
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of hyaluronic acid core–shell nanoparticles via simple microfluidic-assisted nanoprecipitation method for active tumor targeting†
Antonio
Fabozzi
a,
Francesca
Della Sala
b,
Mario
di Gennaro
bc and
Assunta
Borzacchiello
 *b
*b
aALTERGON ITALIA S.r.l., Zona Industriale ASI – 83040 Morra De Sanctis (AV), Italy
bInstitute for Polymers, Composites and Biomaterials, National Research Council, IPCB-CNR, Naples, Italy. E-mail: bassunta@unina.it
cDepartment of Environmental, Biological and Pharmaceutical Sciences and Technologies (DiSTABiF), University of Campania “L. Vanvitelli”, 81100 Caserta, Italy
First published on 22nd September 2022
Abstract
The control of the physicochemical properties of hyaluronic acid core–shell nanoparticles (HA-based NPs) is a suitable strategy to achieve the high reproducibility of biological assays. In particular, the active targeting efficiency of HA-based NPs is a key point for the cancer treatment. This opens a way for the employment of microfluidic technology to overcome the drawbacks related to the synthesis of NPs by means of traditional bulk methods such as lower drug encapsulation efficiency and consequently a lower cancer cell killing effect. For these reasons, blank and irinotecan-loaded HA-based NPs, without any chemical modifications, were synthesized by means of a simple and fast microfluidic method (MM) and compared with those synthesized by the bulk method (BM). In particular, their aggregation behavior and morphology were investigated by dynamic light scattering, ζ-potential, and transmission electron microscopy. The drug encapsulation efficiency and irinotecan kinetic release of irinotecan-loaded HA-based NPs synthesized by both MM and BM were evaluated. All NP formulations synthesized by MM present smaller size, narrower size distribution, controlled morphology, higher yield, and higher drug encapsulation efficiency than those synthesized by BM. Furthermore, the biological assays based on the in vitro biocompatibility and cell uptake using HS578T, human breast carcinoma cells, and L929 cells (healthy fibroblast) were investigated, respectively. In particular, biological assays showed higher cytotoxicity and a higher internalization for HA-based NPs synthesized by MM than those synthesized by BM. Thus, the synthesis of HA-based NPs, without any polymer chemical modifications, by means of new microfluidic technology is demonstrated to be a more effective approach for the cancer treatment.
Introduction
Polymer-based nanoparticles (NPs) have attracted great interest in the cancer therapy field due to their properties related to the capability to encapsulate a variety of anticancer drugs, control their release on tumor sites, and reduced systemic toxicity of cytotoxic agents.1–4 The enhanced permeability and retention (EPR) effect is one of the well-known strategies to promote NP accumulation in cells and tissues of solid tumors.2,5–8 Unfortunately, its efficacy and efficiency are limited by some drawbacks such as the possible failure of NP cell uptake, non-specific targeting,9 and the undesired release of encapsulated drugs before the internalization of NPs.10 To overcome these limitations, functionalized NPs with active targeting moieties, which are able to recognize and subsequently bind tumor cells, obtained by means of proper modifications of NPs surfaces such as nucleic acids,11 antibodies,12 and proteins, have been investigated.13–15 In this framework, hyaluronic acid (HA) has attracted significant attention for tumor-targeted delivery.16,17 This naturally anionic polysaccharide is composed of repeating disaccharide units of β-D-glucuronic acid and N-acetyl-D-glucosamine that specifically binds the CD44 receptor, which is overexpressed in many cancer cells.18 In the literature, a wide set of HA-based drug carriers, obtained by chemical conjugation, have been investigated for tumor targeting;17,19,20 however, chemical modifications on the HA backbone can modify its biological properties.21 Recently, core–shell HA/poly(lactic-co-glycolic acid) (PLGA) NPs composed of a hydrophobic PLGA core sealed by amphiphilic poloxamers (PF68 and PF127, even named PP) and HA shell have been developed by means of nanoprecipitation with subsequent solvent evaporation BM.22,23 Although there are several advantages of HA-based NPs fabricated by BM, some drawbacks such as rare batch-to-batch reproducibility, low drug loading, yield, and low control of the physicochemical properties of NPs exist.24 Moreover, the great variability of the physicochemical properties such as size and size distribution, which are fundamental parameters to achieve biological activity reproducibility, represents a significant obstacle to their applications.25 In the last decade, the microfluidic platform has gained a prominent role in the new synthesis of polymer-based NPs.22,26,27 In contrast to BM, MMs are widely employed in the polymer-based NPs synthetic field due to their properties such as rapid mass transfer, precise control, large reaction interfaces, and high mixing efficiency.28,29 In the microfluidic system, the nucleation and growth steps of NPs can be separated as a function of distance from the position where solution mixing takes place to achieve an accurate control of the NPs physicochemical properties, thus increasing the synthetic batch's reproducibility.30 For these reasons, MM allows to overcome some limitations for the production of NPs, compared to BM, such as industrial scale-up, high yield, high drug encapsulation efficiency, and both synthetic batch-to-batch and biological assays reproducibility.31 Over the past decade, the microfluidic method has enabled the synthesis of a wide set of core–shell micro and nanoparticles.32–34 Microspheres composed of PLGA/alginate core–shell were fabricated using capillary microfluidic devices for controlling Rifampicin release kinetics; the biocompatibility of the PLGA/alginate core–shell microspheres was tested on L929 mouse fibroblasts cell line and no cytotoxicity was observed.35 However, HA-decorated NPs have been used in the diagnostic field; Russo et al. synthesized PEG–Vinyl sulfonate (PEG VS)/thiolate chemically modified HA core–shell NPs by microfluidic-assisted nanoprecipitation process for the encapsulation of gadolinium diethylenetriamine penta-acetic (Gd-DTPA) for magnetic resonance imaging (MRI).36 Tammaro et al. developed a thiolated-HA/PEG-VS core–shell NPs by microfluidic-assisted nanoprecipitation process for the co-encapsulation of Gd-DTPA and ATTO488 simultaneously for MRI/Optical probe imaging.37 Nevertheless, although great efforts have been made based on the application of microfluidic technologies for the production of nanoparticles, the exploitation of microfluidic approaches to support the design of core–shell HA/PLGA NPs without any chemical modification and based on the spontaneous self-arrangement of polymers, FDA approved, for cancer treatment has not been developed. In this context, the present work shows the synthesis of irinotecan (IRI)-loaded HA-based core–shell NPs via microfluidic-assisted nanoprecipitation process, based on electrostatic interactions driven by lipophilicity gradient between polymers, and presents the investigation of the structural, dynamic, and biological properties of the resulting NPs, including cell uptake and cytotoxicity. The aggregation behavior and morphology of HA-based core–shell NPs synthesized by MM are studied at room temperature and compared with those NPs synthesized by BM, which are also investigated as a reference. Dynamic light scattering (DLS), ζ-potential, differential scanning calorimetry (DSC), and transmission electron microscopy (TEM) were used to investigate the aggregation behavior and morphologies of the NPs. Moreover, in vitro cytotoxicity and cell uptake were evaluated on HS578T, human breast carcinoma cells overexpressing CD44 receptor, and in healthy fibroblast L929 cells exposed to NPs. Thus, in the framework of the fast-growing field of polymer-based nanoparticle formulations science and technology, this work represents a valuable example in which a detailed investigation of the new synthetic microfluidic technology, compared to the traditional BM, gives a reliable basis to link the functional behavior of NPs-based formulations to their microstructure and to the fast understanding of their in vitro biological investigations.Materials and methods
HA with a weight-average molecular weight (MW) of 800 kDa was provided by Altergon (Italy). Resomer PLGA (RG504H, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, MW of 38–54 kDa) was purchased from Evonik (Germany). Poloxamers (PEOa-PPOb-PEOa), a group of amphiphilic triblock polymers, designed with variable numbers of oxyethylene (a) and oxypropylene (b) units as stabilizers, were employed. In this study, poloxamer F127 (a = 100 and b = 65) and F68 (a = 76 and b = 29), named PP, obtained from Lutrol (Basf, Germany). Irinotecan hydrochloride, named IRI, acetone, bidistilled water, Nile red (NR) (9-diethylamino-5-benzo[a]phenoxazinone), along with fluorescein isothiocyanate (FITC)-phalloidin and 4′,6-diamidino-2-phenylindole (DAPI), were purchased from Sigma Aldrich (USA). Phosphate buffer saline (PBS) tablets without calcium and magnesium were obtained from MP Biomedicals Inc. Penicillin and streptomycin (10000 U mL−1) from Invitrogen and Life Technologies (Carlsbad, CA) were employed. Trypsin and ethylenediaminetetraacetic acid (EDTA) were purchased from HiMedia (Mumbai, India). Fetal Bovine Serum (FBS) from Lonza (Basel, Switzerland) and Bovine insulin from Gibco EfficientFeed + Supplements (ThermoFisher Scientific, USA) were used.
:50, MW of 38–54 kDa) was purchased from Evonik (Germany). Poloxamers (PEOa-PPOb-PEOa), a group of amphiphilic triblock polymers, designed with variable numbers of oxyethylene (a) and oxypropylene (b) units as stabilizers, were employed. In this study, poloxamer F127 (a = 100 and b = 65) and F68 (a = 76 and b = 29), named PP, obtained from Lutrol (Basf, Germany). Irinotecan hydrochloride, named IRI, acetone, bidistilled water, Nile red (NR) (9-diethylamino-5-benzo[a]phenoxazinone), along with fluorescein isothiocyanate (FITC)-phalloidin and 4′,6-diamidino-2-phenylindole (DAPI), were purchased from Sigma Aldrich (USA). Phosphate buffer saline (PBS) tablets without calcium and magnesium were obtained from MP Biomedicals Inc. Penicillin and streptomycin (10000 U mL−1) from Invitrogen and Life Technologies (Carlsbad, CA) were employed. Trypsin and ethylenediaminetetraacetic acid (EDTA) were purchased from HiMedia (Mumbai, India). Fetal Bovine Serum (FBS) from Lonza (Basel, Switzerland) and Bovine insulin from Gibco EfficientFeed + Supplements (ThermoFisher Scientific, USA) were used.
Synthesis of nanoparticles by microfluidics and bulk method
PLGA/PP NPs were prepared by means of a microfluidic-assisted nanoprecipitation process. In brief, the OP PLGA/PP (1:0.3:0.3) powder was solubilized at 1.25% (w/w) in acetone and 10 μL of Nile Red (1 mg mL−1) was added; on the other hand, the WP was composed of pure bidistilled water, in which 3 mL of a mixture composed of PF68 and PF127, named PP, (0.03% w/w) was added. The same procedure described above was adopted to prepare HA/PLGA/PP NPs in which a second water phase (WP2) solution was composed of HA at 0.08% (w/w) and PP. Before use, the OP, WP, and WP2 solutions were filtered using a 0.45 μm hydrophobic/hydrophilic filter composed of polytetrafluoroethylene (PTFE). In the NPs synthetic MM, an Elveflow Microfluidic Machine equipped with an OB1 pressure controller working at pressures of 2 and 8 bar, and flow sensors working in the range of 0–5000 μL min−1 with an accuracy of 10 μL min−1 were employed. A hydrophilic flow focusing chip composed of 12 mixing stages was used for the fabrication of all NP systems. The geometry of each mixing stage was composed of a large channel of 125 μm × 350 μm depth × width), connected with eight smaller herringbone channels of 50 μm × 125 μm (depth × width). In particular, groups of four of these smaller channels were connected with two parallel channels and then finally recombined with a longer large channel. Briefly, 5 mL of OP, prepared at the same concentration described above, was placed into a 15 mL tube connected to a glass micromixer chip. In particular, the PLGA/PP solution was introduced in the internal channel pumped at 100 μL min−1; the same procedure was adopted to synthesize HA/PLGA/PP NPs (Fig. 1a). For the synthesis of HA/PLGA/PP NPs by MM, the same amount of PLGA, PP, NR, and HA as reported above was used. For the IRI-loaded PLGA/PP and HA/PLGA/PP NPs fabrication, a double step was applied (Fig. 1c). Briefly, the first step was based on the encapsulation of IRI in the OP; irinotecan hydrochloride (2.2 mg mL−1) was solubilized in water and introduced in the internal channel pumped at 100 μL min−1, while the OP was introduced in the external channel OP2. Subsequently, the OP2 was introduced in the internal channel and the WP was introduced in the external channel at the same flow-rate ratio of the PLGA/PP NPs. The same procedure for the IRI-loaded HA/PLGA/PP NPs was adopted. In the BM method, 5 mL of OP composed of PLGA/PP solution was placed into a syringe (23G 1 1/4”, ∅ 0.60 × 30 mm) and pumped by means of a syringe pump at 333 μL min−1 into the WP phase, placed into a beaker, and stirred at 750 rpm (Fig. 1b). The same procedure was adopted to synthesize HA/PLGA/PP NPs. For the IRI-loaded PLGA/PP and HA/PLGA/PP NP synthesis, the IRI was solubilized in water (2.2 mg mL−1) and mixed in the OP by an ultrasonic bath (25 °C, 15 min, 100% power, 59 kHz). Subsequently, the OP2 was pumped through the syringe pump at 333.3 μL min−1 into the WP (Fig. 1d) and the same procedure was adopted to synthesize IRI-loaded HA/PLGA/PP NPs. Afterward, for all the NPs formulations, the organic solvent was evaporated at 25 °C for 35 min by means of a rotary evaporator (Laborota 4010 digital, HEIDOLPH). The obtained NP suspension was washed three times by centrifugation (Hettich Zentrifugen, Germany; 13000 rpm, 15 min) and stored at −80 °C. The size, size distributions, and morphologies of all NP formulations were evaluated by means of dynamic light scattering (DLS), ζ-potential, TEM, and SEM techniques. In vitro cell uptake assays of NPs, synthesized by MM and BM, were performed on L929 and HS578T cell lines.
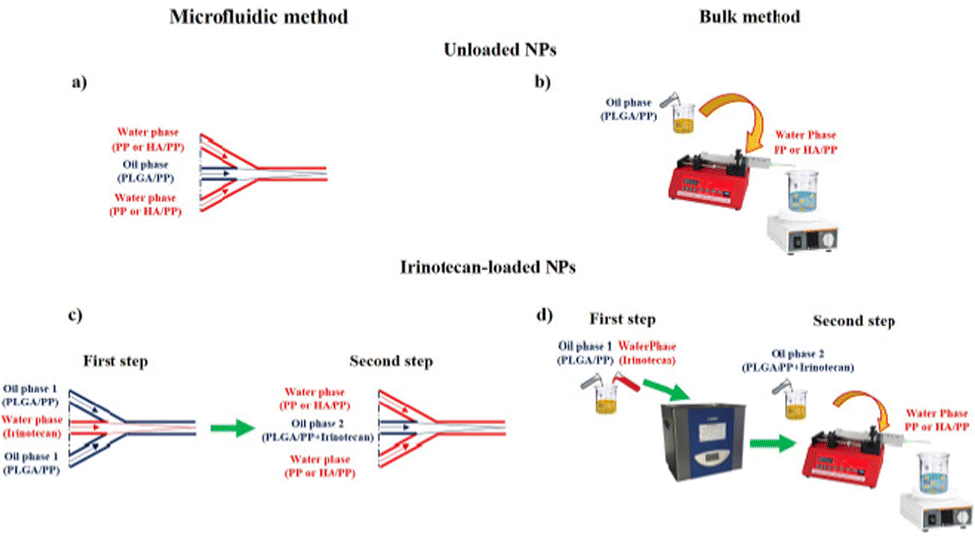 | ||
| Fig. 1 Schematic representation of NPs preparation by single step, (a) microfluidic-assisted nanoprecipitation of blank NPs, (b) BM of blank NPs and double step, (c) microfluidic-assisted nanoprecipitation of irinotecan-loaded NPs, (d) BM of irinotecan-loaded NPs. | ||
Nanoparticles characterization: morphology, size, size distribution, yield, and ζ potential
NPs morphology by means of TEM (FEI Tecnai G12 Spirit Twin) with emission source LaB6 (120 kV, spot size 1) using 400 mesh carbon-coated copper grids at room temperature (RT) was investigated. The carbon-coated copper grid was immersed in ultra-diluted NP suspensions and, after the drying phase, the grid was placed on a rod holder for TEM characterization. Three grids per NP suspension were prepared and a minimum of four micrographs per grid were acquired. Intensity-average hydrodynamic radius and ζ potentials of the NP were determined by means of DLS measurements with a Zetasizer Nano (Malvern Instruments, Malvern, UK). For particle size measurements, NPs were suspended in ultrapure water. To investigate the agglomeration dynamics in bidistilled water at 25 °C, NPs size measurements were acquired for ten days. NP yield was gravimetrically calculated after lyophilization for 24 h (Heto PowerDry PL6000 Freeze Dryer, Thermo Electron Corp., USA; −60 °C, 0.73 hPa). The results were averaged on at least ten measurements.Thermal analyses
The interactions between the polymers in the NP formulations were investigated by thermoanalytical test on PLGA, poloxamers, and HA powders and freeze-dried PLGA, PLGA/PP, and HA/PLGA/PP NPs synthesized by MM. The heat-related transitions of the polymers were detected by a differential scanning calorimeter (DSC discovery TA Instruments, USA) calibrated with pure indium standard. The samples were placed in aluminum pans, which are subsequently subjected to a double scan from −60 °C to 80 °C. In particular, the first scan was acquired to eliminate the thermal history of the samples. Measurements were performed under a nitrogen atmosphere, purged at a flow rate of 50.0 mL min−1, and the heating rate was 5 °C min−1 for all the samples.The heat developed during polymer crystallization and fusion (W g−1) was calculated from the DSC thermograms by integrating the exothermic/endothermic peaks, while the glass transition temperature (Tg) was obtained from thermogram inflection point.
Drug encapsulation efficiency and drug loading
Drug encapsulation efficiency (DEE) was determined by dissolving NPs powder (2 mg) in DMSO (1 mL). The NPs solution was sonicated for 1 h in a water bath at room temperature, 59 kHz, 100% power. IRI amount was quantified by spectrophotometric assay (Multilabel Counter, Victor X3, PerkinElmer) at 370 nm. The linearity of the response was verified on IRI solutions in DMSO (0.04–10 μg mL−1 concentration range; r2 > 0.99). Entrapped IRI percentage was calculated using the following equation. | (1) |
Drug loading (DL) percentage was evaluated using the following equation.38
 | (2) |
In vitro irinotecan kinetic release
The in vitro release curve of IRI from IRI-loaded NPs was determined as follows: 1 mg of the above freeze-dried IRI-loaded NPs was dispersed in an Eppendorf tube containing 1.5 mL of release medium (PBS, pH = 7.4), and then the tube was incubated at 30 rpm in a constant temperature stirrer at 37 °C. Sampling was carried out in three steps using the following procedure: (1) the Eppendorf tube was removed from the stirrer at the predetermined time points; (2) it was centrifuged (VWR, Microstar 17R, ThermoFisher Scientific, Germany) and analyzed through spectrophotometric assay (λ = 268 nm) to quantify the IRI content. Subsequently, the Eppendorf tube was filled with 1 mL of fresh release medium to keep the total amount of the release medium unvaried. The instrument response was linear over the 0.1–50 μg mL−1 concentration range (r2 > 0.99). The experiments were performed in triplicate. The experimental data were interpolated with a model that expresses the release as the sum of a diffusive contribution and of a dissolution contribution.39| Fdiss = Fdiss,∞kdisst0.5 | (3) |
| Fdiss =Fdiss,∞(1 − exp(−kdifft)) = (1 − Fdiss,∞)(1 − exp(−kdifft)) | (4) |
| F = Fdiss + Fdiss = Fdiss,∞kdisst0.5 + (1 − Fdiss,∞)(1 − exp(−kdifft)) | (5) |
Cell culture
Human breast carcinoma cell line (HS578T), kindly gifted by Dr Olga Zeni (IREA-CNR), were used as a model of cells naturally overexpressing CD44 receptor. L929 cells originating from Mouse C34/An connective tissue were obtained from Sigma-Aldrich, (USA) and were used as the control since they have a low degree of CD44 expression 30. Cells were grown in T-75 cell culture flask (Falcon, Italy) in DMEM cell culture medium supplemented with 10% fetal bovine serum and antibiotics (penicillin G sodium 100 U mL−1, streptomycin 100 μg mL−1) at 37 °C and 5% CO2. The medium used for HS578T cells was also enriched with 10 μL mL−1 bovine insulin. When confluent growth was reached, the cells were detached with 0.25% trypsin – EDTA solution and washed twice with PBS. The resulting cell suspensions were centrifuged (5 min, 1000 rpm; BRK55/10 Centrifuge by Centurion Scientific Ltd, UK), the supernatant separated, and the cells re-suspended in fresh culture medium. Viable cells were counted using the TC20 automated Cell Counter (Biorad, USA).Confocal microscopy
Confocal microscopy images were acquired after seeding 2 × 104 cells in 1 mL of medium on 35 mm-diameter Fluoro dish cell culture dish (World Precision Instruments, Inc). Cells were incubated for 24 h at 37 °C in a 5% CO2 atmosphere with a suspension of NPs in the cell culture medium (1 mg mL−1 for the different formulations synthesized by MM and 6.32 mg mL−1 IRI-loaded PLGA/PP, 6.99 mg mL−1 IRI-loaded HA/PLGA/PP synthesized by BM, according to the drug encapsulation efficiency, respectively). After 24 h, non-internalized NPs were removed by washing the samples twice with PBS, fixed with 10% formalin (Sigma-Aldrich) for 1 h, and permeabilized with 0.1% Triton X-100 for 3–5 min. Actin filaments were stained with FITC-phalloidin in PBS for 30 min at 25 °C. After two washes with PBS to remove the unbound phalloidin conjugate, the cell nuclei were stained with DAPI for 10 min at 37 °C. The samples were observed by a confocal microscope (Leica TCS SP8) using a 63× oil immersion objective. Images were acquired with a resolution of 1024 × 1024 pixels.In vitro cytotoxicity
To understand the in vitro cytotoxicity, cells were seeded at a density of 5 × 103 cells per mL on 96 wells (World Precision Instruments, Inc., Sarasota, FL, USA) in triplicate for each well cultured up to 24 h within the different NPs suspensions. Alamar blue assay (AB) was performed by adding AB reagent to the samples (at 10% v/v with respect to the medium) and incubated at 37 °C for 4 h. The absorbance of the samples was measured using a spectrophotometer plate reader (Multilabel Counter, 1420 Victor, PerkinElmer) at 570 nm and 600 nm. AB is an indicator dye incorporating an oxidation–reduction indicator that changes color in response to the chemical reduction in the growth medium, resulting from cell viability. Data are expressed as the percentage difference between the treated and control samples to evaluate the percentage of reduction (Reduction %), which is calculated using the following formula. | (6) |
Quantification of NP uptakes in L929 and HS578T cells
To evaluate the NP intracellular uptake, 2 × 104 cells were seeded in 1 mL of medium for each cell types in a 24 well plate. The NPs were dispersed in cell culture medium at a final concentration of 1 mg mL−1 of NPs, for PLGA/PP MM, HA/PLGA/PP MM, PLGA/PP BM, and HA/PLGA/PP BM, respectively. Cells were incubated with NP suspensions for 1, 3, and 24 h. After incubation, cells were rinsed two times with PBS in order to eliminate all non-internalized nanoparticles and then lysed with 0.1 mL of lysis buffer. Cell lysates were diluted with 0.4 mL of PBS and analyzed by a spectrofluorometer (Multilabel Counter, 1420 Victor, PerkinElmer), at 525–605 nm excitation wavelength. The fluorescence intensity of experimental points was interpolated with the calibration curve obtained using known concentrations of fluorescent NPs for each formulation (0–0.5 mg mL−1). The concentration of internalized NPs was normalized to the number of seeded cells to obtain the number of internalized NP per cell, taking into account the average NP diameter.Statistical analysis
Results are reported as the mean of atleast three replicas ± standard deviation (SD). Statistical analyses were performed using a one-way analysis of variance (ANOVA) and p values < 0.001 were considered statistically significant.Results and discussion
Synthesis and physicochemical characterization of HA-based nanoparticles by means of microfluidic and bulk methods
Blank HA/PLGA/PP, PLGA/PP, and IRI-loaded NPs were prepared through nanoprecipitation with subsequent solvent evaporation method by means of MM and BM used as references (see the Methods section). The nanoprecipitation methods directly produce an aqueous dispersion of the NPs, which are fundamental for biological applications. Briefly, for the MM, the microchip is composed of three inlets, one for the PLGA/PP solution and two for the H2O/PP solution, and one outlet for the self-assembled NPs (Fig. 1a). In particular, PLGA/PP solution (OP) was introduced in the internal channel, while the water phase (WP), composed of H2O/PP, was introduced in the external channel and the flow-rate ratio (i.e., internal flow/external flow) equal to 0.1 was set; the same procedure was adopted to synthesize HA/PLGA/PP NPs.For the IRI-loaded PLGA/PP and HA/PLGA/PP NPs fabrication, a double step was applied (Fig. 1c). Briefly, the first step was based on the encapsulation of IRI in the OP; irinotecan hydrochloride was solubilized in water and introduced in the internal channel while the OP was introduced in the external channel and the flow-rate ratio equal to 0.1 was set (OP2). Subsequently, OP2 was introduced in the internal channel and WP was introduced in the external channel at the same flow-rate ratio as the PLGA/PP NPs. The same procedure for the IRI-loaded HA/PLGA/PP NPs was adopted.
In the BM method, OP composed of PLGA/PP solution was placed into the syringe and pumped by means of a syringe pump into the WP phase under magnetic stirrer (Fig. 1b). The same procedure was adopted to synthesize HA/PLGA/PP NPs. For the IRI-loaded PLGA/PP and HA/PLGA/PP NP synthesis, the IRI was solubilized in water and mixed in the OP in an ultrasonic bath. Subsequently, the OP2 was pumped through the syringe pump into the WP (Fig. 1d) and the same procedure was adopted to synthesize IRI-loaded HA/PLGA/PP NPs.
Afterward, for all NPs formulations synthesized by both MM and BM, the organic solvent was evaporated overnight and then the obtained NP suspensions were purified by centrifugation.
The results of size and ζ potential measurements monitored at room temperature of each NPs formulation, synthesized by MM and BM, are summarized in Table 1.
| Formulation | PDIt=0 | d t=0 (nm) | d t=10 (nm) | ζ potentialt=0 (mV) | ζ potentialt=10 (mV) | Yield (%) |
|---|---|---|---|---|---|---|
| PLGA/PP MM | 0.05 ± 0.01 | 100.5 ± 2.1 | 105.1 ± 5.7 | −32.0 ± 0.64 | −34.1 ± 0.7 | 95.3 |
| IRI-loaded PLGA/PP MM | 0.06 ± 0.01 | 99.6 ± 1.6 | 101.1 ± 2.3 | −31.4 ± 0.86 | −33.6 ± 0.5 | 96.2 |
| HA/PLGA/PP MM | 0.06 ± 0.01 | 123.3 ± 2.2 | 118.1 ± 5.7 | −47.8 ± 1.4 | −46.2 ± 1.2 | 92.4 |
| IRI-loaded HA/PLGA/PP MM | 0.07 ± 0.01 | 121.7 ± 1.2 | 120.4 ± 2.7 | −46.4 ± 1.5 | −44.3 ± 2.1 | 89.7 |
| PLGA/PP BM | 0.11 ± 0.01 | 131.7 ± 3.3 | 139.1 ± 6.2 | −36.6 ± 1.1 | −38.6 ± 1.1 | 58.8 |
| IRI-loaded PLGA/PP BM | 0.09 ± 0.01 | 129.1 ± 1.3 | 130.4 ± 1.8 | −35.1 ± 2.3 | −36.4 ± 2.3 | 52.3 |
| HA/PLGA/PP BM | 0.16 ± 0.01 | 197.7 ± 2.8 | 185.4 ± 4.1 | −55.1 ± 2.2 | −54.9 ± 2.19 | 61.2 |
| IRI-loaded HA/PLGA/PP BM | 0.12 ± 0.01 | 194.7 ± 1.6 | 193.1 ± 2.9 | −53.9 ± 3.4 | −52.3 ± 1.17 | 59.8 |
DLS measurements show a monomodal size distribution of all formulations synthesized from both MM and BM (Fig. S1, ESI†). The diameter d of PLGA/PP and HA/PLGA/PP NPs synthesized by MM were 100.5 ± 2.01 nm and 123.3 ± 2.17 nm, with a narrow size distribution (polydispersion index of 0.05 and 0.06), respectively. However, in the case of NPs synthesized by BM, the size distributions of PLGA/PP and HA/PLGA/PP were 131.7 ± 3.31 nm and 197.7 ± 2.78 nm with a broad size distribution (polydispersion index of 0.11 and 0.16), respectively. In particular, NPs composed of PLGA/PP, synthesized by both BM and MM, show a size almost constant over time (ten days). On the contrary, NPs composed of HA/PLGA/PP show a size decrease, higher for NPs synthesized by BM than NPs synthesized by MM, likely due to the detachment of the HA, which occurs over time, from the NPs surface.
The addition of HA in the BM synthesis of NPs resulted in a decrease of ζ potential from ∼−36.6 mV for PLGA/PP NPs to ∼−55.1 mV for HA/PLGA NPs. The same trend but with lower values was observed for PLGA/PP (∼−32 mV) and HA/PLGA/PP (∼−47.8 mV) NPs, as synthesized by MM. A less marked decrease of the ζ potential trend of the NPs synthesized by MM is likely due to the lower surface area exposed by NPs. Moreover, for NPs synthesized by MM, a decrease in the yield from 95.3% was observed, and for PLGA/PP NPs to 92.4% for HA/PLGA/PP NPs. In any case, the yield values of all the NPs formulations synthesized by MM resulted in higher values than that for the PLGA/PP (58.8%) and HA/PLGA/PP (61.2%) NPs synthesized by BM. The decrease in the yield from PLGA/PP to HA/PLGA/PP NPs of both the synthetic methods is due to HA residues that do not interact with the poloxamers. Indeed, the core–shell HA-based NPs are composed of a hydrophobic core, PLGA and poloxamers, which act as a bridge and allow the interaction between the core and the hydrophilic shell, HA, on the surface of NPs (Fig. S2, ESI†). Moreover, for the drug-loaded NPs, no variations in the terms of diameter d and ζ potential were observed both for the NPs synthesized by MM and BM (Table 1). Furthermore, the morphological investigation of NPs by TEM analysis was performed. The selected TEM micrographs of spherical PLGA/PP and HA/PLGA/PP NPs are reported in Fig. 2.
 | ||
| Fig. 2 Selected TEM micrographs of (a) HA/PLGA/PP MM; (b) PLGA/PP MM; (c) HA/PLGA/PP BM; (d) PLGA/PP BM. | ||
The TEM micrographs show a regular spherical shape for both HA/PLGA/PP and PLGA/PP NPs with a diameter d of ∼118 nm and ∼95 nm, respectively (Fig. 2a and b). On the contrary, from the inspection of TEM micrographs of HA/PLGA/PP and PLGA/PP NPs, synthesized by BM, a dimensional distribution with a diameter d from ∼160 nm to ∼200 nm and from ∼130 nm to ∼170 nm with an irregular shape is observed, respectively (Fig. 2c and d), and even confirmed by SEM analysis (Fig. S3a and b, ESI†). The above results clearly highlight that the MM is a simple way of preparing NPs with a small size, narrow size distribution, and controlled morphology. In particular, the morphological investigation confirms that the BM method, providing inhomogeneous reaction conditions, works at the centimeter level, resulting in the random packing of NPs, showing a wide size distribution. On the contrary, the MM improves the mixing at a high flow rate, within the microchannels, leading to a stronger packing of PLGA/PP and HA/PLGA/PP NPs, resulting in smaller and controlled size NPs than those synthesized by BM. From the TEM micrographs for PLGA/PP and HA/PLGA/PP NPs, a spherical shape was observed; in particular, for PLGA/PP and HA/PLGA/PP NPs, core–shell structures were detected, in which their surface expose hydrophilic or PEO or HA, respectively. To demonstrate the core–shell structures of nanoparticles, thermal characterization was employed. For this reason, PLGA NPs, without using PP, were fabricated. The results of thermal analyses are reported in Fig. 3 and summarized in Table 2.
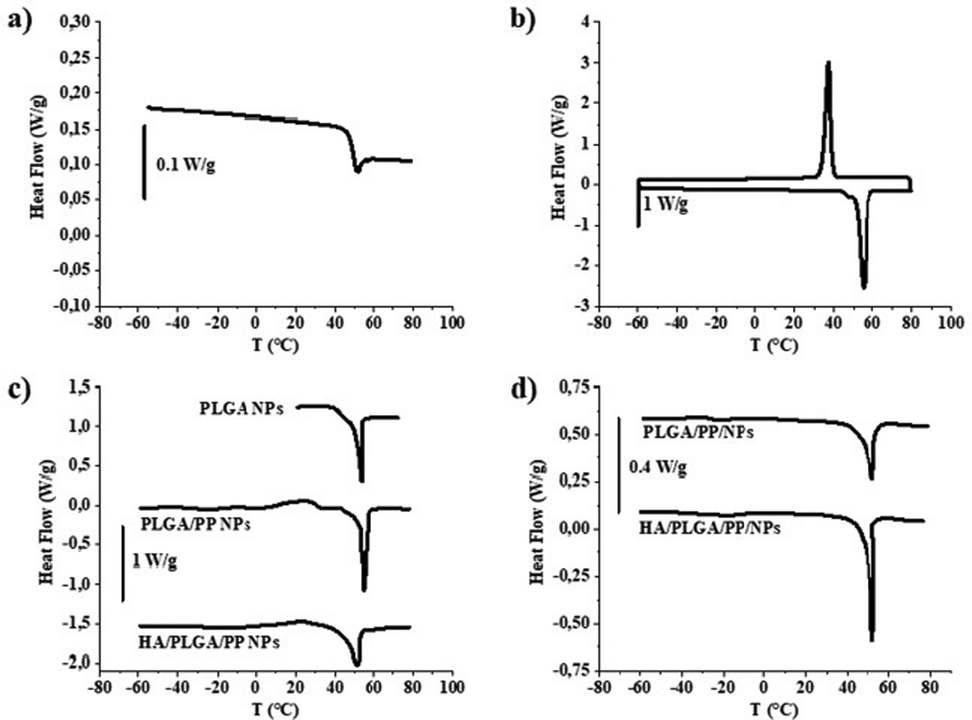 | ||
| Fig. 3 DSC thermograms of PLGA powder (a); PP powder (b); PLGA, PLGA/PP and HA/PLGA/PP NPs first scan (c); PLGA/PP and HA/PLGA/PP NPs, second scan (d). Results were obtained from the last three independent experiments. | ||
| T g (°C) | T f (°C) | ΔHf (J g−1) | T c (°C) | ΔHc (J g−1) | |
|---|---|---|---|---|---|
| PLGA powder | 48.6 ± 1.5 | — | — | — | — |
| Poloxamers | — | 55.3 ± 0.1 | 135 ± 2 | 37.3 ± 0.2 | 121 ± 1 |
| PLGA NPs | 45.3 ± 0.5 | — | — | — | — |
| PLGA/PP NPs | 46.3 ± 2.2 | 56.4 ± 1.7 | 28.4 ± 0.4 | 29.3 ± 1.3 | 29.8 ± 1.9 |
| HA/PLGA/PP NPs | 45 ± 1 | 55.8 ± 1.5 | 31.1 ± 0.1 | 27.4 ± 1.3 | 24.9 ± 2.1 |
The thermogram of PLGA powder shows a glass transition temeperature (Tg) of ∼ 48 °C (Fig. 3a), while, for the poloxamers (Fig. 3b), the crystallization and melting temperatures of 37 °C and 55 °C were detected, respectively. In the case of HA powder, the DSC thermogram revealed no thermodynamic transition in the temperature range examined. For PLGA NPs (Fig. 3c), a Tg lower than PLGA powder was observed, ∼45 °C instead of ∼48 °C, respectively, thus indicating a likely plasticizing effect of the poloxamers employed as surfactants in the WP. Furthermore, the Tg values were very similar even when the PP have been mixed in the OP of both PLGA/PP and HA/PLGA/PP NPs. Moreover, for PLGA/PP NPs, a broadening of PP crystallization peak and a decrease in their crystallization heat (ΔHc) and temperature (Tc) were detected. This suggests an interaction between the polymeric chains of PLGA and PP, solubilized in the OP, employed to fabricate the NPs. As reported in the literature, the hydrophobic PO moieties were oriented toward the PLGA core of the NPs because of the amphiphilic nature of PP, while the hydrophilic EO moieties were oriented toward the aqueous phase.40
The use of HA in the NP formulations does not affect the crystallization and melting compared to PLGA/PP NPs. Furthermore, during the heating in the first ramp, the Tg related to PLGA present in the NPs is detectable, while after PP melting, in the second scan (Fig. 3d), the peaks of both PLGA and PP are not detectable. This phenomenon is likely due to the absent direct interaction between PLGA and HA; in particular, the PLGA constitutes the core, the PP with their amphiphilic moieties acting as a bridge between the inner core and the shell, while the HA is superficially exposed composing the outer shell. After PP melting, this polymeric network is not detected and a polymeric mixture, with different thermal properties from the starting polymers, is obtained.
In Table 3, the polymer-to-drug weight ratio and drug encapsulation efficiency for IRI-loaded NPs are reported.
| Formulation | Polymer:drug ratio |
Drug encapsulation efficiency (%) | Drug loading (%) |
|---|---|---|---|
| PLGA/PP MM | 2.2 ± 0.1 | 86.5 ± 1.1 | 27.5 ± 1.0 |
| HA/PLGA/PP MM | 2.1 ± 0.1 | 87.8 ± 1.6 | 22.7 ± 1.2 |
| PLGA/PP BM | 1.8 ± 0.2 | 66.5 ± 2.3 | 18.5 ± 1.4 |
| HA/PLGA/PP BM | 1.8 ± 0.1 | 64.3 ± 1.3 | 16.6 ± 1.5 |
Using microfluidics, ∼87.15% of the IRI was encapsulated, while the average encapsulation was about ∼65.4% for the conventional BM. The DL percentage of NPs fabricated by MM was on average ∼25.1%, while for the NPs fabricated by BM, it was on average ∼17.5%. These differences are due to the lack of laminar flow conditions and longer mixing time obtained in traditional BMs. IRI-loaded PLGA/PP and HA/PLGA/PP NPs fabricated by MM resulted in greatly improved IRI encapsulation efficiency and a higher DL than those fabricated by conventional BM; the polymer-to-drug ratio is about 2 for all formulations.
In vitro release kinetic of Irinotecan
Drug release tests were accomplished in order to evaluate the release capacity of the IRI-loaded NPs. The complete release of the drug was observed in 15 days for all the formulations (Fig. 4). The experimental points were interpolated by means of a mathematical model that expresses the release as the sum of two contributions—a diffusional contribution and a dissolution contribution. The parameters obtained from the interpolation are reported in Table 4.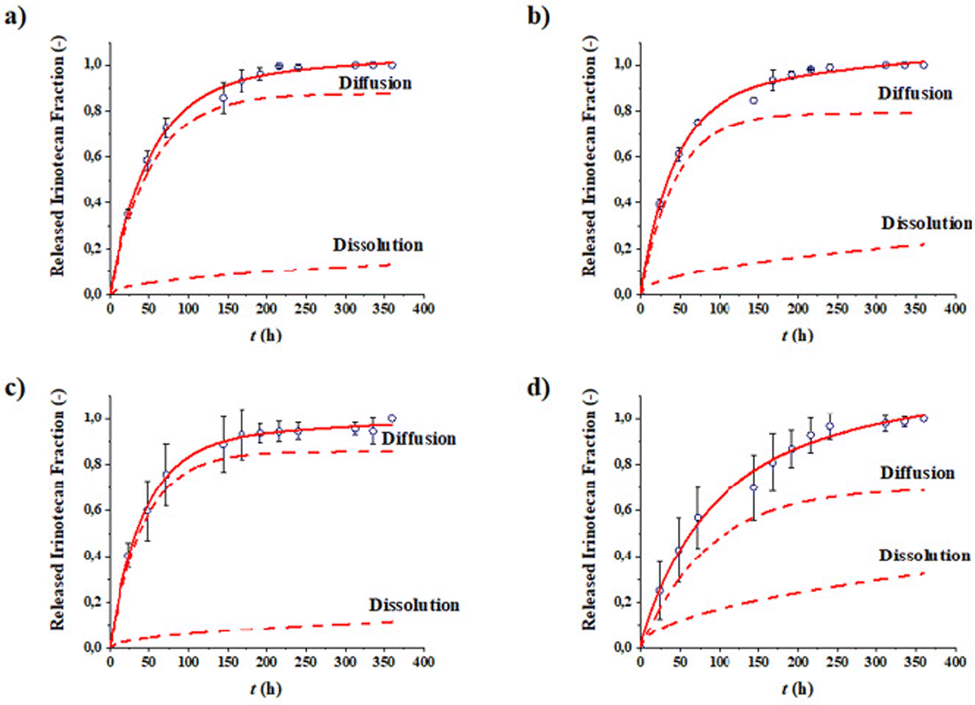 | ||
| Fig. 4 Release profiles of IRI over time of PLGA/PP MM (a), HA/PLGA/PP MM (b), PLGA/PP BM (c), and HA/PLGA/PP BM (d). | ||
| Formulation | F diss,∞ (−) | k diss (h−1/2) | F diff,∞(−) | kdiff (h−1) |
|---|---|---|---|---|
| IRI-loaded PLGA/PP MM | 0.14 ± 0.05 | 0.043 ± 0.006 | 0.86 ± 0.05 | 0.0230 ± 0.0012 |
| IRI-loaded HA/PLGA/PP MM | 0.3 ± 0.2 | 0.057 ± 0.004 | 0.7 ± 0.2 | 0.012 ± 0.002 |
| IRI-loaded PLGA/PP BM | 0.12 ± 0.07 | 0.057 ± 0.004 | 0.88 ± 0.07 | 0.0192 ± 0.0013 |
| IRI-loaded HA/PLGA/PP BM | 0.21 ± 0.07 | 0.056 ± 0.003 | 0.79 ± 0.07 | 0.023 ± 0.002 |
All formulations analyzed exhibit an IRI release curve profile in which the diffusion of the drug encapsulated the NP cores is the predominant mechanism. HA/PLGA/PP NPs, synthesized by both MM and BM, exhibited a higher IRI released fraction due to dissolution at equilibrium Fdiss,∞ than the PLGA/PP NPs. This result is in agreement with the DLS data, which shows a reduction in the size of HA-based NPs due to the solubilization of part of the polymer that forms the outer shell. These differences in behavior are not observed from the values of the kinetic constants, which resulted in very similar formulations. This outcome is probably due to the release rate influenced by factors that the model does not take into account, such as the drug encapsulation efficiency and the size of the NPs. Furthermore, since the polymeric matrices are the same for all the formulations, no evident differences were found in the release kinetic profile curves of IRI.
Biological evaluation of HA-based nanoparticles fabricated by means of mcrofluidic and bulk methods
In vitro biological studies results of the MM and BM NPs formulation are presented in Fig. 5. | ||
| Fig. 5 Cytotoxicity assay results. Percentage of cell viability of HS578T and L929 after 24 and 48 h of incubation with NPs MM (a and b) and NPs BM (c and d). Cell viability was calculated with respect to the non-treated control cells (p < 0.0001). | ||
The results of the in vitro biological studies of the MM and BM NPs formulation are presented in Fig. 5. For both formulations, it is possible to observe that blank NPs are not cytotoxic; indeed the percentage of viability, compared to the control, for HA/PLGA/PP and PLGA/PP for both MM and BM, is almost 100%. In the case of HS578T cells, it can be seen from Fig. 5a that the blank NPs are also not cytotoxic. Differently, in the case of IRI-loaded NPs, the behaviors of HA/PLGA/PP and PLGA/PP NPs are statistically significantly different (p < 0.0001), both at 24 and 48 h. In particular, the percentage of viability is about 38% after 24 h and 80% after 48 h for HA/PLGA/PP and PLGA/PP NPs, respectively, therefore indicating that HA-coated NPs can inhibit cellular growth more effectively compared to PLGA/PP NPs. Differently, for MM NPs (Fig. 5b) in L929 cells, for both the IRI-loaded particles, the percentage of viability is about 80% after 24 h and about 70% after 48 h. This result indicates that IRI-loaded PLGA/PP and HA/PLGA/PP particles are able to inhibit the cell growth and the HA coating does not influence this ability. A similar result was obtained for the BM NPs; for L929 (Fig. 5d), the percentage of viability is about 80% for both HA/PLGA/PP and PLGA/PP NPs after 24 h and 60% for HA/PLGA/PP. On the other hand, for HS578T, the percentage of viability is drastically decreased to about 45% for the HA/PLGA/PP NPs. Taken together, the results suggest that HA tropism for tumor cells CD44+ is marked compared to healthy cells; thus, the HA/PLGA/PP NPs, both MM and BM, are able to deliver more drugs into cancer cells, resulting in a stronger cell cytotoxicity effect. More specifically, it is possible to note that the IRI-NPs formulated by MM showed higher cytotoxicity effect compared to BM, probably due to the better stability of the NPs; the endocytosis mediated by the HA receptor was found to be more effective, thus allowing a better cellular absorption of NP in CD44+ cells.19Fig. 6 shows the representative morphology of the tumor (HS578T) and healthy (L929) cell lines after exposure to different types of NPs fabricated by MM.
 | ||
| Fig. 6 Representative confocal images of the L929 and HS578T cell morphologies after 24 of incubation of HA/PLGA/PP, PLGA/PP and IRI-HA/PLGA/PP and IRI-PLGA/PP fabricated by MM. Actin filaments stained by phalloidin-FITC (green) and DAPI (blue)-stained nuclei cells. Scale bar: 50 μm. Red arrow indicates the apoptotic cells that display the typically round-shaped morphologies, as shown after the incubation with IRI-loaded NPs and, in particular, NPs with HA superficial decoration. Images were acquired with a resolution of 1024 × 1024 pixels with a 40× oil immersion objective. | ||
In particular, the NPs without IRI showed the normal morphology of the cells, which is attributed respectively to L929 fibroblasts and HS578T breast cancer cells.41 The exposure of IRI-loaded NPs for MM formulations showed cells with typical morphological events of the apoptosis process, such as the shrinkage of the cell and fragmentation into membrane-bound apoptotic bodies.42
NPs uptake by cells are important features affecting the therapeutic effect of cancer chemotherapy. One of the major challenges of nanomedicine is to build nanosystems able to deliver drugs directly and specifically to tumor cells. To understand if the different methods are able to affect the efficacy of NPs interactions with cells and how they are internalized, the NP uptake studies were performed. MM and BM Fluorescent NR-loaded NPs were used to follow the NPs tracking over the cell membrane. Fig. 7a displays the qualitative cellular uptake of both MM and BM NPs after 24 h of incubation in HS578T and L929 cells. NPs are represented in the images as red dots; nuclei were stained by DAPI, while the cytoskeleton was stained using FITC-labeled phalloidin and it appears green. In both the cell lines, the NPs were localized inside the cell cytoplasm and near the perinuclear region: red-stained NPs appear clearly as larger dots for the BM NPs, indicating possible greater aggregations in the cytoplasm, while the MM NPs appear to have a more homogeneous distribution as small diffuse spots in the cell cytoplasm. The results of quantitative NPs uptake are shown in Fig. 7b for HS578T cells and Fig. 7c for the L929 cells. NP internalization increased with the exposure time until 24 h, and it was higher for HS578T tumor cells than the healthy L929 cells in all the cases, highlighting the tropism of the HA, which externally decorated the NPs surface in both MM and BM formulations for the CD44 receptor overexpressed on HS578T membranes. From the comparison of the uptakes of the MM and BM NPs, it is clear that the MM NPs were better internalized than the BM NPs in all the cases. Specifically, the internalization for tumor cells was two orders of magnitude higher after 3 h and up to three orders of magnitude higher after 24 h. Overall, a greater internationalization for MM NPs externally decorated with HA was observed. These data indicated that the NPs produced with the MM have better performing technological features in terms of the stability and homogeneity of the dimensions, and PDI has a superior efficiency of interaction with the cell membranes.43
 | ||
| Fig. 7 Confocal images (a) of HS578T and L929 cells exposed to HA/PLGA/PP MM, PLGA/PP MM and HA/PLGA/PP BM, PLGA/PP BM NPs after 24 h incubation. Maximum projection of Z-stack. Nuclei (DAPI) are shown in blue; actin filaments (phalloidin) in green; NPs in red. Scale bar: 50 μm. NP uptakes histograms of HA/PLGA/PP MM, PLGA/PP MM and HA/PLGA/PP BM, PLGA/PP BM NPs at short exposure time (1, 3, and 24 h), expressed as concentration of NPs [mg mL−1] internalized in HS578T (b) and L929 (c). | ||
Conclusions
IRI-loaded HA-based NPs based on the nanoprecipitation with a subsequent solvent evaporation process was developed by means of new technology named microfluidic method and compared with those fabricated by traditional bulk method, respectively. To the best of our knowledge, this is the first example of hyaluronic acid/PLGA core–shell nanoparticles, without any polymer chemical modifications, based only on their electrostatic interactions driven by lipophilicity gradient, fabricated by the microfluidic method. Compared with the NPs prepared via the bulk method, the HA-based NPs produced by the microfluidic method have notable advantages in terms of physicochemical properties such as smaller particle size, narrower size distribution, lower polydispersion index, and higher drug encapsulation efficiency than those fabricated by the bulk method. Due to the higher control of the physicochemical properties, the NPs prepared via microfluidics showed higher cytotoxicity, resulting in a stronger cell-killing effect and higher internalization toward CD44 overexpressing the breast tumor cells. All our results highlighted that the high control of the physicochemical properties of the polymer-based nanoparticles synthesized by the microfluidic method guarantees a higher reproducibility of the biological assays than those synthesized by traditional methods. In conclusion, our findings suggest that NPs fabricated by the microfluidic method are promising candidates for the delivery of a variety of hydrophilic/hydrophobic anticancer drugs for cancer treatment.Author contributions
Antonio Fabozzi: investigation, formal analysis, visualization, methodology, writing – original draft, & editing. Francesca Della Sala: formal analysis, writing – original draft. Mario di Gennaro: formal analysis, visualization, methodology. Assunta Borzacchiello: funding acquisition, project administration, resources conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the research project “Advise Drugs and Anti-tumoral Vaccines From The Sea-Por Campania FESR 2014–2020 AND WITH THE AXIS 1 OO.SS. 1.2.2/1.1 CUP B43D18000240007”.Notes and references
- J. M. Caster, S. K. Yu, A. N. Patel, N. J. Newman, Z. J. Lee, S. B. Warner, K. T. Wagner, K. C. Roche, X. Tian, Y. Z. Min and A. Z. Wang, J. Nanomed. Nanotechnol., 2017, 13, 1673–1683 CrossRef CAS.
- Y. C. Wang, P. W. Li, T. T. D. Tran, J. Zhang and L. X. Kong, Nanomaterials, 2016, 6(2), 26 CrossRef PubMed.
- H. Zhang, Y. Liu, G. Chen, H. Wang, C. Chen, M. Li, P. Lu and Y. Zhao, Sci. Bull., 2020, 65, 380–388 CrossRef CAS.
- A. Maus, L. Strait and D. Zhu, Engineered Regeneration, 2021, 2, 31–46 CrossRef.
- S. Acharya and S. K. Sahoo, Adv. Drug Delivery Rev., 2011, 63, 170–183 CrossRef CAS PubMed.
- L. M. Pan, J. A. Liu and J. L. Shi, Chem. Soc. Rev., 2018, 47, 6930–6946 RSC.
- N. Yang, Y. P. Ding, Y. L. Zhang, B. Wang, X. Zhao, K. M. Cheng, Y. X. Huang, M. Taleb, J. Zhao, W. F. Dong, L. R. Zhang and G. J. Nie, ACS Appl. Mater. Interfaces, 2018, 10, 22963–22973 CrossRef CAS PubMed.
- Q. Long, Z. Liu, Q. Shao, H. Shi, S. Huang, C. Jiang, B. Qian, Y. Zhong, X. He and X. Xiang, Adv. Sci., 2022, 2200856 CrossRef CAS PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- Y. Nakamura, A. Mochida, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2016, 27, 2225–2238 CrossRef CAS PubMed.
- T. R. Tian, D. X. Xiao, T. Zhang, Y. J. Li, S. R. Shi, W. Y. Zhong, P. Gong, Z. Liu, Q. Li and Y. F. Lin, Adv. Funct. Mater., 2021, 31(5), 2007342 CrossRef CAS.
- A. Ahmad, F. Khan, R. K. Mishra and R. Khan, J. Med. Chem., 2019, 62, 10475–10496 CrossRef CAS.
- R. J. Banga, S. A. Krovi, S. P. Narayan, A. J. Sprangers, G. L. Liu, C. A. Mirkin and S. T. Nguyen, Biomacromolecules, 2017, 18, 483–489 CrossRef CAS PubMed.
- P. Grenier, I. M. D. Viana, E. M. Lima and N. Bertrand, J. Controlled Release, 2018, 287, 121–131 CrossRef CAS PubMed.
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernandez, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- G. L. Huang and H. L. Huang, J. Controlled Release, 2018, 278, 122–126 CrossRef CAS PubMed.
- Z. J. Luo, Y. Dai and H. L. Gao, Acta Pharm. Sin. B, 2019, 9, 1099–1112 CrossRef PubMed.
- S. Amorim, D. S. da Costa, D. Freitas, C. A. Reis, R. L. Reis, I. Pashkuleva and R. A. Pires, Sci. Rep.-UK, 2018, 8, 16058 CrossRef PubMed.
- F. Della Sala, A. Fabozzi, M. di Gennaro, S. Nuzzo, P. Makvandi, N. Solimando, M. Pagliuca and A. Borzacchiello, Macromol. Biosci., 2021, 2100304, DOI:10.1002/Mabi.202100304.
- K. T. Kim, J. Y. Lee, D. D. Kim, I. S. Yoon and H. J. Cho, Pharmaceutics, 2019, 11(6), 280 CrossRef CAS PubMed.
- S. Tiwari and P. Bahadur, Int. J. Biol. Macromol., 2019, 121, 556–571 CrossRef CAS PubMed.
- S. Giarra, C. Serri, L. Russo, S. Zeppetelli, G. De Rosa, A. Borzacchiello, M. Biondi, L. Ambrosio and L. Mayol, Carbohydr. Polym., 2016, 140, 400–407 CrossRef CAS PubMed.
- L. Wang, Y. J. Hu, Y. W. Hao, L. Li, C. X. Zheng, H. J. Zhao, M. Y. Niu, Y. Y. Yin, Z. Z. Zhang and Y. Zhang, J. Controlled Release, 2018, 286, 74–84 CrossRef CAS PubMed.
- C. I. C. Crucho and M. T. Barros, Mater. Sci. Eng., C, 2017, 80, 771–784 CrossRef CAS PubMed.
- J. Zhang, H. Tang, Z. F. Liu and B. A. Chen, Int. J. Nanomed., 2017, 12, 8483–8493 CrossRef CAS PubMed.
- L. Sun, F. Bian, Y. Wang, Y. Wang, X. Zhang and Y. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4527–4532 CrossRef CAS PubMed.
- Y. Liu, Y. Cheng, C. Zhao, H. Wang and Y. Zhao, Adv. Sci., 2022, 9, 2104272 CrossRef CAS PubMed.
- T. S. Kaminski and P. Garstecki, Chem. Soc. Rev., 2017, 46, 6210–6226 RSC.
- D. Zhang, W. Li, Y. Shang and L. Shang, Engineered Regeneration, 2022, 3, 258–261 CrossRef.
- Y. C. Huang, T. T. Han, J. Xuan, H. Xu, Y. L. Wang and L. Zhang, J. Micromech. Microeng., 2018, 28, 105021 CrossRef.
- A. Fabozzi, F. Della Sala, M. di Gennaro, N. Solimando, M. Pagliuca and A. Borzacchiello, Polym. Chem.-UK, 2021, 12, 6667–6687 RSC.
- T. Si, Z. Q. Zhu and X. R. Xu, Lab Chip, 2015, 15, 646–649 RSC.
- R. R. Hood and D. L. DeVoe, Small, 2015, 11, 5790–5799 CrossRef CAS PubMed.
- S. Y. M. Lu, Y. P. Ho, C. L. Grigsby, K. W. Leong and T. J. Huang, ACS Nano, 2014, 8, 10026–10034 CrossRef PubMed.
- T. K. J. Wu, K. W. Kwok Yeung, H. C. Shum, K. M. Chee Cheung, L. Wang and M. Kai Tsun, Acta Biomater., 2013, 9, 7410–7419 CrossRef PubMed.
- M. Russo, P. Bevilacqua, P. A. Netti and E. Torino, Sci. Rep.-UK, 2016, 6, 37906 CrossRef CAS.
- A. C. di Polidoro, O. Tammaro, E. Romano, P. A. Netti and E. Torino, Sci. Rep.-UK, 2020, 10, 6028 CrossRef PubMed.
- A. G. Tripathi and R. Saraf, Int. J. PharmTech Res., 2010, 2, 2116–2123 CAS.
- L. Mayol, M. Biondi, L. Russo, B. M. Malle, K. Schwach-Abdellaoui and A. Borzacchiello, Carbohydr. Polym., 2014, 102, 110–116 CrossRef CAS PubMed.
- L. Mayol, C. Serri, C. Menale, S. Crispi, M. T. Piccolo, L. Mita, S. Giarra, M. Forte, A. Saija and M. Biondi, Eur. J. Pharm. Biopharm., 2015, 93, 37–45 CrossRef CAS PubMed.
- F. Della Sala, T. Silvestri, A. Borzacchiello, L. Mayol, L. Ambrosio and M. Biondi, Colloids Surf., B, 2022, 210, 112240 CrossRef CAS PubMed.
- G. Häcker, Cell Tissue Res., 2000, 301, 5–17 CrossRef PubMed.
- E. Chiesa, A. Greco, F. Riva, R. Dorati, B. Conti, T. Modena and I. Genta, Pharmaceuticals, 2022, 15, 103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj03279a<tab /> |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |