
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic and optical properties of C16S8 and C16S4Se4 molecules and crystals
Artem V.
Kuklin
 *a,
Diana I.
Saykova
bc,
Rahul
Suresh
b,
Lyudmila V.
Begunovich
b,
Gleb V.
Baryshnikov
*de,
Nataliya
Karaush-Karmazin
e,
Svetlana V.
Saikova
c and
Hans
Ågren
a
*a,
Diana I.
Saykova
bc,
Rahul
Suresh
b,
Lyudmila V.
Begunovich
b,
Gleb V.
Baryshnikov
*de,
Nataliya
Karaush-Karmazin
e,
Svetlana V.
Saikova
c and
Hans
Ågren
a
aDepartment of Physics and Astronomy, Uppsala University, Box 516, SE-751 20 Uppsala, Sweden. E-mail: artem.icm@gmail.com
bInternational Research Center of Spectroscopy and Quantum Chemistry - IRC SQC, Siberian Federal University, 79 Svobodny pr., 660041 Krasnoyarsk, Russia
cDivision of Physical and Inorganic Chemistry, Institute of Non-ferrous Metals, Siberian Federal University, 79 Svobodny pr., 660041 Krasnoyarsk, Russia
dLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, 60174, Norrköping, Sweden. E-mail: glib.baryshnikov@liu.se
eDepartment of Chemistry and Nanomaterials Science, Bohdan Khmelnytsky National University, 18031 Cherkasy, Ukraine
First published on 20th June 2022
Abstract
Hetero[8]circulenes have been proposed as promising fluorescent emitters for organic light-emitting diodes and as emerging materials in the construction of organic solar cells. Among them, octathio[8]circulene (C16S8) and its derivative tetrathiotetraseleno[8]circulene (C16S4Se4) crystals have been highlighted as efficient charge transport materials in field-effect devices. Using density functional theory, we investigate in this paper the electronic and optical properties of the C16S8 and C16S4Se4 molecules and crystals in order to revise previously reported controversial experimental data and report highly reproducible new results. We find that formation of the crystals results in a significant band gap decrease (∼0.6–0.7 eV) caused by relatively strong intermolecular interactions. A partial replacement of S atoms with Se atoms also leads to a small band gap reduction. The C16S8 crystal demonstrates a band gap of 3.32 eV, while a band gap of 3.20 eV is found for C16S4Se4. Both C16S8 and C16S4Se4 compounds are optically transparent in the visible region, confirming the absence of red coloration reported previously in one of experimental papers. The C16S8 and C16S4Se4 crystals demonstrate anisotropic electronic, mechanical and optical properties. These findings might initiate future experimental studies of the red color origin for C16S8 and provide insight for the design and engineering of C16S8 and C16S4Se4 based charge transport and optical devices.
Introduction
In recent years, polycyclic organic compounds such as [n]circulenes have gained significant attention due to their unusual symmetry and π-conjugation features. [n]circulenes are macrocyclic arenes composed of several hydrocarbon cycles linked together in the shape of a flower. The central n-sided polygon is completely surrounded and fused by benzenoids. Due to their high stability, symmetry, and optical properties [n]circulenes can be used in the field of nanoelectronics as promising fluorescent emitters for organic light-emitting diodes and in the construction of solar cells.1–3[n]circulenes are typically semiconductors with tunable band gaps depending on the stacking features. Moreover, circulenes can be coupled through post-transformation into one- (1D) or two-dimensional (2D) materials which demonstrate useful semiconducting or semimetallic properties.4 Recently it was shown that tetraoxa[8]circulene-based 2D polymers can exhibit strong topological states caused by spin–orbit coupling effects,5 which is not typical for materials based on light elements (C, H, O). In addition, 2D-polymers of tetraoxa[8]circulene modified with s-block metals, such as Li, Na, Ca, exhibit metallic properties. Moreover, decoration by Ca atoms may result in superconducting properties.6 The introduction of heteroatoms (for example, S or Se) into the periphery of the circulene frame can induce intercolumnar interactions and improve the mobility of charge carriers in organic semiconductors.7
Heteroatomic circulenes, and in particular octathio[8]circulene (also called “sulflower”), have been of great research interest due to their atypical electronic properties since their first mentioning in 2006 by Nenajdenko et al.8 Over a decade, much research has been conducted to define sulphur-rich polycyclic aromatic hydrocarbons due to their significant charge transfer and photovoltaic properties.9–11 Subsequently, Dong et al.12 synthesized the second generation of “sulflower” with all sulphur-terminated edges. Many studies have been reported on the different derivatives of oligothiophenes13–15 and their properties over the past few decades such as vibrational spectra,16,17 optical properties,18 fundamental structure–property relationships,19 as well as their applications in organic light-emitting diodes (OLEDs),20 photovoltaics and field-effect transistors.21,22 However, the tunable fluorescence and phosphorescence properties of circulenes and their electronic structure in the crystalline phase have only been scarcely studied despite the crucial role of such characteristics for OLED applications.23,24
In 2008, a group of researchers, separated two forms of “sulflower” by a sublimation method – a white amorphous film and red polycrystals, and identified that the white amorphous film turns slightly pink when left under normal conditions, referring to the transition from the amorphous to the polycrystalline phase.25 Their experimental absorption spectra for the polycrystalline form revealed prominent absorption peaks at 416 and 527 nm which were assigned to the strong intermolecular interactions in the crystal because these peaks were absent in the white amorphous form. TD-DFT calculations26 predicted the most intense peak at 239 nm for the amorphous form which is in good agreement with the experimental results for the main band at 244 nm.25 The unstructured shoulder observed in the experiment for C16S8 hexane solution in the range 250–300 nm is most likely caused by weakly allowed transitions to 1E1u states (calculated at 262 nm) and by the symmetry-forbidden singlet–singlet transitions (in the framework of the D8h symmetry point group) that gain some non-zero intensity through reducing molecular symmetry.27 However, Fujimoto et al.28,29 reported the synthesis of colorless octathio[8]circulene with no absorption peak in the UV-Vis spectra contradictory to the previous reports despite the similar method of preparation of the material.29 Moreover, Arago et al.30 supported this finding with detailed theoretical calculations and the absence of the absorption peak in the UV-Vis region referring to the high symmetry and loss of π-conjugation.31 Though the red colour of the polycrystalline sulflower is justified by the interference effect of the thin film,28 Bukalov et al.25 reported the appearance of a red colour sulflower even in a 10 μ thickness ruling out the interference effect.
Dadvand et al.32 synthesized a selenium analogue of sulflower as an organic semiconductor (OSC), namely tetraselenotetrathio[8]circulene, also known as selenosulflower. Unlike sulflower, selenosulflower is colourless, both in the amorphous and crystalline states.26 DFT calculations26 indicated a broadening of the absorption spectra for tetraselenotetrathio[8]circulene through the reduction of molecular symmetry and thus appearance of additional symmetry-allowed transitions. Gahungu and Zhang performed a series of DFT calculations for different configurations of terminal atoms in circulenes intended for use in field-effect transistors (FETs)33 and were the first to explain the essential properties of C16Se8. Later on, in 2015, Yin et al. coined the term “selflower” for C16Se8 and calculated the charge transport properties of selenosulflower and selflower.34
In this work, we report DFT studies on the electronic and optical properties of octathio[8]circulene mentioned as “sulflower” and crystals tetrathiotetraseleno[8]circulene mentioned as “selenosulflower” to resolve previous inconsistencies related to their optical properties published in experimental reports.
Computational details
The geometry, electronic structures and optical properties of octathio[8]circulene and tetrathiotetraseleno[8]circulene were investigated using the Vienna ab initio simulation package (VASP)35–38 and utilizing density functional theory (DFT) in the form of the generalized gradient approximation (GGA).39,40 The structures were relaxed with the PBE functional41 proposed by Perdew, Burke, Ernzerhof (PBE) while electronic and optical properties were calculated by the HSE06 functional.42 The empirical Grimme D3 correction43,44 was used to correctly describe weak dispersion interactions. The first Brillouin zone was divided into 4 × 4 × 12 gamma-centered k-points according to the Monkhorst–Pack scheme.45 The cutoff energy for the basis set of plane waves was set to 600 eV after a convergence test. The criteria for convergence of interatomic forces and electronic energy iterations were set to 10−2 eV Å−1 and 10−4 eV, respectively. The VESTA software46 was used to visualize atomic structures. The “vaspkit” software47 was used for pre- and post-processing.Results and discussion
The crystal structures of octathio[8]circulene and tetrathiotetraseleno[8]circulene were adopted from the reported experimental XRD data.8 The optimized lattice parameters (Fig. 1) are a = 11.15, b = 16.60 and c = 3.97 Å (a = 11.28, b = 16.66 and c = 4.14 Å) for C16S8 (C16S4Se4). Each unit cell consists of 2 molecules related to the Ci space group with three types of contacts between each molecule. In octathio[8]circulene, the molecules are held together by two types of contacts, π–π stacking (3.96 Å) and S–S intermolecular contacts (3.23, 3.33 and 3.58 Å) whereas in tetrathiotetraseleno[8]circulene there are 4 contacts – π–π stacking (4.15 Å), Se–Se (3.58 Å), Se–S (3.26 Å) and S–S (3.40 Å). From the geometrical features, it is evident that the π–π stacking is weaker compared to other interactions for both octathio[8]circulene and tetrathiotetraseleno[8]circulene.25 | ||
| Fig. 1 Top and side views of the crystal structure of (a) octathio[8]circulene and (b) tetrathiotetraseleno[8]circulene. Black, yellow and green colors correspond to carbon, sulfur and selenium atoms, respectively. The unit cells are denoted by black lines. | ||
Intermolecular interactions in crystals play a crucial role in fusing and holding separate molecules in a bulk. To elucidate strength of binding between molecules in the octathio[8]circulene and tetrathiotetraseleno[8]circulene crystals we calculated the formation Gibbs energies as ΔG = ΔE + ΔZPE − TΔS, where ΔE is the difference in the total energy of product and reagents. ΔZPE and ΔS are the differences in zero-point energy and entropy between the crystalline solid and the gas phase molecules. Thermodynamic properties were calculated using the PHONOPY code.48 The calculated formation energies without thermodynamic contribution equal −3.48 and −3.92 eV for C16S8 and C16S4Se4 respectively, that correspond to ∼ −0.073 and −0.082 eV atom−1. Such energy values demonstrate strong intermolecular bonding in the crystals above typical van der Waals energies confirming also a more complex interaction between the molecules in the crystals. Including thermodynamic characteristics, at temperature of 300 K, the respective free formation Gibbs energies are predicted to be −0.057 and −0.068 eV atom−1 for C16S8 and C16S4Se4. These energy values are still a bit above the upper limit of the van der Waals energy.
The strong intermolecular bonding in the crystals can result in interesting mechanical properties. It is also important to assess the effect of lattice distortion on structural stability. The stiffness tensors were calculated based on the Strain versus Energy method (Table 1). Following the Born–Huang criteria,49 all elastic stability criteria as seen in ref. 50 have been met, therefore the crystals are mechanically stable. The mechanical properties along the x and y directions are similar because of the relatively isotropic character of the crystals along those directions, while they are significantly lower along the z direction due to π–π stacking which is weaker compared to the other directions. The calculated average moduli and Poisson ratios are compared to those reported for other organic crystals.51,52 From the computed mechanical properties it is evident that partially Se substituted C16S4Se4 crystal is softer compared to pure C16S8.
| Crystal | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | E | B | G | ν |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C16S8 | 26.68 | 8.53 | 4.99 | 27.12 | 10.11 | 16.75 | 6.62 | 3.20 | 16.14 | 17.83 | 12.62 | 7.05 | 0.27 |
| C16S4Se4 | 23.41 | 5.69 | 5.97 | 22.01 | 12.65 | 13.80 | 6.56 | 2.73 | 12.54 | 13.47 | 11.62 | 5.15 | 0.31 |
Density of states (DOS) plots of C16S8 and C16S4Se4 are shown in Fig. 2. The variations in the occupancy of the states are plotted in the graph between energy and DOS.53,54 A small shift in the occupancy of DOS in the conduction band on Se substitution for both molecules and crystal structures can be seen in Fig. 2. The substitution of S with Se atoms leads to a shift of the conduction band towards lower energies. The bandwidth of the conduction band is clearly larger than that of the valence one that results in good electron mobility reported in ref. 7. The valence band of both crystals is nearly the same, while the conduction band of C16S4Se4 is shifted toward lower energies due to the weaker C–Se covalent bonding character compared to the C–S one.
 | ||
| Fig. 2 Density of states of octathio[8]circulene and tetrathiotetraseleno[8]circulene (a) molecules and (b) crystals. The Fermi levels are set to 0 eV. | ||
HOMO and LUMO energies of the octathio[8]circulene molecule are found to be −4.95 and −0.98 eV and for the tetrathiotetraseleno[8]circulene molecule, HOMO and LUMO are found to be −4.75 and −0.93 eV. This result is consistent with the decrease of HOMO and LUMO through the substitution of S by Se. When comparing HOMO and LUMO energies for both molecules with that of the crystals, one finds a large shift in the energies of the HOMO and LUMO levels. HOMO and LUMO are 2.28 and 5.60 eV for octathio[8]circulene crystal and 2.05 and 5.25 eV for the tetrathiotetraseleno[8]circulene crystal. The replacement of S by Se leads to a slight decrease in the energy gap in both molecules and crystals. Comparing the isolated molecules and respective crystals, one can conclude that the crystal field effect is quite pronounced and leads to a decrease of the band gaps in order of 0.6–0.7 eV, that, as a result, should affect the optical properties of the materials.
In order to accurately describe the electronic properties of the crystals, the band structures were calculated (Fig. 3). Both crystals demonstrate semiconducting properties with indirect band gaps. Octathio[8]circulene and tetrathiotetraseleno[8]circulene show band gaps of 3.32 and 3.20 eV, respectively. Therefore, the replacement of 50% of S atoms with Se reduces the band gap by 0.12 eV, which might be caused by the stronger interactions of the Se atoms due to their large polarizability but also by that the covalent bond character is weaker as compared to sulflower. The conduction band (CBM) minimum is located between the B and Γ points in both crystals, while the valence band maximum (VBM) in octathio[8]circulene resides at point Y_2, however, the points Γ, Z, and C_2 are close in energy and, therefore, can also be involved in inter-band transitions. The VBM of tetrathiotetraseleno[8]circulene is located at the A point. The existence of both flat and dispersive bands indicates that the charge transport properties of the crystals are anisotropic with strong carrier mobility due to the strong band dispersion around the B point.55
 | ||
| Fig. 3 Band structures of (a) octathio[8]circulene and (b) tetrathiotetraseleno[8]circulene. The Fermi level is set to 0 eV. | ||
Optical spectra were calculated and plotted for crystals and molecules of octathio[8]circulene (Fig. 4a) and tetrathiotetraseleno[8]circulene (Fig. 4b). One can see that there is no major difference between the absorption spectra of the octathio[8]circulene crystal and the molecule, whereas there is a considerable blue shift in the absorption spectra of the tetrathiotetraseleno[8]circulene molecule with respect to the crystal. This shift can be explained by a more pronounced crystal field effect in C16S4Se4. The octathio[8]circulene crystal starts to absorb at ∼370 nm, while the respective molecule demonstrates absorption at ∼290 nm. The C16S4Se4 crystal and molecule reveal absorption at ∼390 and 310 nm, respectively. This means that the materials either in crystalline or amorphous forms should not absorb visible light because there are no strong absorption peaks above 400 nm. The main absorption peaks of the C16S8 crystal are located at 210, 240 nm and there is a shoulder around 300 nm. The C16S4Se4 crystal reveals some redshift of peaks (185, 220, 266 and 305 nm) compared to those of C16S8.
 | ||
| Fig. 4 Optical absorption spectra of (a) octathio[8]circulene and (b) tetrathiotetraseleno[8]circulene molecules and crystals, (c) experimental UV-vis spectra of octathio[8]circulene taken from 1 – [28] and 2 – [25]. | ||
Comparing our theoretical absorption spectra of octathio[8]circulene with the experimental results in ref. 25 and Fig. 4c (red line), one can conclude that typical peaks at 207, 240, and 298 nm for the crystal phase are in fair agreement with the present results excluding peaks at 416 and 527 nm which are not reproduced by the theoretical calculations. These two peaks result in the coloration of the 1R phase (red crystals) but it was earlier concluded that they are caused only by strong intermolecular interactions in the crystal.25 Our calculations disprove this conclusion. Though the band gap decrease caused by crystallization is significant, the intermolecular interactions in the crystal are not so large to induce absorption in the visible range. At the same time, Fujimoto et al.28 did not observe any absorption in the visible region for a 10 nm thick C16S8 thin film (Fig. 4c, blue line). The calculated absorption spectrum of the C16S8 crystal almost perfectly matches the one for the crystal obtained by Fujimoto et al.28 something that confirms validity of our results. We believe that this result is more reasonable because an experimental redshift from 302 nm in solution to 527 nm in a red crystal would result in almost a 1.7 eV band gap decrease, which goes far beyond intermolecular interactions. Therefore, we conclude that intermolecular interactions in the crystal cannot be a reason for the red coloration of crystals produced by Bukalov et al.25 We speculate that the observed red-colored crystals could be the result of the formation of ion radical pairs in the crystalline phase which could be formed under sublimation conditions or due to the presence of some low-symmetrical oligothiophene impurities. Experimental cyclic voltammogram measurements28 demonstrated faint red colour at 0.0 eV and dark red colour at 0.8 eV. The reported theoretical calculations of octathio[8]circulene cation demonstrate an absorption peak around 550 nm that potentially confirms our assumption. These findings might initiate future experimental studies of the origin of the red colour for C16S8. Our predictions on the spectral behaviour of C16S4Se4 do not have experimental validation yet but can hopefully be tested in future experiments.
To elucidate the direction-depended optical absorption, the imaginary part of the frequency-dependent dielectric function along different axes was plotted for two crystals (Fig. 5). Due to the anisotropic character, the crystals are expected to demonstrate tunable optical properties that depend on a certain axis. As stated previously both crystals are transparent in the infrared and visible regions due to relatively wide band gaps. It is also evident that C16S4Se4 has more prominent absorption at 4 eV as compared to C16S8. There is an obvious anisotropy between polarization along x(y) and z directions caused by the significant structural differences, while for in-plane polarization, the difference is not so strong because the molecular packing along x and y directions is similar, something that also is confirmed by the elastic constants. Since these molecular crystals are relatively soft, a moderate applied strain should not much change the VBM and CBM positions and therefore the optical absorption should be stable under deformation of the crystals.
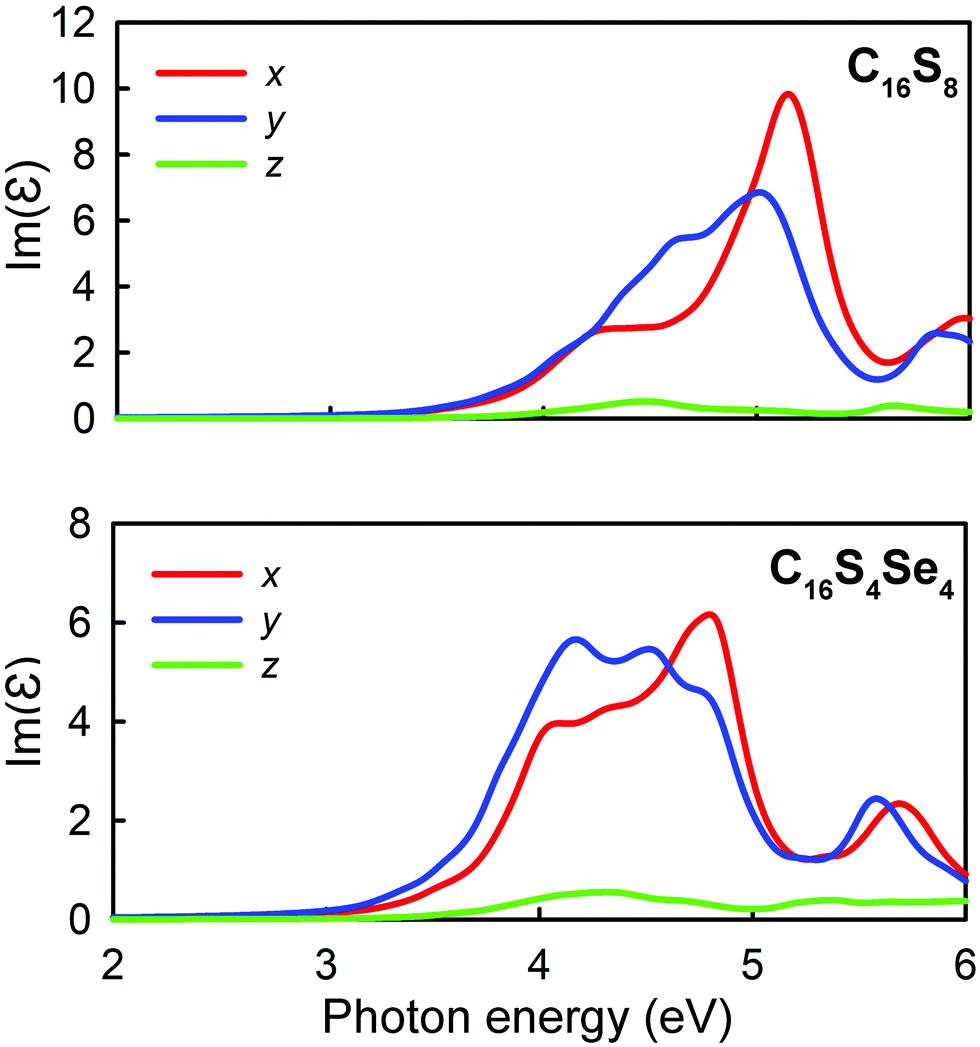 | ||
| Fig. 5 Direction decomposed imaginary part of dielectric function of C16S8 (top panel) and C16S4Se4 (bottom panel) crystals. | ||
Conclusion
In this paper, the electronic and optical properties of octathio- and tetrathiotetraseleno[8]circulenes molecules and crystals were studied using the TD-HSE approach. The optimized geometrical parameters reveal that the π–π interactions are relatively weak compared to the other interactions between the molecules in a crystal. The band structure analysis demonstrates band gaps of 3.32 and 3.20 eV for C16S8 and C16S4Se4, respectively, resulting in a band gap reduction by 0.12 eV on partial replacing S atoms with Se atoms. At the same time, the formation of crystals from molecules leads to a ∼0.6–0.7 eV band gap decrease revealing strong intermolecular interactions in the crystals. The UV-Vis absorption spectra do not confirm the occurrence of red color in the C16S8 1R crystal as reported in one of the previous experimental studies. Though the crystals possess a significant redshift of 80 nm as compared to the isolated molecules, the interaction is not as strong as suggested previously. We find that the C16S8 and C16S4Se4 crystals demonstrate anisotropic electronic, mechanical, and optical properties. We believe the present fundamental results can provide valuable data for the practical design of charge transport and optical devices based on octathio- and tetrathiotetraseleno[8]circulenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. I. S., R. S. and L. V. B acknowledge support from the Russian Science Foundation (Project 19-73-10015). G. V. B. and N. K-K. acknowledge the Ministry of Education and Science of Ukraine (Project 0121U107533). The authors thank the Swedish National Infrastructure for Computing (SNIC 2021-3-22) at the National Supercomputer Centre of Linköping University (Sweden) partially funded by the Swedish Research Council through grant agreement no. 2018-05973.References
- N. N. Karaush-Karmazin, G. V. Baryshnikov and B. F. Minaev, Chemistry, 2021, 3, 1411–1436 CrossRef CAS.
- S. K. Pedersen, K. Eriksen, H. Ågren, B. F. Minaev, N. N. Karaush-Karmazin, O. Hammerich, G. V. Baryshnikov and M. Pittelkow, J. Am. Chem. Soc., 2020, 142, 14058–14063 CrossRef CAS PubMed.
- Y. Matsuo, T. Tanaka and A. Osuka, Chem. – Eur. J., 2020, 26, 8144–8152 CrossRef CAS PubMed.
- P. W. Fritz, T. Chen, T. Ashirov, A.-D. Nguyen, M. Dincă and A. Coskun, Angew. Chem., Int. Ed., 2022, 61, e202116527 CrossRef CAS PubMed.
- A. V. Kuklin, G. V. Baryshnikov, B. F. Minaev, N. Ignatova and H. Ågren, J. Phys. Chem. C, 2018, 122, 22216–22222 CrossRef CAS.
- L. V. Begunovich, A. V. Kuklin, G. V. Baryshnikov, R. R. Valiev and H. Ågren, Nanoscale, 2021, 13, 4799–4811 RSC.
- N. N. Karaush-Karmazin, G. V. Baryshnikov, A. V. Kuklin, D. I. Saykova, H. Ågren and B. F. Minaev, J. Mater. Chem. C, 2021, 9, 1451–1466 RSC.
- K. Y. Chernichenko, V. V. Sumerin, R. V. Shpanchenko, E. S. Balenkova and V. G. Nenajdenko, Angew. Chem., Int. Ed., 2006, 45, 7367–7370 CrossRef CAS PubMed.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- O. Yemul, M. Ujihara, N. Maki and T. Imae, Polym. J., 2005, 37, 82–93 CrossRef CAS.
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed.
- R. Dong, M. Pfeffermann, D. Skidin, F. Wang, Y. Fu, A. Narita, M. Tommasini, F. Moresco, G. Cuniberti, R. Berger, K. Müllen and X. Feng, J. Am. Chem. Soc., 2017, 139, 2168–2171 CrossRef CAS PubMed.
- V. G. Nenajdenko, V. V. Sumerin, K. Y. Chernichenko and E. S. Balenkova, Org. Lett., 2004, 6, 3437–3439 CrossRef CAS PubMed.
- M. Miyasaka, A. Rajca, M. Pink and S. Rajca, J. Am. Chem. Soc., 2005, 127, 13806–13807 CrossRef CAS PubMed.
- R. Malavé Osuna, R. Ponce Ortiz, V. Hernández, J. T. López Navarrete, M. Miyasaka, S. Rajca, A. Rajca and R. Glaser, J. Phys. Chem. C, 2007, 111, 4854–4860 CrossRef.
- C. Ehrendorfer and A. Karpfen, J. Phys. Chem., 1995, 99, 5341–5353 CrossRef CAS.
- F. Negri and M. Z. Zgierski, J. Chem. Phys., 1994, 100, 2571–2587 CrossRef CAS.
- E. Van Keuren, H. Möhwald, S. Rozouvan, W. Schrot, V. Belov, H. Matsuda and S. Yamada, J. Chem. Phys., 1999, 110, 3584–3590 CrossRef CAS.
- R. Breslow, Acc. Chem. Res., 1973, 6, 393 CrossRef CAS.
- I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281–2305 CrossRef CAS.
- Y. Sun, Y. Liu and D. Zhu, J. Mater. Chem., 2005, 15, 53–65 RSC.
- G. Barbarella, M. Melucci and G. Sotgiu, Adv. Mater., 2005, 17, 1581–1593 CrossRef CAS.
- Y. Matsuo, F. Chen, K. Kise, T. Tanaka and A. Osuka, Chem. Sci., 2019, 10, 11006–11012 RSC.
- F. Chen, Y. S. Hong, D. Kim, T. Tanaka and A. Osuka, ChemPlusChem, 2017, 82, 1048–1051 CrossRef CAS PubMed.
- S. S. Bukalov, L. A. Leites, K. A. Lyssenko, R. R. Aysin, A. A. Korlyukov, J. V. Zubavichus, K. Y. Chernichenko, E. S. Balenkova, V. G. Nenajdenko and M. Y. Antipin, J. Phys. Chem. A, 2008, 112, 10949–10961 CrossRef CAS PubMed.
- G. V. Baryshnikov, B. F. Minaev, V. A. Minaeva and V. G. Nenajdenko, J. Mol. Model., 2013, 19, 4511–4519 CrossRef CAS PubMed.
- B. Minaev, E. Jansson, H. Ågren and S. Schrader, J. Chem. Phys., 2006, 125, 234704 CrossRef PubMed.
- T. Fujimoto, M. M. Matsushita, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2008, 130, 15790–15791 CrossRef CAS PubMed.
- T. Fujimoto, R. Suizu, H. Yoshikawa and K. Awaga, Chem. – Eur. J., 2008, 14, 6053–6056 CrossRef CAS PubMed.
- J. Aragó, P. M. Viruela and E. Ortí, J. Mol. Struct. THEOCHEM, 2009, 912, 27–31 CrossRef.
- S. Hotta, in Mathematical Physical Chemistry, Springer Singapore, Singapore, 2020, pp. 125–150 Search PubMed.
- A. Dadvand, F. Cicoira, K. Y. Chernichenko, E. S. Balenkova, R. M. Osuna, F. Rosei, V. G. Nenajdenko and D. F. Perepichka, Chem. Commun., 2008, 5354–5356 RSC.
- G. Gahungu and J. Zhang, Phys. Chem. Chem. Phys., 2008, 10, 1743–1747 RSC.
- J. Yin, K. Chaitanya and X. H. Ju, J. Mater. Chem. C, 2015, 3, 3472–3481 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse, J. Non-Cryst. Solids, 1995, 192–193, 222–229 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 4978 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Ehrlich, J. Moellmann and S. Grimme, Acc. Chem. Res., 2013, 46, 916–926 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- V. Wang, N. Xu, J. C. Liu, G. Tang and W. T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- M. Born and K. Huang, Dynamical theory of crystal lattices, Clarendon Press, 1954 Search PubMed.
- F. Mouhat and F.-X. Coudert, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- E. Kiely, R. Zwane, R. Fox, A. M. Reilly and S. Guerin, CrystEngComm, 2021, 23, 5697–5710 RSC.
- I. A. Fedorov, C. V. Nguyen and A. Y. Prosekov, ACS Omega, 2021, 6, 642–648 CrossRef CAS PubMed.
- R. Suresh, R. Shankar and S. Vijayakumar, J. Mol. Model., 2020, 26, 1–25 CrossRef PubMed.
- R. Suresh, V. S. Anithaa, R. Shankar and V. Subramaniam, J. Mol. Model., 2021, 27, 1–14 CrossRef PubMed.
- Y. A. Duan, H. Bin Li, Y. Geng, Y. Wu, G. Y. Wang and Z. M. Su, Org. Electron., 2014, 15, 602–613 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |