
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShort and efficient synthesis of alkylresorcinols: a route for the preparation of cannabinoids†
Rosaria
Villano
 *a,
Hannah
Straker
b and
Vincenzo
Di Marzo
ac
*a,
Hannah
Straker
b and
Vincenzo
Di Marzo
ac
aIstituto di Chimica Biomolecolare, CNR, Via Campi Flegrei 34, 80078 Pozzuoli, Napoli, Italy. E-mail: rosaria.villano@icb.cnr.it
bGW Pharma part of Jazz Pharmaceuticals, Kent Science Park, Sittingbourne, Kent ME9 8AG, UK
cCanada Excellence Research Chair on the Microbiome-Endocannabinoidome Axis in Metabolic Health, Faculty of Medicine and Faculty of Agricultural and Food Sciences, Centre de Recherche de l’Institut de Cardiologie et Pneumologie de l’Université et Institut sur la Nutrition et les Aliments Fonctionnels, Centre NUTRISS, Université Laval, QC G1V 4G5, Quebec City, Canada
First published on 14th October 2022
Abstract
Alkylresorcinols (ARs) are phenolic lipids present in several plants and, from a synthetic point of view, are also versatile key intermediates for the production of many biologically active molecules. In this work, we describe a general and efficient method for the preparation of ARs, also in deuterated form. Finally, this methodology was conveniently exploited for a very rapid approach to synthesize cannabidiol (CBD) and cannabidivarin (CBDV), two non-psychotropic cannabinoids with a bicyclic scaffold present in Cannabis sativa.
Introduction
Alkylresorcinols (ARs) or 1,3-dihydroxy-5-n-alkylbenzenes are phenolic lipids present in several plants, bacteria and marine sponges.1 Some of these molecules, with alkyl chains in the range of C15 to C25, are found in whole grain and bran products and consequently they are used as objective dietary biomarkers that reflect the intake of certain foods.2 Others, with alkyl chains of different length, have been tested for various in vitro bioactivities (colon cancer cell growth inhibition, antioxidant activity, lipase activity inhibition, antibacterial activity, etc.)2,3 with interesting results and in fact, ARs can be used as adjuvants for antimicrobial and anticancer treatment.3bARs, in addition to being target products, are also very common structural subunits in natural and pharmaceutically active products. As such, they are extensively used in organic chemistry as versatile starting materials for the synthesis of a broad range of complex molecular scaffolds: coumarins,4 2,8-dioxabicyclo[3.3.1]nonanes,5 isoflavones,6 benzopyrans7 and cannabinoids8 (Scheme 1).
 | ||
| Scheme 1 Synthesis of complex molecular scaffolds starting from ARs 1. | ||
In the light of these considerations, the development of effective and versatile protocols for their synthesis is highly desirable. In this context, the design of practical synthetic strategies for the production of several structurally different ARs using a minimum number of steps and readily available inexpensive starting materials, as well as the elaboration of methodologies that can also be exploited for the synthesis of deuterated analogues, is a fascinating challenge.
Generally, traditional approaches for the synthesis of ARs involve Grignard and related reactions, palladium-catalysed Suzuki coupling, reductive alkylation followed by oxidative decarboxylation or Wittig reaction (Scheme 2).1,9–13 Some of these strategies are characterized by a rather high number of steps that inevitably lead to a reduction in the total yield of the process, expensive reagents, highly reactive organometallic species which require an accurate control of experimental parameters and very dry conditions. A further disadvantage is that only some of such strategies have also been exploited for the synthesis of the corresponding deuterated analogues (above all with short saturated alkyl chain).14–16
 | ||
| Scheme 2 Some synthetic strategies for the production of ARs reported in the literature. | ||
We report here a simple methodology for the preparation of ARs, as such or chemoselectively deuterated, in high yields.
Finally, in order to explore further the synthetic utility of this strategy, ARs produced through this optimized procedure were used as starting materials to synthesize two non-psychotropic bicyclic cannabinoids17,18 (CBD and CBDV) and their deuterium labelled analogues via conventional terpenylation. The elaboration of new efficient synthetic procedures for the production of cannabinoids, in order to produce adequate quantities of products for clinical uses, is strongly desirable for their promising therapeutic potential (migraine and headaches, glaucoma, convulsions, etc.).19–22 In particular, the development of protocols potentially applicable to automated processes (flow synthesis) represents a future challenge. In this regard, the first automation of the terpenylation step23 of the synthesis of CBD and CBDV for possible large-scale industrial applications opens up new perspectives and renders the development of simple and efficient methodologies for the production of ARs even more important. In addition, having versatile methodologies that can also be used for the synthesis of deuterated ARs and, consequently, deuterated cannabinoids suitable as internal standards for mass spectrometric analyses, or as starting materials for the production of deuterated metabolites for the study of cannabinoid metabolism and pharmacokinetics in animals and humans, is also important.
Results and discussion
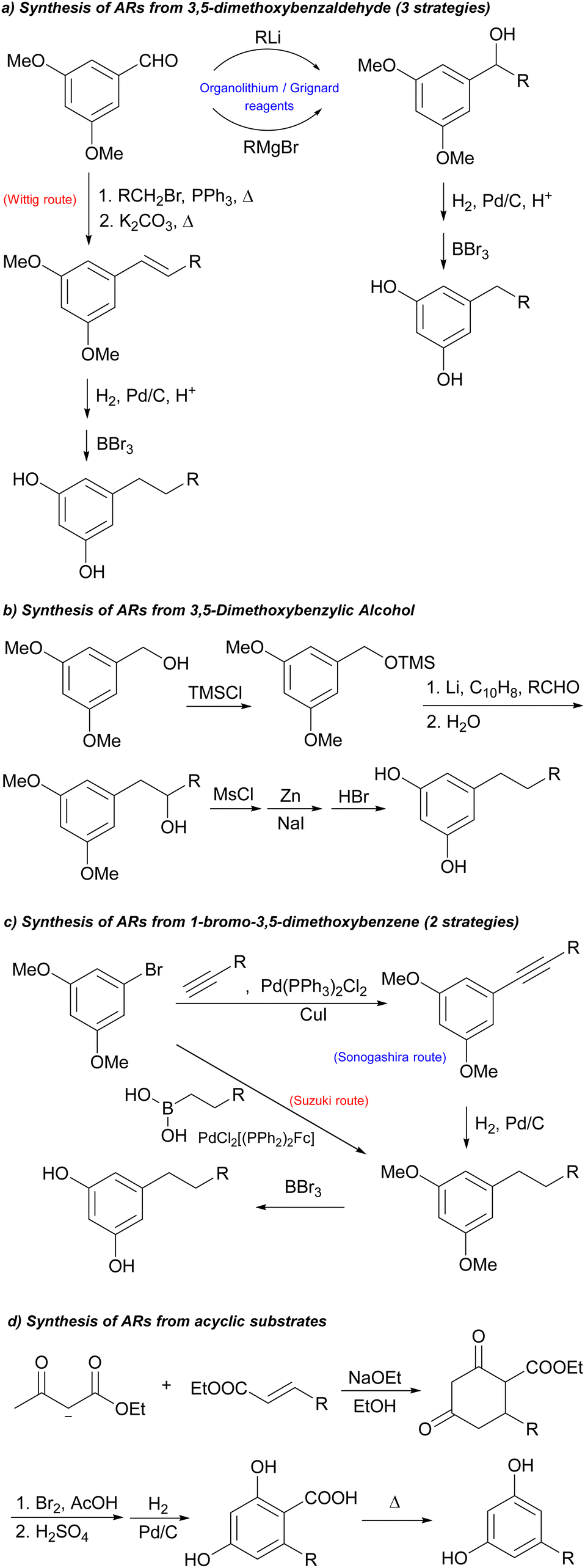
As delineated in Scheme 3, our strategy for the synthesis of ARs 1 started from the reaction between the commercially available 1-ethynyl-3,5-dimethoxybenzene 2 and an appropriate alkyl iodide 3 in the presence of LDA. This methodology proved to be effective with three different alkyl iodides (also the deuterated 3c), giving only products 4 in quantitative yields and without the formation of side-products, so the crude product was used without any further purification. For short chain alkyl halides, LDA was indispensable to obtain the desired product. Alternative bases such as nBuLi24 or KOH in DMSO25 also gave rise to competitive alkylation of the aromatic ring with production of complex reaction mixtures that were difficult to separate. | ||
| Scheme 3 Synthetic sequence for the production of ARs. | ||
The following Pd/C-catalysed reduction of the triple bond in 4 with H2 in H2O/THF or alternatively in EtOH provided the product 5 with the suitable saturated side chain (C3 in 5a and 5c or C5 in 5b) and its subsequent demethylation16 by BBr3 in CH2Cl2 gave the ARs 1a–c.
Therefore, deuterated ARs could be prepared with the same synthetic strategy in Scheme 3 by using deuterated alkyl iodides 3, but also in a very convenient and economic way. The triple bond of the product 4 was not only able to be easily and selectively hydrogenated but also deuterated thus allowing the introduction of four deuterium atoms in the alkyl chain. This approach is particularly useful and convenient when the appropriate deuterated alkyl iodide is not commercially available or is too expensive.
In fact, when the Pd/C-catalysed reduction of the triple bond with H2 was realized in D2O/THF instead of H2O/THF,26,27 the complete deuteration of the triple bond afforded the deuterated products 5d and 5e in quantitative yields (Scheme 4). Therefore, the Pd/C-catalysed H2–D2 exchange reaction using H2–D2O provided an efficient, cheap and environmentally friendly way for the in situ preparation of D2. Gratifyingly, these reactions were efficient, with >95% deuterium incorporation,28 no competitive deuteration of the aromatic ring observed, and a site-selective incorporation of deuterium on the alkyl chain achieved.
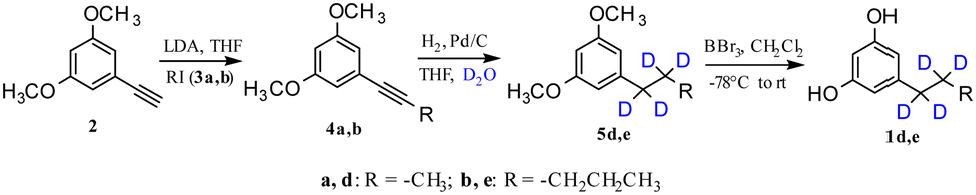 | ||
| Scheme 4 Synthetic sequence for the production of deuterated ARs via deuteration of the triple bond. | ||
All reactions of this multistep synthetic sequence (Schemes 3 and 4) were cleaned and characterized by very simple workups; ARs 1a–e (deuterated or not) were isolated in high yields (≥ 87% overall yield from 2, higher than those reported in the literature using different synthetic strategies23) over only 3 steps without isolation of the intermediate products 4 and 5, thus allowing a beneficial reduction in the amount of used organic solvents.
Importantly, the methodology is very versatile and it could be potentially used to prepare new ARs with different length or completely deuterated side chains, simply by appropriately selecting the alkyl halide 3 for the alkylation reaction (first step) and the solvent for the triple bond reduction (second step). Finally, this protocol was readily scalable (up to 10 mmol) without substantial change in efficiency.
To further explore the synthetic value of this strategy, we applied the methodology described here to prepare two bicyclic cannabinoids (CBD and CBDV) and their deuterated analogues.
As depicted in Scheme 5, all ARs 1a–e were condensed with commercially available (1S,4R)-4-isopropenyl-1-methyl-2-cyclohexen-1-ol 6 in the presence of 33 mol% wet pTSA to afford the trans bicyclic cannabinoids as a mixture of normal and abnormal scaffolds.8 The products 7 (up to 26% yield) and 8 (up to 38% yield) were isolated after a chromatographic column and, importantly, under these experimental conditions, no tricyclic structure was produced. The yields of this last reaction of terpenylation were comparable with those reported in the literature.8
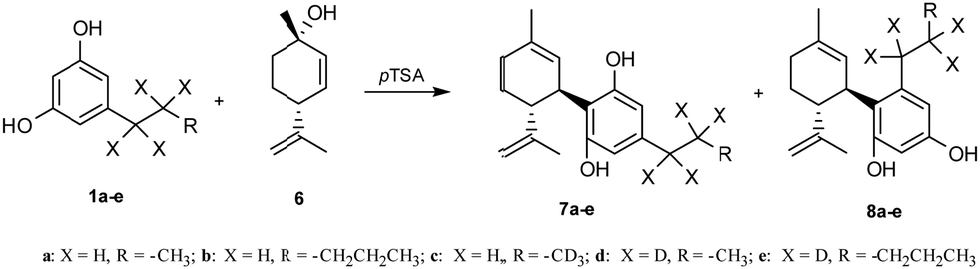 | ||
| Scheme 5 Synthesis of bicyclic cannabinoids (normal and abnormal scaffolds) via terpenylation. | ||
The coupling of ARs 1b and 1e with 6 afforded CBD 7b and its deuterated analogue 7e, respectively, as confirmed by the 1H-NMR spectra. The 1H NMR spectra of botanical purified CBD supplied by GW Research Ltd, now a part of Jazz Pharmaceuticals, Inc., and synthetic CBD 7b matched perfectly (Fig. 1, blue vs. red) confirming that the structure and the relative configuration of the stereogenic centers were the same (trans). The absolute configuration of natural CBD was elucidated by Mechoulam as R at both C-3 and C-4.29 Also, in the synthetic CBD 7b the absolute configuration at C-4 was R because it was derived from the chiral building block 6 (chiral pool approach). Consequently, the configuration at C-3 was also assigned as R, as in botanical CBD. In parallel, the spectrum of 7e, while confirming all the previous considerations, shows loss of signals at 2.4–2.5 ppm and 1.6 ppm due to the deuterium contributions (Fig. 1, green).
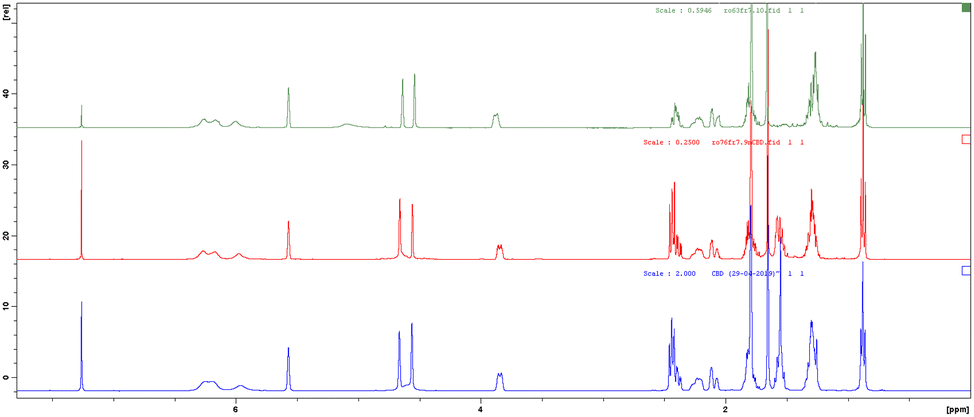 | ||
| Fig. 1 1H NMR spectra of pure botanical CBD supplied by GW Research Ltd, now a part of Jazz Pharmaceuticals, Inc. (blue), synthetic CBD 7b (red) and its deuterated analogue 7e (green). | ||
A similar NMR comparison was also realized for pure botanical CBDV supplied by GW Research Ltd, now a part of Jazz Pharmaceuticals, Inc., (Fig. 2, blue), synthetic CBDV 7a (Fig. 2, red) and its deuterated analogues 7c and 7d (Fig. 2, green and purple respectively) with similar results to those highlighted for CBD.
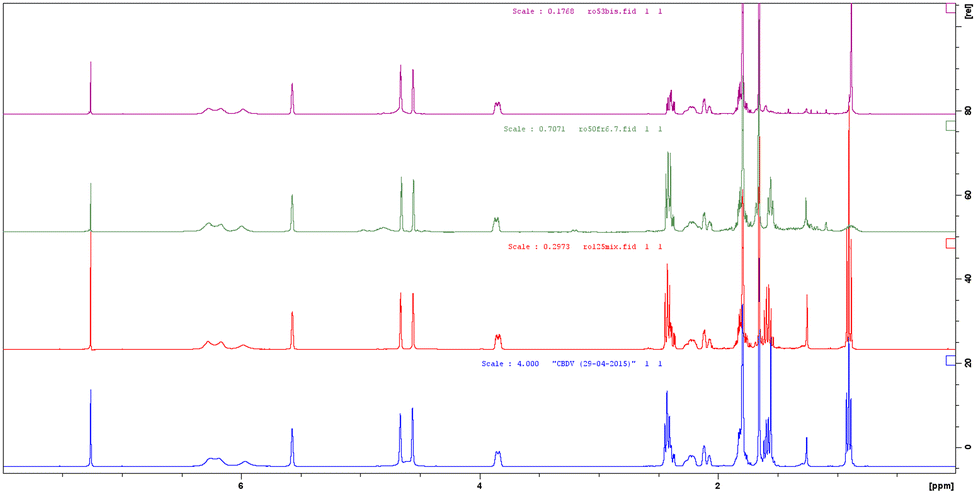 | ||
| Fig. 2 1H NMR spectra of pure botanical CBDV supplied by GW Research Ltd, now a part of Jazz Pharmaceuticals, Inc. (blue), synthetic CBDV 7a (red) and its deuterated analogues 7c (green) and 7d (purple). | ||
Conclusions
In summary, the methodology described in this paper offers an easy and efficient approach to the synthesis of an interesting class of molecules known as alkylresorcinols (ARs). This methodology is general and versatile, and suitable for the construction of a library of ARs and their deuterated analogues in only 3 steps. The deuterium labelled analogues can easily be synthesized using a deuterated building block (alkyl iodide) but also by the complete deuteration of the triple bond via Pd/C-catalysed exchange reaction between H2 and D2O.Key advantages of this synthetic sequence are that all reactions led to the corresponding products with high yields, without the formation of side-products and requiring only column chromatographic purification of the final AR, with a reduction in the amount of solvent and energy needed for separation and purification.
All these features make this strategy particularly useful for the production of ARs on a medium scale, allowing them to be used as starting materials in the synthesis of complex molecular scaffolds (as exemplified here by the application of the method to two bicyclic cannabinoids and their deuterium labelled analogues), under batch but also, in perspective, in flow chemistry conditions for possible large-scale industrial applications.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank GW Research Ltd, now a part of Jazz Pharmaceuticals, Inc., for providing us the botanically derived highly-purified CBD and CBDV and for financial support. The authors gratefully acknowledge Dr Emanuela G. Azara (CNR-ICB, Sassari) for MS analyses.Notes and references
- A. Kozubek and J. H. P. Tyman, Resorcinolic lipids, the natural non-isorenoid phenolic amphiphiles and their biological activity, Chem. Rev., 1999, 99, 1–26 CrossRef CAS PubMed and references therein.
- (a) R. Landberg, M. Marklund, A. Kamal-Eldin and P. Aman, An update on alkylresorcinols – Occurrence, bioavailability, bioactivity and utility as biomarkers, J. Funct. Foods, 2014, 7, 77–89 CrossRef CAS; (b) A. B. Ross, Present Status and Perspectives on the Use of Alkylresorcinols as Biomarkers of Wholegrain Wheat and Rye Intake, J. Nutr. Metab., 2012, 1–12 Search PubMed.
- (a) J. H. Kraal, A. A. Hussain, S. B. Gregorio and E. Akaho, Exposure time and effect of hexylresorcinols on bacterial aggregates, J. Dent. Res., 1979, 58, 2125–2131 CrossRef CAS PubMed; (b) A. A. Zabolotneva, O. P. Shatova, A. A. Sadova, A. V. Shestopalov and S. A. Roumiantsev, An Overview of Alkylresorcinols Biological Properties and Effects, J. Nutr. Metab., 2022, 4667607 CAS.
- J. M. Timonen, R. M. Nieminen, O. Sareila, A. Goulas, L. J. Moilanen, M. Haukka, P. Vainiotalo, E. Moilanen and P. H. Aulaskari, Synthesis and anti-infammatory effects of a series of novel 7-hydroxycoumarin derivatives, Eur. J. Med. Chem., 2011, 46, 3845–3850 CrossRef CAS PubMed.
- Y. Zhu, Z. Yao and F. Xu, Cationic-lanthanide-complex-catalysed reaction of 2-hydroxychalcones with naphthols: Facile synthesis of 2,8-dioxabicyclo[3.3.1]nonanes, Tetrahedron, 2018, 74, 4211–4219 CrossRef CAS.
- H. Singh and R. Pratap, A convenient one-pot synthesis of 7-hydroxy-isoflavones from resorcinol with substituted phenylacetic acids, Tetrahedron Lett., 2006, 47, 8161–8163 CrossRef CAS.
- S. M. Radwan, E. A. Bakhite and A. M. K. El-Dean, Synthesis and some reactions of new benzo[b]pyran derivatives, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 101, 207–211 CrossRef CAS.
- R. K. Razdan, The total synthesis of cannabinoids, in The total synthesis of Natural Products, ed. J. ApSimon, John Wiley & Sons, 1981, ch. 2, vol. 4, pp. 185–262 Search PubMed.
- J. W. Huffman, J. Liddle, S. G. Duncan, S. Yu, B. R. Martin and J. L. Wiley, Synthesis and pharmacology of the isomeric methylheptyl-Δ8-tetrahydrocannabinols, Bioorg. Med. Chem., 1998, 6, 2383–2396 CrossRef CAS PubMed.
- (a) R. S. Marmor, An improved synthesis of 5-alkylresorcinols, J. Org. Chem., 1972, 37, 2901–2904 CrossRef CAS; (b) M. Sisa, M. Dvorakova and T. Vanek, Concise access to primin, miconidin and related natural resorcinols, Tetrahedron, 2017, 73, 5297–5301 CrossRef CAS.
- Y. Zhu, D. N. Soroka and S. Sang, Synthesis and inhibitory activities against colon cancer cell growth and proteasome of alkylresorcinols, J. Agric. Food Chem., 2012, 60, 8624–8631 CrossRef CAS PubMed.
- E. Alonso, D. J. Ramon and M. Yus, Simple synthesis of 5-substituted resorcinols: a rivisited family of interesting bioactive molecules, J. Org. Chem., 1997, 62, 417–421 CrossRef CAS PubMed.
- A. A. Durrani and J. H. P. Tyman, Long chain phenols. Part 16. A. Novel synthesis of homologous orsellinic acids and their methyl esters, J. Chem. Soc. Perkin Trans. I, 1980, 1658–1666 RSC.
- A. R. Banijamali, N. Abou-Taleb, C. J. Van der Schyf, A. Charalambous and A. Makriyannis, Synthesis of deuterium labeled cannabinoids, J. Label. Comp. Radiopharm., 1987, 25(1), 73–82 CrossRef.
- S. P. Nikas, G. A. Thakur and A. Maktiyannis, Synthesis of side chain specifically deuterated (-)-Δ9-tetrahydrocannabinols, J. Labelled Compd. Radiopharm., 2002, 45, 1065–1076 CrossRef CAS.
- S. P. Nikas, G. A. Thakur and A. Maktiyannis, Regiospecifically deuterated (-)-Δ9-tetrahydrocannabivarins, J. Chem. Soc., Perkin Trans. 1, 2002, 2544–2548 RSC.
- M. A. ElSohly and D. Slade, Chemical Constituents of Marijuana: The Complex Mixture of Natural Cannabinoids, Life Sci., 2005, 78(5), 539–548 CrossRef CAS PubMed.
- M. A. ElSohly and W. Gul, Constituents of Cannabis sativa, in Handbook of Cannabis, ed. R. G. Pertwee, Oxford University Press, New York, NY, 2014, pp. 3–22 Search PubMed.
- A. Ligresti, L. De Petrocellis and V. Di Marzo, From phytocannabinoids to cannabinoid receptors and endocannabinoids: Pleiotropic physiological and pathological roles through complex pharmacology, Physiol. Rev., 2016, 96, 1593–1659 CrossRef CAS PubMed.
- D. Friedman and O. Devinsky, Cannabinoids in the treatment of Epilepsy, N. Engl. J. Med., 2015, 373(11), 1048–1058 CrossRef CAS PubMed.
- L. Cristino, T. Bisogno and V. Di Marzo, Cannabinoids and the expanded endocannabinoid system in neurological disorders, Nat. Rev. Neurol., 2020, 16(1), 9–29 CrossRef PubMed and references therein.
- K. B. Walsh, A. E. McKinney and A. E. Holmes, Minor cannabinoids: biosynthesis, molecular pharmacology and potential therapeutic uses, Front. Pharmacol., 2021, 12, 777804 CrossRef CAS PubMed.
- E. Chiurchiù, S. Sampaolesi, P. Allegrini, D. Ciceri, R. Ballini and A. Palmieri, A novel and pratical continuous flow chemical synthesis of cannabidiol (CBD) and its CBDV and CBDB analogues, Eur. J. Org. Chem., 2021, 1286–1289 CrossRef.
- D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, Indole synthesis via Rhodium catalyzed oxidative coupling of acetanilides and internal alkynes, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef CAS PubMed.
- R. A. W. Johnstone, D. Tuli and M. E. Rose, A simple method for C-alkylation, J. Chem. Res., 1980, 9, 3593–3597 Search PubMed.
- H. Sajiki, T. Kurita, H. Esaki, F. Aoki, T. Maegawa and K. Hirota, Complete replacement of H2 by D2via Pd/C-catalyzed H/D exchange reaction, Org. Lett., 2004, 6, 3521–3523 CrossRef CAS PubMed.
- T. Kurita, F. Aoki, T. Mizumoto, T. Maejima, H. Esaki, T. Maegawa, Y. Monguchi and H. Sajiki, Facile and convenient method of deuterium gas generation using a Pd/C-catalyzed H2-D2 exchange reaction and its application to synthesis of deuterium-labeled compounds, Chem. – Eur. J., 2008, 14, 3371–3379 CrossRef CAS PubMed.
- Deuterium incorporation was determined by 1H NMR spectroscopy.
- Y. Gaoni and R. Mechoulam, The isolation and structure of Δ1-Tetrahydrocannabinol and other neutral cannabinoids from hashish, J. Am. Chem. Soc., 1971, 93, 217–224 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj03547b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |