydroxylamine (50% (aq), Sigma-Aldrich), magnesium sulfate (ACS reagent, Fisher Scientific), calcium chloride (ACS reagent, Fisher Scientific), titanium oxide oxalate dihydrate (98%, Sigma-Aldrich), CHES (2-(cyclohexylamino) ethanesulfonic acid, Alfa Aesar), sodium hydroxide (AnalaR, Sigma-Aldrich), hydrochloric acid (36%, BDH), thiamine hydrochloride (>98.5%, Duchefa Biochemie), D-glucose (Melford), ammonium chloride (AnalaR, BDH), D(+)-biotin (>97.5%, Duchefa Biochemie), ethylene diammine tetra-acetic acid (EDTA, >99.0%, Sigma-Aldrich), iron(III) chloride hexahydrate (ACS reagent, Fisher Scientific), zinc chloride (ACS reagent, Fisher Scientific), copper(II) chloride dihydrate (ACS reagent, Fisher Scientific), cobalt(II) chloride hexahydrate (ACS reagent, Fisher Scientific), boric acid (>99.5%, Sigma-Aldrich), magnesium(II) chloride hexahydrate (ACS reagent, Fisher Scientific) and sulfuric acid (95%, BDH), nails (150 mm, RH150B500, ForgeFix) were used as received. De-ionised water (ELGA Purelab) was used in all experiments and plastic spatulate were used to transfer solid reagents. Carbonate buffer solutions were made up in boiled water (to remove dissolved CO2).24pH measurements were carried out using a HANNA pH 211 instrument that had previously been calibrated at pH 7.00 and pH 9.00. UV/Vis fixed wavelength measurements were carried out on a JENWAY 6315 UV/Vis spectrophotometer and UV/Vis scanning measurements were carried out on a Agilent Cary 100 UV/Vis spectrophotometer. Dissolved O2 concentrations were measured using a calibrated Hach Lange LD0101 (di)oxygen probe. The cultures of E. coli were grown in a NBS Innova 42 shaker. Anti-bacteria assays were carried out in a biological safety cabinet (Class II Type KS 9, Herasafe KS) and the E. coli populations were measured through OD 600 using a microplate reader (FLUOstar Omega, BMG LABTECH). X-Ray Diffraction (XRD) was operated in Bragg-Brentano geometry with a PANalyticak X’Pert Pro diffractometer equipped with a X’Celerator detector and Ni-filtered Cu Kα radiation (λ = 1.5418 Å). Data were gathered in the 2θ range of 5–120° with a step size of 0.0167° and an equivalent counting time of 200 s per step. X-Ray photoelectron spectroscopy (XPS) was carried with a Thermo Fisher Nexsa Surface Analysis system.
Preparation of M9 5X medium
NaH2PO4 (16.95 g, 141.2 mmol), KH2PO4 (7.500 g, 55.15 mmol), NaCl (1.250 g, 21.39 mmol) and NH4Cl (2.500 g, 46.74 mmol) were dissolved in deionised water (375 mL) and the solution was sterilised by autoclaving at 121 °C for 15 min.25Preparation of trace elements solution
EDTA (5.00 g, 17.1 mmol) was dissolved in deionised water (800 mL) and adjusted to pH 7.50 using NaOH. FeCl3·6H2O (0.830 g, 3.00 mmol), ZnCl2 (0.084 g, 0.616 mmol), CuCl2·2H2O (0.013 g, 0.076 mmol), CoCl2·6H2O (0.010 g, 0.077 mmol), H3BO3 (0.010 g, 0.16 mmol) and MnCl2·6H2O (0.0016 g, 0.0068 mmol) were added and made up the solution with deionised water (1.00 L). The solution was filtered through 0.2 μm filter for sterilisation.Preparation of minimal media
Thiamine (0.0005 g, 0.002 mmol), biotin (0.0005 g, 0.002 mmol), NH4Cl (0.5000 g, 9.347 mmol), glucose (2.0000 g, 11.101 mmol), MgSO4 (0.500 mL, 1 M, 0.500 mmol) and CaCl2 (0.150 mL, 1 M, 0.150 mmol) were dissolved in deionised water, and made up to volume (24.5 mL), and then filtered for sterilisation. Trace elements solution (0.5 mL), sterilised deionised water (100 mL) and M9 5X medium (375 mL) were added to prepare the minimal media (500 mL).Anti-bacterial performance testing
A starter culture with a single colony of E. coli (strain JM109, Promega) was incubated overnight in Luria-Bertani (LB) broth at 37 °C. In a typical anti-bacterial assay, the cells were washed with water and resuspended in minimal media to obtain a starting OD600 of 0.1. Prior to the dilution, samples containing different components (NH2OH, Tiron, phosphate, MnCl2·4H2O) from the in situ generation of H2O2 system were prepared in 25 mL centrifuge tubes to investigate the role of these components in bacteriostatic processes. A start OD 600 of E. coli was placed in the centrifuge tube and then five replicated samples (100 μL) were incubated in a 96 well cell culture plate. The reactions were incubated at 37 °C in a microplate reader and the OD 600 of each sample was recorded at various times.Anti-corrosion testing
The influence of the in situ generated H2O2 formulation on anti-corrosion performance was evaluated via iron nail corrosion assays, which were carried in vials (30 mL) half-filled with solution to ensure the surface of the nail was in contact with both solution and air at 20 °C. The anti-corrosion performance of the formulations was tested by visually recording the corrosion progress of the nails after two weeks, one month and six months, and characterising the surface of the nails after six months using XPS and XRD.Measurement of dissolved O2 and H2O2
In a typical experiment, carbonate buffer (pH 9.00, 500 mM, 5.00 mL), 50% aq. NH2OH (1.53 mL, 25.0 mmol) and Tiron (0.0249 g, 0.0750 mmol) were made up in a volumetric flask (50.0 mL) using deionised water and transferred to a beaker that was contained in a temperature-controlled water bath. O2 readings were taken every 30 s at 20 ± 1 °C for 16 min with the probe suspended in the unstirred solution. MnCl2·4H2O (0.500 mL, 5.00 mM) was added at t = 16 min (or at the start for slower runs) and readings were then taken every 10 s. Once the readings were stable, readings were then taken every 5 min. Additionally, aliquots (0.100 mL) of the reaction solution were removed at 15 min intervals and added to aq. acidified Ti(IV) solution (2.00 mL) to measure the concentration of in situ generated H2O2.26 For deuterium isotope studies NH2OH·HCl (0.139 g, 0.200 mmol) and Na2CO3 (0.0530 g, 0.500 mmol) were dissolved in D2O (10.0 mL) and added to a vial (30 mL) containing Tiron (4.98 mg, 0.0150 mmol) and aq. MnCl2·4H2O (0.100 mL, 5.00 mM). Adjustments of pD were made with DCl or NaOD. O2 measurements were commenced immediately and then every 10 s; this procedure was repeated using H2O in place of D2O. All experimental runs were repeated three times, and the average of the data was obtained.Results and discussion
Removal of dissolved O2
Fig. 2 shows the removal of O2 from aqueous solution at 20 ± 1 °C in the presence of Tiron (1.50 mM) and NH2OH (500 mM) at pH 9.00 (carbonate buffer) when MnCl2·4H2O (50.0 μM) was added (t = 16 min). The concentration of dissolved O2 fell rapidly from ∼9.5 mg L−1 (∼0.30 mM) to ∼0 mg L−1 in ∼30 s at a rate of ∼20 mg L−1 min−1 (black line), with no measurable change in the concentration of dissolved O2 in the presence of Tiron and NH2OH but no added Mn(II) (red line). A slower consumption of dissolved O2 was observed in the presence of Mn(II) and NH2OH, with no Tiron added (blue line). It has previously been reported that the electron-withdrawing sulfonate groups (–SO3−) on the catechol ring of Tiron not only make this compound water soluble,27 but can also promote electron transfer from NH2OH to O2. | ||
Fig. 2 The change of dissolved O2 concentration in the formulation in the presence and absence of Mn(II), Tiron, and NH2OH. Mn(II)/Tiron/NH2OH (–■–), Mn(II)/NH2OH, no Tiron ( ), no Mn(II)/Tiron/NH2OH ( ), no Mn(II)/Tiron/NH2OH ( ). Experiment conditions: [Tiron] = 1.50 mM, [NH2OH] = 500 mM, [MnCl2·4H2O] = 50.0 μM, initial pH: 9.0, [carbonate] = 50.0 mM, temperature: 20 ± 1 °C. ). Experiment conditions: [Tiron] = 1.50 mM, [NH2OH] = 500 mM, [MnCl2·4H2O] = 50.0 μM, initial pH: 9.0, [carbonate] = 50.0 mM, temperature: 20 ± 1 °C. | ||
The fall in the concentration of dissolved O2 in the solution containing Mn(II) and NH2OH (but without Tiron) was initially quite fast but then slowed significantly suggesting that Tiron acts as a co-catalyst to promote the consumption of dissolved O2 enabling the efficient in situ generation of H2O2. The concentration of dissolved O2 remained at effectively zero over a period ∼17 h. During this time there was a continuous increase in the concentration of in situ formed H2O2 to ∼12 mM, even after the measured concentration of dissolved O2 had reached ∼0 mg L−1 and this was probably due to the slow diffusion of additional O2 into the unstirred solution which was open to the atmosphere (Fig. S1, ESI†). The lack of any measurable dissolved O2 in the solution during this period shows that there is rapid reduction of the additional diffused O2 to H2O2 on a time-scale that is faster than can be measured by the dissolved O2 probe. No disproportionation of the in situ formed H2O2 to O2 was observed.
Before the addition of MnCl2·4H2O (t = 16 min) the concentration of dissolved O2 decreased very slowly in the absence of Tiron (Fig. 2) which suggests that the reduction of O2 by NH2OH was catalysed by the presence of adventitious Mn(II). This reaction can be effectively stopped by the addition of ethylene diammine tetra-acetic acid (H4EDTA, 1.00 mM), that acts as a Mn(II) sequestering agent (Fig. S2, ESI†).
When the temperature was increased from 20 °C to 50 °C, there was no noticeable effect on the rapid removal of dissolved O2 (Fig. S3, ESI†), indicating that this is a robust catalytic system that can be used in hot water systems for the removal of O2 and thus prevent corrosion and also potentially kill legionella bacteria by the in situ generation of H2O2. There was a higher consumption rate of dissolved O2 before MnCl2·4H2O was added at higher temperatures. Similar trends were also observed by Triki et al.28 and Medina et al.,29 in which O2 and formic acid were used for the in situ generation of H2O2.
It is assumed that the catalyst [MnIII(1,3-(SO3)2Cat)2]5− (where Cat = 4,5-dihydoxybenzene), is rapidly assembled in basic aqueous solution by the coordination of two deprotonated Tiron anions to Mn(II) which is presumably kept in the Mn(II) oxidation state by the presence of excess NH2OH that reduces Mn(III) to Mn(II).27 Enhanced rates were observed when the buffer was changed from phosphate (pH 7.5–8.5) to carbonate (pH 9.0–11.0), and especially when the pH was raised from 9.0 to 10.5 (Fig. S4, ESI†).
The presence of intramolecular hydrogen bonds between the bound substrate molecules (NH2OH and O2) and the oxygen and protonated oxygen atoms on the catecholate ligands are very likely to be important in stabilising NH2OH and O2, and also in facilitating the electron and proton transfers between the substrate molecules. The crystal structure for [Na]5[MnIII(3,5-(SO3)2Cat)2]·10H2O shows a square planar arrangement of two doubly deprotonated catecholate anions around Mn(III).30 In the pH range of 9–11, it would be expected that one of the catecholate oxygen atoms in Tiron will be protonated (pKa1 = 7.57, pKa2 = 12.5)31 and this would allow for a proton transfer from the catecholate oxygen to the coordinated superoxide ion (O2−), that is formed from the 1-electron reduction of coordinated O2 by coordinated NH2OH (with both occupying axial positions), that is facilitated through redox changes at the Mn(II) complex. A further 1-electron and proton transfer from coordinated “NHOH” would generate the coordinated hydroperoxide ion (HO2−) and azanone (nitroxyl, HNO) which then undergoes further fast reduction by NH2OH to produce N2 and water, while protonation of HO2− generates H2O2 and a protonated bound catecholate to enable the cycle to be repeated (Scheme 1). A pH of ∼10.5 was observed to be optimal, presumably because of the need to maintain the delicate balance of protonated catecholate to stabilise the binding of substrate molecules, and facilitate proton transfer in this enzyme-like system. The O2 removal rate reduced when the pH was above 10.5 (Fig. S4, ESI†), which suggests that the deprotonation of the second hydrogen on the catecholate oxygen atom weakens the hydrogen bonds between NH2OH and Tiron, and hinders the proton transfer.
 | ||
| Scheme 1 Proposed mechanism for the [MnII(1,3-(SO3)2Cat)2]6− catalysed reduction of O2 to H2O2 by NH2OH. | ||
Deuterium isotope studies indicate no differences in rate when using ND2OD in place of NH2OH (Fig. S5, ESI†), suggesting that there is no deuterium isotope effect in the expected direction for proton transfer (D–O/N bonds have a higher bond strength than H–O/N bonds),32 so it can be implied that the initial electron-transfer is the determining step in the reduction of O2.33 A similar mechanism has been proposed by Machan for the electrocatalytic reduction of O2 to H2O2 by a bipyridine Schiff-base ligand in the presence of a coordinated phenolate moiety where the proton transfer originates from the protonated phenolate molecule.34
Anti-bacterial investigation
The in situ generation of H2O2 and the consumption of O2 can be considered to be particularly important in antibacterial formulations.35,36E. coli was selected for this investigation, since it is sensitive to dissolved O2 concentrations.37 Fig. S6 (ESI†) shows the growth curve of E. coli in minimal media (MM) and the overall inhibitory effect on the bacterial growth of this formulation.The reproduction of E. coli can be divided into four periods: the lag period, the logarithmic period, the stationary period, and the death period (not shown due to time limit of the experiment).38 The lag period represents the stage when the bacteria are initially added to the broth and propagation does not take place due to the adaptation of the bacteria to the medium. After the lag period, the bacteria begin to reproduce rapidly. The growth curve at this stage is like a logarithmic function, so it is called the logarithmic period. During the stationary period, propagation slows down as a result of nutrient consumption, and a dynamic equilibrium exists between the growth and death of bacteria. The death outpaces the growth of bacteria when the death period begins, and therefore the population begins to decline. An obvious inhibitory effect on the bacterial growth could be observed at the very beginning of the culture when the formulation was dosed.
H2O2 is a well-known bacteriostatic agent. Recent studies suggest that low concentrations of intracellular H2O2 can bring certain select biosynthetic processes to a halt due to the H2O2 damage to the mitochondrial DNA.39–41 Different concentrations of H2O2 were directly added, and the results show [H2O2] < 2.00 mM make little difference to the bacterial growth, but concentrations > 5.00 mM, in contrast, have a severe inhibitory effect (Fig. 3). Using the formulation, concentrations of generated H2O2 reach ∼10 mM after 30 min of reaction,42 and therefore this formulation exhibits a beneficially bacteriostatic effect due to the in situ generated H2O2.
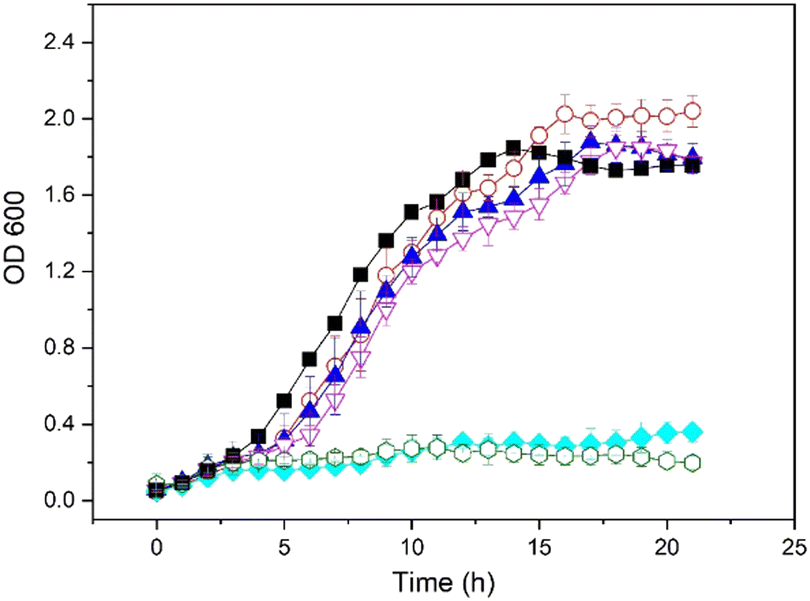 | ||
Fig. 3 The inhibitory effect of different concentrations of added H2O2 on bacterial growth. Culture conditions: ( ) without added H2O2, ( ) without added H2O2, ( ) [H2O2] = 0.500 mM, ( ) [H2O2] = 0.500 mM, ( ) [H2O2] = 1.00 mM, ( ) [H2O2] = 1.00 mM, ( ) [H2O2] = 2.00 mM, ( ) [H2O2] = 2.00 mM, ( ) [H2O2] = 5.00 mM, ( ) [H2O2] = 5.00 mM, ( ) [H2O2] = 10.00 mM, Unbuffered, temperature: 37 ± 1 °C. ) [H2O2] = 10.00 mM, Unbuffered, temperature: 37 ± 1 °C. | ||
When NH2OH alone was added, the growth of bacteria was severely inhibited, because NH2OH is very toxic to bacteria (Fig. 4(a)).43 The components phosphate (pH 8.0), Tiron and MnCl2·4H2O do not inhibit the bacterial growth (black square, Fig. 4(b)). In addition, no obvious difference was observed between the experiments with or without in situ generated H2O2 if NH2OH was added (magenta inverted triangle, blue triangle and red dot, Fig. 4(b)), indicating compared to the high toxicity of NH2OH, the in situ generated H2O2 and the consumption of O2 only play a minor part in the anti-bacterial contribution of the formulation, even if anaerobic conditions do supresses the growth rate of E. coli to some extent.44
 | ||
Fig. 4 (a) The inhibitory effect of NH2OH on bacterial growth, ( ) MM + bacteria, ( ) MM + bacteria, ( ) MM + bacteria + NH2OH, ( ) MM + bacteria + NH2OH, ( ) MM + bacte ria + Tiron, ( ) MM + bacte ria + Tiron, ( ) MM + bacteria + phosphate, ( ) MM + bacteria + phosphate, ( ) MM + bacteria + MnCl2·4H2O, (b) bacterial growth on removing single component during the in situ generation of H2O2, ( ) MM + bacteria + MnCl2·4H2O, (b) bacterial growth on removing single component during the in situ generation of H2O2, ( ) MM + bacteria + Tiron + phosphate + MnCl2·4H2O, ( ) MM + bacteria + Tiron + phosphate + MnCl2·4H2O, ( ) MM + bacteria + Tiron + phosphate + NH2OH + MnCl2·4H2O, ( ) MM + bacteria + Tiron + phosphate + NH2OH + MnCl2·4H2O, ( ) MM + bacteria + Tiron + phosphate + NH2OH, ( ) MM + bacteria + Tiron + phosphate + NH2OH, ( ) MM + bacteria + phosphate + NH2OH + MnCl2·4H2O, ( ) MM + bacteria + phosphate + NH2OH + MnCl2·4H2O, ( ) MM + bacteria. Culture conditions: [Tiron] = 1.50 mM, [NH2OH] = 500.0 mM, [phosphate] = 50.0 mM, [MnCl2·4H2O] = 50.0 μM, temperature: 37 ± 1 °C. ) MM + bacteria. Culture conditions: [Tiron] = 1.50 mM, [NH2OH] = 500.0 mM, [phosphate] = 50.0 mM, [MnCl2·4H2O] = 50.0 μM, temperature: 37 ± 1 °C. | ||
Anti-corrosion investigation
The corrosion of iron samples under different aeration conditions can produce iron oxides such as magnetite (Fe3O4), lepidocrocite (γ-FeOOH), akaganeite (β-FeOOH) and goethite (α-FeOOH),45 with the oxidation state of iron increasing as the corrosion progresses.46 In this work, iron nails were selected as samples for immersion experiments. The corrosion products were analysed by XPS and XRD after six months of immersion to evaluate the extent of corrosion, with the appearances of the nails recorded at fixed intervals of two weeks, one month and six months (Fig. 5). | ||
| Fig. 5 The corrosion of iron nails in different conditions after two weeks, one month and six months of immersion. Assay I: Deionised water (pH 7.0); Assay II: [Tiron] = 1.50 mM, [NH2OH] = 500 mM, [MnCl2·4H2O] = 50.0 μM, [phosphate] = 50.0 mM (pH 8.0); Assay III: Deionised water (pH 7.0), removal of the dissolved O2 using N2 and then the vial was sealed with a lid; Assay IV: [H2O2] = 50 mM (pH 7.0); Assay V: [Tiron] = 1.50 mM, [NH2OH] = 500 mM, [MnCl2·4H2O] = 50.0 μM, [carbonate] = 50.0 mM (pH 9.0); Assay VI: [Tiron] = 1.50 mM, [MnCl2·4H2O] = 50.0 μM, [phosphate] = 50.0 mM (pH 8.0). | ||
Clear corrosion appeared in the nail of assay I (deionised water), which can be considered to be the initial formation of iron oxy-hydroxides (FeOOH).47 Magnetite (Fe3O4) was generated at the interface of the metal and iron oxy-hydroxides as the immersion time was extended. As magnetite is less porous, O2 cannot easily reach the metal surface, and therefore the corrosion was slowed.46 After six months of immersion, microscopic cracks formed on the surface of the corrosion product and O2 would again be in contact with the metal surface resulting in severe corrosion. The whole corrosive process in assay I can be described by eqn (2)–(4).48
| 4Fe0 + 3O2 + 2H2O → 4γ-FeOOH | (2) |
| 8γ-FeOOH + Fe0 + 3O2 → 3Fe3O4 + 4H2O | (3) |
| 4Fe3O4 + O2 + 6H2O → 12γ-FeOOH | (4) |
No corrosion scale developed during the whole process of assay II (formulation, phosphate, pH 8.0), which may be explained by the consumption of dissolved O2 with the in situ generation of H2O2 and the presence of NH2OH that acts as a reducing agent.
In the XPS spectra, the peak positions of Fe-2p reflect the ionic states of Fe,49 and these peaks are the basis for the qualitative determination of different states of iron. The Fe-2p XPS spectra for the iron sample in assay I shows that the peak of Fe-2p2/3 which is located around 710.9 eV can be divided into two peaks, at 710.6 eV and 708.6 eV, corresponding to Fe3+ and Fe2+ oxides respectively (Fig. 6).50
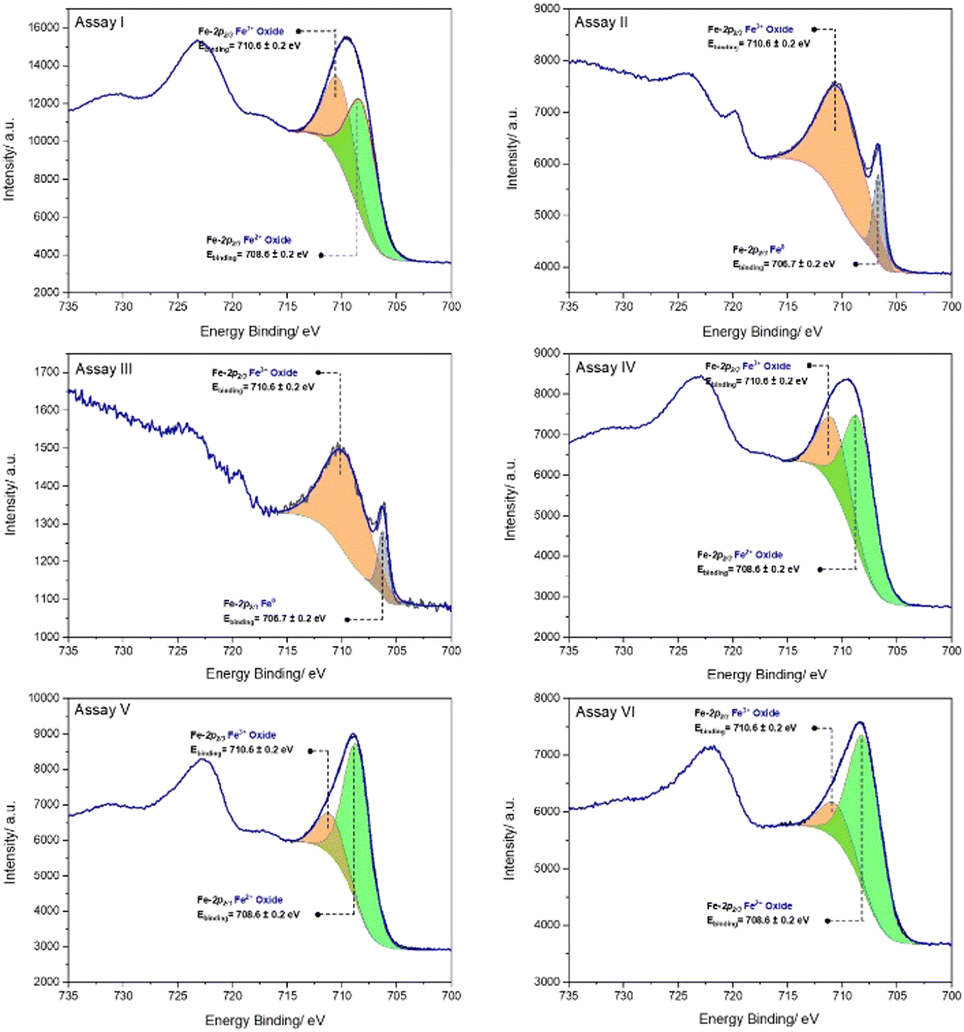 | ||
| Fig. 6 Fe-2p XPS spectra of iron nails in different assays after six months of immersion. Assay I: Deionised water (pH 7.0); Assay II: [Tiron] = 1.50 mM, [NH2OH] = 500 mM, [MnCl2·4H2O] = 50.0 μM, [phosphate] = 50.0 mM (pH 8.0); Assay III: Deionised water (pH 7.0), removal of the dissolved O2 using N2 and then the vial was sealed with a lid; Assay IV: [H2O2] = 50 mM (pH 7.0); Assay V: [Tiron] = 1.50 mM, [NH2OH] = 500 mM, [MnCl2·4H2O] = 50.0 μM, [carbonate] = 50.0 mM (pH 9.0); Assay VI: [Tiron] = 1.50 mM, [MnCl2·4H2O] = 50.0 μM, [phosphate] = 50.0 mM (pH 8.0). | ||
An extra peak at ∼706.7 eV in the assay II Fe-2p XPS spectrum can be attributed to Fe0, indicating this iron sample suffered a smaller degree of corrosion compared to the sample in assay I. Fig. 7 presents the XRD diffractograms after six months of immersion. The peaks identified in the JCPDS (Joint Committee on Powder Diffraction Standards) database are labelled to indicate the identified phases. The phase identified in assay I is magnetite, while the best match in assay II was found to be Fe0, which is consistent with the results observed in the XPS spectra.
 | ||
| Fig. 7 XRD patterns of iron nails in different assays after six months of immersion. Assay I: Deionised water (pH 7.0); Assay II: [Tiron] = 1.50 mM, [NH2OH] = 500 mM, [MnCl2·4H2O] = 50.0 μM, [phosphate] = 50.0 mM (pH 8.0); Assay III: Deionised water (pH 7.0), removal of the dissolved O2 using N2 and then the vial was sealed with a lid; Assay IV: [H2O2] = 50 mM (pH 7.0); Assay V: [Tiron] = 1.50 mM, [NH2OH] = 500 mM, [MnCl2·4H2O] = 50.0 μM, [carbonate] = 50.0 mM (pH 9.0); Assay VI: [Tiron] = 1.50 mM, [MnCl2·4H2O] = 50.0 μM, [phosphate] = 50.0 mM (pH 8.0). | ||
Assays III (anaerobic deionised water) and IV (added H2O2) were conducted to verify the contribution of the consumption of O2 and the in situ generated H2O2 to the anti-corrosion performance. No corrosion was observed in assay III due to the removal of dissolved O2, and its XPS and XRD spectra reveal that Fe0 is still observable on the surface of the nail. The nail in assay IV only shows clear signs of corrosion after six months. Significantly, no corrosion occurred after two weeks (Fig. 7), which might be ascribed to a different corrosion mechanism which firstly involves slow cathodic reduction of H2O2 and anodic oxidation of Fe0 at the two poles of the nail, as can be seen after 1 month. It is reported that Fe0 and H2O2 react under acidic conditions by the Fenton reaction that generates hydroxyl radicals (eqn (5)–(6)).51,52
| Fe0 + H2O2 + 2H+ → Fe2+ + 2H2O | (5) |
| Fe2+ + H2O2 → Fe3+ + ˙OH + OH− | (6) |
At the beginning of the corrosive process in assay IV, Fe3+ leaching takes place (eqn (6)) rather than corrosion scale formation. Then rust, consisting of ferric oxyhydroxides (mainly γ-FeOOH, Fig. 7), is formed. In addition, an initial lag period is reported to exist and it can be prolonged when increasing the ratio of H2O2/Fe0.53 The lag phase may be because the formation of pits, which represent reactive sites on iron samples, is slow in the presence of H2O2.54
Carbonate buffer is not suitable for this formulation, since corrosion occurred in assay V (formulation, carbonate, pH 9.0) after only two weeks. Fe(OH)2, Fe3O4 and FeCO3 make up the majority of the corrosion products of iron samples (Fig. 7).55 Previous work by us demonstrated that carbonates enhance the oxidative ability of this formulation by generating a high-valent manganese complex (Mn(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and therefore corrosion still occurs even if dissolved O2 is removed from the system.42 Under these conditions, α-FeOOH is the main product (Fig. 7, assay V). Fe2+ ions were more susceptible to leaching in assay V than assay II according to the UV/Vis spectral analysis (Fig. 8). The peak at 368 nm in assay V is similar to that found for [FeIII(cat)2]−.56
O) and therefore corrosion still occurs even if dissolved O2 is removed from the system.42 Under these conditions, α-FeOOH is the main product (Fig. 7, assay V). Fe2+ ions were more susceptible to leaching in assay V than assay II according to the UV/Vis spectral analysis (Fig. 8). The peak at 368 nm in assay V is similar to that found for [FeIII(cat)2]−.56
 | ||
Fig. 8 UV/Vis spectra of different assay solutions two weeks after iron nails were immersed in them (( ) assay II, ( ) assay II, ( ) assay V, ( ) assay V, ( ) assay VI) and one month (( ) assay VI) and one month (( ) assay II, ( ) assay II, ( ) assay V, ( ) assay V, ( ) assay VI). ) assay VI). | ||
No peak at 368 nm was observed in assay II after two weeks of immersion. Only the mono-Tironate complex, [MnIII(1,3-(SO3)2Cat)2]−, with a peak at 395 nm was observed in assay II after one month of immersion. The peak representing [FeIII(1,3-(SO3)2Cat)2]5− in assay V disappeared after one month of immersion, which indicates the FeIII was separated from the [FeIII(1,3-(SO3)2Cat)2]5− complex and formed corrosion scale.
The slightly yellow solution that appeared in assay VI after two weeks could be ascribed to the oxidation of deprotonated Tiron to the corresponding quinone by traces of air. No metal leaching was observed in assay VI, but the peak at 465 nm after one month of immersion represents the formation of [NiII(1,3-(SO3)2Cat)2]6−,57 and the peak at 417 nm represents the formation of iron(II) mono-Tironate [FeII(1,3-(SO3)2Cat)]2−,56 which suggests corrosion has occurred. The occurrence of corrosion can be further confirmed by the XPS spectrum of assay VI, in which no Fe0 can be observed, and the corrosive extent of assays II and III is similar according to their XPS spectra and XRD patterns. Therefore, the consumption of dissolved O2 in the in situ generation of H2O2 is a key factor for the anti-corrosion performance of this formulation.
Conclusions
MnII (50.0 μM), in the presence of Tiron (1.50 mM) form a very efficient and robust catalytic system for the removal of dissolved O2 from aqueous solution and the in situ generation of H2O2 in the presence of NH2OH as a reducing substrate. The chemical formulation described here has the potential not only to prevent corrosion in central heating/hot water systems and cooling waters by the efficient removal of dissolved O2 but also to kill bacteria such as Legionella pneumophila and address two major global environmental and health issues. The role of Tiron as a specific chelator of Mn(II) is interesting in increasing the efficiency of dissolved O2 removal from aqueous solutions and in the rapid formation of H2O2 through stabilising the substrates at the catalytic centre and facilitating efficient electron- and proton-transfer between the substrate molecules in this enzyme-like system. As a redox active, non-innocent, ligand Tiron forms an electron-sink to facilitate multi-electron transfer with 1st row transition metals, in this case manganese.58,59 The antibacterial effect of this formulation is mainly attributed to the addition of NH2OH, while the in situ generated H2O2 and the consumption of dissolved O2 provides an additional inhibitory effect. The results presented here show that this formulation can effectively inhibit corrosion by consuming dissolved O2 under near neutral and basic conditions. This formulation is not applicable as an anti-corrosion formulation under acidic conditions since protons and the in situ generated H2O2 can facilitate the leaching of Fe2+ through Fenton reactions. Carbonate buffer is not suitable for this formulation due to the formation of high-valent manganese complex that promotes corrosion. The ability of the in situ formed [MnII(1,3-(SO3)2Cat)2]6− pre-catalyst to efficiently remove dissolved O2 could have applications for some circulated water systems such as central heating and cooling waters and may also help extend the service life of metal pipe lines. This formulation can also be used where O2-free conditions are required with the use of in situ generated H2O2 under anaerobic conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ye Cao (201907000136), Yue Xu (201908060367) and Qi Li (202006020019) hold China Scholarship Council Studentship (CSC) Awards at Queen Mary University of London. The authors would like to thank the Nuffield Foundation for providing a research placement bursary to Syeda K. Imaand. We would also wish to thank Anouk van Beurden, Martyna Dackiewicz, Zainul-Abedin Natha and Syeda K. Imaand for generating some of the preliminary data.Notes and references
- G. Zhang, B. Li, J. Liu, M. Luan, L. Yue, X.-T. Jiang, K. Yu and Y. Guan, Microbiome, 2018, 6, 222 CrossRef PubMed.
- Y. Cui, S. Liu, K. Smith, H. Hu, F. Tang, Y. Li and K. Yu, J. Environ. Sci., 2016, 48, 79–91 CrossRef CAS PubMed.
- I. Annus, A. Vassiljev, N. Kändler and K. Kaur, Journal of Water Supply: Research and Technology-Aqua, 2019, 69, 201–209 CrossRef.
- A. Saatchi, C. Dehghanian and M. J. M. P. Esfandiari, Mater. Perform., 2003, 42, 20–24 CAS.
- M. K. DeSantis, S. Triantafyllidou, M. R. Schock and D. A. Lytle, Environ. Sci. Technol., 2018, 52, 3365–3374 CrossRef CAS.
- C.-Y. Peng, G. V. Korshin, R. L. Valentine, A. S. Hill, M. J. Friedman and S. H. Reiber, Water Res., 2010, 44, 4570–4580 CrossRef CAS.
- L. Ma, J. Wang, D. Zhang, Y. Huang, L. Huang, P. Wang, H. Qian, X. Li, H. A. Terryn and J. M. C. Mol, Chem. Eng. J., 2021, 404, 127118 CrossRef CAS.
- F. Varela, M. Y. J. Tan and M. Forsyth, Electrochim. Acta, 2015, 186, 377–390 CrossRef CAS.
- Y. Qiang, H. Li and X. Lan, J. Mater. Sci. Technol., 2020, 52, 63–71 CrossRef.
- H. Tamura, Corros. Sci., 2008, 50, 1872–1883 CrossRef CAS.
- J. Jin, G. Wu, Z. Zhang and Y. Guan, Bioresour. Technol., 2014, 165, 162–165 CrossRef CAS.
- G. T. Vladisavljević, Sep. Purif. Technol., 1999, 17, 1–10 CrossRef.
- A. K. Pabby and A. M. Sastre, J. Membr. Sci., 2013, 430, 263–303 CrossRef CAS.
- F. J. C. Roe, G. A. Grant and D. M. Millican, Nature, 1967, 216, 375–376 CrossRef CAS.
- R. Lebeuf, Y. Zhu, V. Nardello-Rataj, J.-P. Lallier and J.-M. Aubry, Green Chem., 2012, 14, 825–831 RSC.
- L. Valgimigli, R. Amorati, M. G. Fumo, G. A. DiLabio, G. F. Pedulli, K. U. Ingold and D. A. Pratt, J. Org. Chem., 2008, 73, 1830–1841 CrossRef CAS PubMed.
- R. Lebeuf, V. Nardello-Rataj and J.-M. Aubry, Chem. Commun., 2014, 50, 866–868 RSC.
- R. Lebeuf, V. Nardello-Rataj and J.-M. Aubry, Adv. Synth. Catal., 2017, 359, 268–278 CrossRef CAS.
- R. Lebeuf, V. Nardello-Rataj and J.-M. Aubry, Top. Catal., 2013, 56, 933–938 CrossRef CAS.
- J. Lee, S.-M. Baek, C. Boo, A. Son, H. Jung, S. S. Park and S. W. Hong, J. Cleaner Prod., 2020, 277, 124049 CrossRef CAS.
- D. F. Evans and T. S. Sheriff, J. Chem. Soc., Chem. Commun., 1985, 1407–1408 RSC.
- C. Dacarro, A. M. Picco, P. Grisoli and M. Rodolfi, J. Appl. Microbiol., 2003, 95, 904–912 CrossRef CAS.
- Z. Pan, L. Mølhave and S. K. Kjaergaard, Indoor Air, 2000, 10, 237–245 CrossRef CAS PubMed.
- G. Delory and E. J. B. J. King, Biochem. J., 1945, 39, 245 CrossRef CAS PubMed.
- N. K. Tripathi, A. Shrivastva, K. C. Biswal and P. V. L. Rao, Ind. Biotechnol., 2009, 5, 179–183 CrossRef CAS.
- T. S. Sheriff, S. Miah and K. L. Kuok, RSC Adv., 2014, 4, 35116–35123 RSC.
- T. S. Sheriff, J. Chem. Soc., Dalton Trans., 1992, 1051–1058 RSC.
- M. Triki, S. Contreras and F. Medina, J. Sol-Gel Sci. Technol., 2014, 71, 96–101 CrossRef CAS.
- M. S. Yalfani, S. Contreras, F. Medina and J. Sueiras, Appl. Catal., B, 2009, 89, 519–526 CrossRef CAS.
- T. S. Sheriff, P. Carr and B. Piggott, Inorg. Chim. Acta, 2003, 348, 115–122 CrossRef CAS.
- G. A. L'Heureux and A. E. Martell, J. Inorg. Nucl. Chem., 1966, 28, 481–491 CrossRef.
- S. Scheiner and M. Čuma, J. Am. Chem. Soc., 1996, 118, 1511–1521 CrossRef CAS.
- A. K. Colter, A. G. Parsons and K. Foohey, Can. J. Chem., 1985, 63, 2237–2240 CrossRef CAS.
- S. L. Hooe, A. L. Rheingold and C. W. Machan, J. Am. Chem. Soc., 2018, 140, 3232–3241 CrossRef CAS.
- J. Mukherjee, S. Majumdar and T. Scheper, Appl. Microbiol. Biotechnol., 2000, 53, 180–184 CrossRef CAS PubMed.
- X. Xu, D. Chen, Z. Yi, M. Jiang, L. Wang, Z. Zhou, X. Fan, Y. Wang and D. Hui, Langmuir, 2013, 29, 5573–5580 CrossRef CAS PubMed.
- R. H. W. Lam, M.-C. Kim and T. Thorsen, Anal. Chem., 2009, 81, 5918–5924 CrossRef CAS PubMed.
- A. Akbal and H. H. Balik, Pol. J. Environ. Stud., 2013, 22, 1589–1594 Search PubMed.
- M. Sobota Jason, M. Gu and A. Imlay James, J. Bacteriol., 2014, 196, 1980–1991 CrossRef CAS PubMed.
- A. Anjem and J. A. Imlay, J. Biol. Chem., 2012, 287, 15544–15556 CrossRef CAS PubMed.
- A. Sen and J. A. Imlay, Front. Immunol., 2021, 12, 667343 CrossRef CAS PubMed.
- Y. Cao and T. S. Sheriff, Chemosphere, 2022, 286, 131792 CrossRef CAS PubMed.
- S. S. Mohammadi, A. Pol, T. van Alen, M. S. M. Jetten and H. J. M. Op den Camp, Front. Microbiol., 2017, 8, 1901 CrossRef PubMed.
- Y. Matsuoka and H. Kurata, Biotechnol. Biofuels, 2017, 10, 183–198 CrossRef PubMed.
- T. Misawa, K. Hashimoto and S. Shimodaira, Corros. Sci., 1974, 14, 131–149 CrossRef CAS.
- D. Neff, P. Dillmann, L. Bellot-Gurlet and G. Beranger, Corros. Sci., 2005, 47, 515–535 CrossRef CAS.
- J. B. Memet, P. Girault, R. Sabot, C. Compère and C. Deslouis, Electrochim. Acta, 2002, 47, 1043–1053 CrossRef CAS.
- Y. H. Huang and T. C. Zhang, Water Res., 2005, 39, 1751–1760 CrossRef CAS PubMed.
- T. Yamashita and P. Hayes, J. Electron Spectrosc. Relat. Phenom., 2006, 152, 6–11 CrossRef CAS.
- D. Pally, P. Le Bescop, M. L. Schlegel, F. Miserque, L. Chomat, D. Neff and V. L’Hostis, Corros. Sci., 2020, 170, 108650 CrossRef CAS.
- Y. Wu, S. Zhou, F. Qin, K. Zheng and X. Ye, J. Hazard. Mater., 2010, 179, 533–539 CrossRef CAS PubMed.
- L. Chu, J. Wang, J. Dong, H. Liu and X. Sun, Chemosphere, 2012, 86, 409–414 CrossRef CAS PubMed.
- R. Ling, J. P. Chen, J. Shao and M. Reinhard, Water Res., 2018, 134, 44–53 CrossRef CAS.
- S. A. M. Refaey, S. S. Abd El-Rehim, F. Taha, M. B. Saleh and R. A. Ahmed, Appl. Surf. Sci., 2000, 158, 190–196 CrossRef CAS.
- L. J. Simpson and C. A. Melendres, J. Electrochem. Soc., 1996, 143, 2146–2152 CrossRef CAS.
- M. J. Sever and J. J. Wilker, Dalton Trans., 2004, 1061–1072 RSC.
- S. Sandhu, K. Jn and J. Singh, J. Indian Chem. Soc., 1976, 53, 114–117 CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- W. Kaim, Dalton Trans., 2019, 48, 8521–8529 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj03806d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |