
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Niobium and Tantalum complexes derived from the acids Ph2C(X)CO2H (X = OH, NH2): synthesis, structure and ROP capability†
Xin
Zhang
,
Timothy J.
Prior
 and
Carl
Redshaw
*
and
Carl
Redshaw
*
Plastics Collaboratory, Department of Chemistry, The University of Hull, Cottingham Rd, Hull HU6 7RX, UK. E-mail: c.redshaw@hull.ac.uk
First published on 1st July 2022
Abstract
Reaction of benzilic acid, Ph2C(OH)(CO2H), L1H2, with equimolar amounts of M(OR)5 (M = Nb, Ta) led, following work-up, to the tetranuclear complexes [Nb4(OEt)8(L1)4(μ-O)2] (1) or [Ta4(OEt)8(L1)4(μ-O)2]·0.5MeCN (2·0.5MeCN), respectively. Similar use of 2,2′-diphenylglycine, Ph2C(NH2)(CO2H), L2H3, led to the isolation of the dinuclear complexes [Nb2(OEt)4(L2H2)4(μ-O)]·2MeCN (3·2MeCN) or [Ta2(OEt)4(L2H2)4(μ-O)]·2.25MeCN (4·2.25MeCN). The molecular structures of 1–4 are reported. Complexes 1–4 have been screened for their potential to act as catalysts for the ring opening polymerization (ROP) of ε-caprolactone (ε-CL) and rac-lactide (r-LA), with or without benzyl alcohol (BnOH) present. In the case of ε-CL, complex 1 displayed best activity with >99% conversion at 100 °C, whilst 3 and 4 were virtually inactive under the same conditions. All complexes show moderate activities towards the ROP of r-LA at 160 °C, with 1–3 producing heterotactic enriched PLA while 4 afforded isotactic enriched PLA. Copolymerization studies revealed the most efficient system involved the initial addition of r-LA (for 6 h) followed by ε-CL (for 42 h), which led to 99% conversion for each of the monomers. Block copolymers of PLA-b-CL and PCL-b-LA and random copolymers PLA-co-CL were successfully synthesized by adjusting the feed sequence.
Introduction
In recent years, there has been increasing research attention paid to accessing polyesters, as both a biodegradable and sustainable alternative to polyolefins.1 Indeed, polyesters are now popular materials employed in various fields, including the food and packaging industries as well as in medicinal and pharmaceutical fields.2–5 Homo- and copolymers that are of particular interest include those derived from ε-caprolactone (ε-CL) and lactide (LA).6–10 One of the main routes for accessing such materials is via ring opening polymerization (ROP) catalyzed by a metal-based alkoxide.11–18 We note that the industrial production of PCL and PLA relies on the use of a tin(II)-based catalyst, namely tin(octanoate),19 but such an approach is affected by the toxicity of the tin present. Given this, the development of alternative catalysts for the ROP of cyclic esters based on non-toxic metals remains highly desirable. With this in mind, we have initiated a study of ROP catalysts based on the group V metals niobium and tantalum. Results involving chelating phenoxide ligation revealed that for the ROP of ε-caprolactone, activities were >96% at temperatures in excess of 100 °C over either 20 h when conducted in toluene or 1 h when conducted in the absence of solvent.20 In other systems, we have also been exploring the use of ligands derived from the acids Ph2C(X)CO2H, where X = OH, or NH2, and have reported multimetallic ROP systems based on lithium or zinc as well as rare earth complexes.21–23 Herein, we extend our investigations on such acids and report the synthesis and characterisation of new niobium and tantalum complexes bearing ligands derived from 2,2′-diphenylglycine or benzilic acid (see Scheme 1). The molecular structures are reported together with the catalytic activity of these new niobium and tantalum systems for the ROP of cyclic esters (ε-caprolactone (ε-CL) and rac-lactide (r-LA)). | ||
| Scheme 1 Complexes 1–4 prepared herein. | ||
Results and discussion
Synthesis of benzilic acid derived complexes
Reaction of [Nb(OEt)5] with one equivalent of benzilic acid, Ph2C(OH)CO2H, (L1H2) in refluxing toluene afforded, after extraction into acetonitrile, colourless prisms of the complex [Nb4(OEt)8(L1)4(μ-O)2] (1). Crystals suitable for an X-ray diffraction study were grown from a saturated solution of acetonitrile at ambient temperature. The molecular structure is shown in Fig. 1, with selected bond lengths and angles given in the caption; for an alternative view see Fig. S1, ESI.† Complex 1 adopts a centrosymmetric structure in the monoclinic space group C2/c. Each Nb(V) ion is six coordinate: one benz2− anion is chelating through the alkoxide and one oxygen of the carboxylate; there are two cis ethoxide anions, one μ2-oxide oxygen atom that forms a bridge between Nb(1) and Nb(1)i or Nb(2) and Nb(2)i (where i = 1 − x, y, ½ − z), and the final coordination site is occupied by the carbonyl oxygen of a benz2− bound at the other symmetry-unique metal ion. The five-membered chelate appears to be fairly strained with bond angles of 75.07(6) and 74.41(6)° at Nb(1) and Nb(2) respectively. It is notable that the C···O distances in each carboxylate are rather similar. The C–O bond lengths are 1.271(3) Å for C(1)–O(1) and 1.244(3) Å for C(1)–O(2); for C(15)–O(4) and C(15)–(O5) the lengths are 1.266(2) and 1.250(3) Å respectively. The small difference between the two C···O distances suggests these are best visualised as delocalised carboxylates rather than carbonyl and alkoxide moieties. The bonds Nb(1)–O(5)i (where i = 1 − x, y, ½ − z) and Nb(2)–O(2) are 2.2460(15) and 2.2401(15) Å are rather longer than all of the other Nb–O bonds suggesting a weaker interaction consistent with significant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O character. There is minor disorder in the orientation of the ethoxide alkyl chains, but this was readily modelled using standard procedures.
O character. There is minor disorder in the orientation of the ethoxide alkyl chains, but this was readily modelled using standard procedures.
 | ||
| Fig. 1 (a) Asymmetric unit of 1 with atoms drawn as 50% probability ellipsoids. (b) Core of 1 (twice the asymmetric unit). Hydrogen atoms and phenyl groups have been omitted for clarity. Small-scale disorder is not represented. Symmetry equivalent atoms are generated by i = 1 − x, y, ½ − z. Selected bond lengths (Å) and angles (°): Nb(1)–O(1) 2.1205(15), Nb(1)–O(3) 1.9517(15), Nb(1)–O(51) 2.2460(15), Nb(1)–O(7) 1.9152(4), Nb(1)–O(9) 1.8572(16), Nb(1)–O(10) 1.8575(16), Nb(2)–O(2) 2.401(15), Nb(2)–O(4) 2.1334(14), Nb(2)–O(6) 1.9442(4), Nb(2)–O(8) 1.9081(14), Nb(2)–O(11) 1.8647(15), Nb(2)–O(12) 1.8502(15); O(1)–Nb(1)–O(3) 75.07(6), O(9)–Nb(1)–O(10) 97.42(7), O(4)–Nb(2)–O(6) 74.41(6), O(8)–Nb(2)–O(11) 91.27(6). | ||
The space group symmetry assembles pairs of these units into tetranuclear molecules that are non-centrosymmetric and contain four niobium atoms, four benz2−, eight ethoxide ligands and two μ2-oxide anions bridging between pairs of niobium atoms.
Similar use of [Ta(OEt)5] led to the isolation of the complex [Ta4(OEt)8(L1)4(μ-O)2]·0.5MeCN (2·0.5MeCN). Crystals suitable for an X-ray diffraction study were grown from a saturated solution of acetonitrile at ambient temperature. The molecular structure is shown in Fig. 2, with selected bond lengths and angles given in the caption; for an alternative view see Fig. S2, ESI.† This is isostructural with the Nb compound and differs only in the presence of a small amount of uncoordinated solvent that was modelled using a solvent mask (Squeeze) in Olex2.
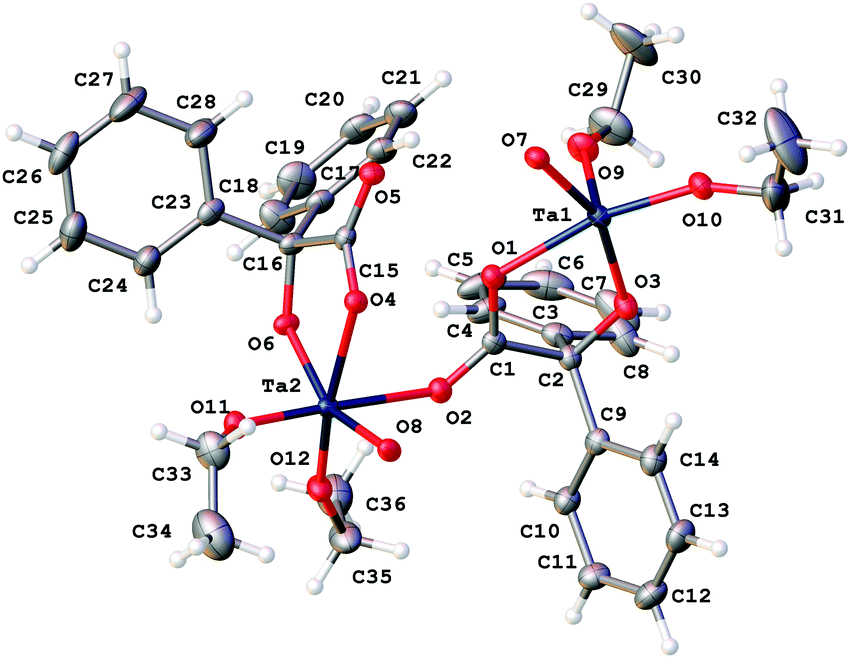 | ||
| Fig. 2 Asymmetric unit of 2·0.5MeCN. Non coordinated solvent molecules have been removed for clarity. Selected bond lengths (Å) and angles (°): Ta(1)–O(1) 2.1135(16), Ta(1)–O(3) 1.9573(17), Ta(1)–O(5) 2.2246(16), Ta(1)–O(7) 1.9175(14), Ta(1)–O(9) 1.8629(18), Ta(1)–O(10) 1.8643(18), Ta(2)–O(2) 2.2139(17), Ta(2)–O(4) 2.1283(16), Ta(2)–O(6) 1.9506(16), Ta(2)–O(8) 1.9091(4), Ta(2)–O(11) 1.8759(17), Ta(2)–O(12) 1.8547(17); O(1)–Ta(1)–O(3) 75.19(7), O(9)–Ta(1)–O(10) 96.60(8), O(4)–Ta(2)–O(6) 74.38(6), O(8)–Ta(2)–O(11) 91.32(7). | ||
The 1H NMR spectrum is consistent with the formulation with peaks assigned to aromatic protons (20H), as well as methylene (8H) and methyl (12H) groups of the ethoxide ligands, respectively. The IR spectra do not contain a sharp band at 3394 cm−1 corresponding to the ν(OH) stretching vibration of the parent L2H2 acid. The presence of new peaks at 486, 462 cm−1 suggest the formation of M–O bonds (Fig. S3, ESI†). In the mass spectrum of 1, peaks at m/z 1487.40 corresponds to loss of four OEt and H+ from the parent ion (Fig. S4, ESI†); the ESI-MS of 2·0.5MeCN is shown in Fig. S5, ESI.†
Synthesis of diphenylglycine derived complexes
Reaction of [Nb(OEt)5] with one equivalent of 2,2′-diphenylglycine, Ph2C(NH2)CO2H (L2H3), in refluxing toluene afforded, after extraction into acetonitrile, colourless prisms of the complex [Nb2(OEt)4(L2H2)4(μ-O)]·2MeCN (3·2MeCN). Crystals suitable for an X-ray diffraction study were grown from a saturated solution of acetonitrile at ambient temperature. The molecular structure is shown in Fig. 3, with selected bond lengths and angles given in the caption; for an alternative view see Fig. S6, ESI.† Complex 3·2MeCN crystallises in the centrosymmetric monoclinic space group C2/c with two unique Nb atoms in the asymmetric unit that are linked by an oxo bridge. The dinuclear molecules comprise two niobium atoms, four L2H2 ligands, two ethoxide ligands and one μ2-oxide anion. Each Nb(V) ion is seven coordinate in a roughly pentagonal bipyramidal geometry. In the plane around each Nb ion are two cis chelating L2H2− anions and one oxide. Above and below the plane in axial positions lie ethoxide anions. The oxide bridge is close to symmetric (bond lengths are 1.8942(12) and 1.9154(12) Å for Nb(1) and Nb(2) respectively) and the dimer is nearly centrosymmetric. The bound NH2 group makes hydrogen bonds to the carboxylate bound at the second metal centre in the cluster.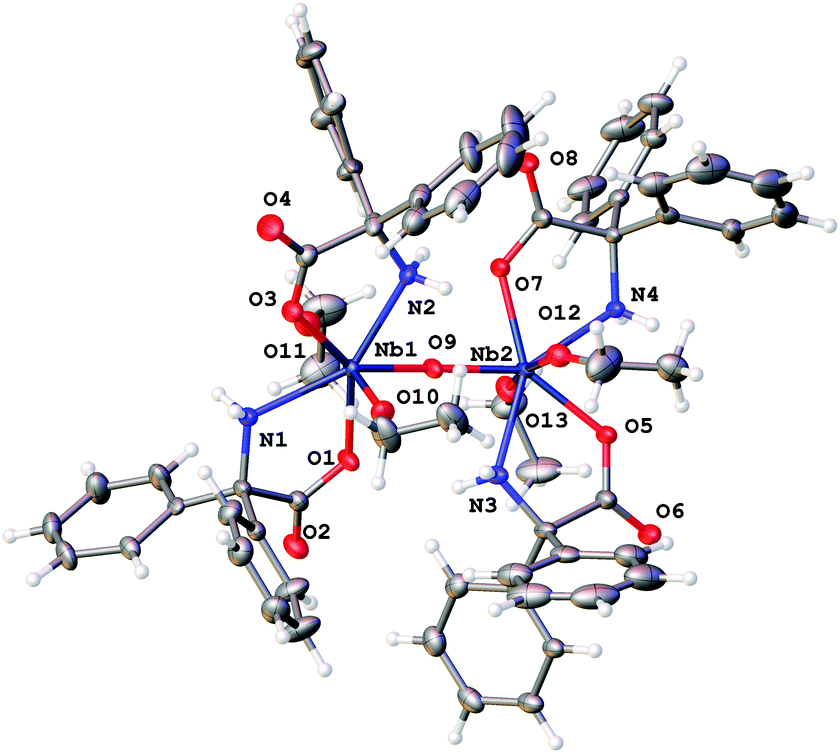 | ||
| Fig. 3 Molecular structure of [Nb2(OEt)4(L2H2)4(μ-O)]·2MeCN (3·2MeCN). Non coordinated solvent molecules have been removed for clarity. Selected bond lengths (Å) and angles (°): Nb(1)–O(1) 2.1123(12), Nb(1)–O(3) 2.1060(12), Nb(1)–O(9) 1.8942(12), Nb(1)–O(10) 1.8791(14), Nb(1)–O(11) 1.9022(14), Nb(1)–N(1) 2.3077(15), Nb(1)–N(2) 2.2919(14), Nb(2)–O(5) 2.1093(12), Nb(2)–O(7) 2.1130(12), Nb(2)–O(9) 1.9154(14), Nb(2)–O(12) 1.8907(14), Nb(2)–O(13) 1.8647(14), Nb(2)–N(3) 2.2967(14), Nb(2)–N(4) 2.3068(14); Nb(1)–O(9)–Nb(2) 165.47(9), O(1)–Nb(1)–N(1) 69.92(5), O(9)–Nb(1)–N(2) 69.93(5), O(5)–Nb(2)–N(3) 69.98(5), O(7)–Nb(2)–N(4) 69.84(5). | ||
There is minor disorder in the ethoxide and also the orientation of the phenyl groups. This was dealt with satisfactorily using standard techniques within Olex2. A small amount of unbound solvent was modelled using a solvent mask (Squeeze) within Olex2.
Similar use of [Ta(OEt)5] led to the isolation of the complex [Ta2(OEt)4(L2H2)4(μ-O)]·2.25MeCN (4·2.25MeCN). Crystals suitable for an X-ray diffraction study were grown from a saturated solution of acetonitrile at ambient temperature. The molecular structure is shown in Fig. 4, with selected bond lengths and angles given in the caption; for an alternative view see Fig. S7, ESI.† The Ta compound is isostructural to 3 and differs only in the amount of solvent present.
 | ||
| Fig. 4 Molecular structure of [Ta2(OEt)4(L2H2)4(μ-O)]·2.25MeCN (4·2.25MeCN). Non coordinated solvent molecules have been removed for clarity. Selected bond lengths (Å) and angles (°): Ta(1)–O(1) 2.101(4), Ta(1)–O(3) 2.094(4), Ta(1)–O(9) 1.899(4), Ta(1)–O(10) 1.895(4), Ta(1)–O(11) 1.904(4), Ta(1)–N(1) 2.298(5), Ta(1)–N(2) 2.283(5), Ta(2)–O(5) 2.101(4), Ta(2)–O(7) 2.109(4), Ta(2)–O(9) 1.920(4), Ta(2)–O(12) 1.894(4), Ta(2)–O(13) 1.891(4), Ta(2)–N(3) 2.299(5), Ta(2)–N(4) 2.297(5); Ta(1)–O(9)–Ta(2) 164.9(3), O(1)–Ta(1)–N(1) 70.25(16), O(9)–Ta(1)–N(2) 70.07(17), O(5)–Ta(2)–N(3) 70.10(16), O(7)–Ta(2)–N(4) 70.26(16). | ||
The 1H NMR spectrum supports the formulation of 3·2MeCN and 4·2.25MeCN with the multiple aromatics peaks between 7.03–7.75 ppm, and quartets at 3.50–2.52 ppm and a triplet at 1.06–1.10 ppm assigned to the ethoxide groups; the signal for the NH2 groups appear as a broad peak at ca. 2.2 ppm. In the IR spectrum of the parent acid L2H3, there are two absorptions near 3441 and 3267 cm−1 (assigned to symmetric and asymmetric ν(N–H) stretching), and two weak ν(N–H) absorptions are found at near 3440 and 3314 cm−1 for complexes 3·2MeCN and 4·2.25MeCN (Fig. S8, ESI†). In the mass spectrum of complex 3·2MeCN, the peaks at m/z 1366 corresponds to loss of 3H+ from the parent ion (Fig. S9, ESI†); the ESI-MS of 4·2.25MeCN is given in Fig. S10, ESI.†
Catalytic behaviour toward ring opening polymerization (ROP) of ε-caprolactone (ε-CL)
Complexes 1–4 were screened for the ROP of ε-CL and the results are presented in Table 1. The benzilic acid derived complexes 1 and 2·0.5MeCN exhibited conversions of >99% and 50% over 24 h, respectively (for a plot of conversion and time (min) for the polymerization of ε-CL using 1 and 2, see Fig. S11, ESI†). By contrast, complexes 3·2MeCN and 4·2.25MeCN derived from 2,2′-diphenylglycine were inactive towards the ROP of ε-CL under the same conditions (Table 1, entries 1–4). The results suggest the presence of the amine-containing Ph2C(NH2)CO2− moiety is unfavourable in terms of ROP of ε-CL in such complexes. We note that during metal-free studies, we also observed a similar trend, i.e. L1H2 was active whereas L2H3 was not.23| Run | Cat. | [CL]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[cat]0:[BnOH]a :[cat]0:[BnOH]a |
Conv.b (%) | M n (calc.) | M n (obs.) | PDId |
|---|---|---|---|---|---|---|
| a [CL] = 10.00 mmol, [catalyst 1 or 2] = 0.01 mmol, [3 or 4] = 0.02 mmol, [BnOH] = 0.04 mmol (0.01 M in toluene), 100 °C, 24 h. b Determined by 1H NMR spectroscopy. c M n(calc.) = 114.14 × [CL]0/[Nb/Ta]0 × conv.% + MBnOH, assuming one propagation chain per metal atom. d M n(obs) and PDI obtained by GPC in THF relative to polystyrene standards corrected by the Mark–Houwink correction factor Mn(obs) = MnGPC raw data × 0.56. e Run 9, the reaction was happened at 160 °C. | ||||||
| 1 | 1 | 250:1:1 |
>99 | 7170 | 12000 |
1.2 |
| 2 | 2 | 250:1:1 |
50 | 3675 | 9000 | 1.9 |
| 3 | 3 | 250:1:1 |
2 | — | — | — |
| 4 | 4 | 250:1:1 |
2 | — | — | — |
| 5 | 1 | 250:1:0 |
76 | 5468 | 9000 | 1.2 |
| 6 | 2 | 250:1:0 |
38 | 2757 | 6000 | 1.3 |
| 7 | 3 | 250:1:0 |
1 | — | — | — |
| 8 | 4 | 250:1:0 |
1 | — | — | — |
| 9 | 4 | 250:1:0 |
87 | 12459 |
10000 |
1.3 |
| 10 | 1 | 125:1:1 |
99 | 3639 | 7000 | 1.4 |
| 11 | 1 | 500:1:1 |
91 | 13091 |
28000 |
1.3 |
| 12 | 1 | 1000:1:1 |
20 | 5815 | 9000 | 1.4 |
Complex 1 afforded PCL with high molecular weight (Mn = 12000) and narrow molecular distribution (1.2) (Table 1, entry 1). When conducting the polymerizations in the absence of BnOH, the polymerization activity and molecular weight decreased slightly (Table 1, entries 5–8). When the mole ratio CL:Nb was increased from 125 to 500, the molecular weight increased from 7000–28000 (PDI = 1.2–1.4) with the conversion rates and distributions remaining stable. Increasing the ratio to 1000:1 proved to be detrimental to the conversion rate (Table 1, entry 12).
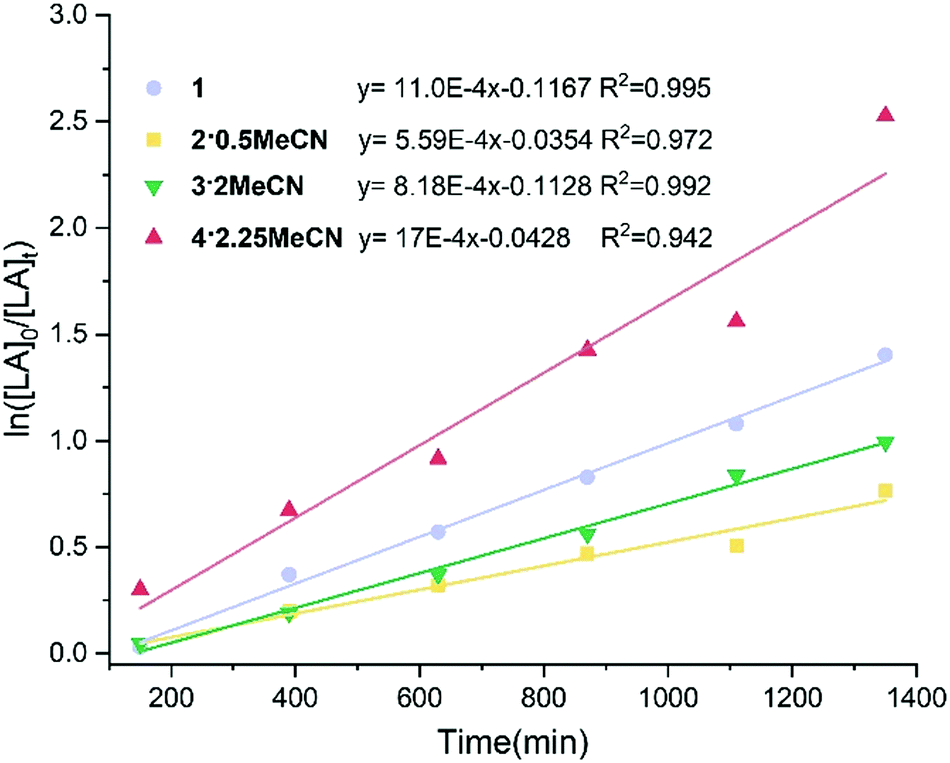
A kinetics study for the polymerization of ε-CL using 1 and 2·0.5MeCN in the ratio [CL]0:[Nb/Ta]0:[BnOH] 250:1:1 at 100 °C was performed (Fig. 5). From the plot, there is an induction period (ca. 60 min.) for 1. The calculated slope of the linear section of the curve is equal to the apparent polymerization rate constant.24
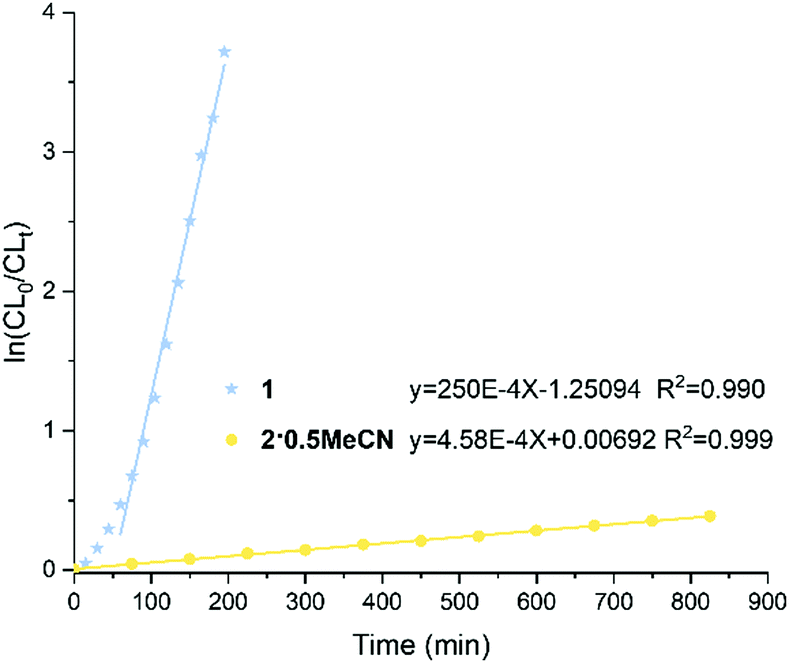 | ||
| Fig. 5 Plots of ln([CL]0/[CL]t) versus time catalyzed by complexes 1 and 2·0.5MeCN; reaction conditions: [CL]:[Nb/Ta]:[BnOH] = 250:1:1, [CL] = 10.00 mmol, [catalyst] = 0.01 mmol, [BnOH] = 0.04 mmol (0.01 M in toluene), 100 °C. | ||
For complex 2·0.5MeCN, there is a first-order dependence of the rate of the polymerization without any induction period. The rate constants (Kobs) are 250 × 10−4 min−1, R = 0.990 and 4.58 × 10−4 min−1, R = 0.999 for 1 and 2·0.5MeCN, respectively. From the rate constants, it is inferred that the polymerization rate is significantly faster for the Nb complex versus the Ta complex. In previous work,20 we have utilized tetraphenolate niobium and tantalum complexes for the ROP of ε-CL. Under the same conditions, such niobium complexes exhibited excellent conversions and afforded higher molecular weight products compared with the analogous tantalum complexes.
In the MALDI-TOF spectrum (Fig. 6), the PCL formed by 1 exhibits two major families of peaks. The main peaks are assigned to polymer chains with PhCH2O– end groups which agreed with the 1H NMR spectra (Fig. S12, ESI†). The results suggested the formation of Nb-OCH2Ph species acted as the initiator for the ROP via the coordination–insertion mechanism;25 signals at 7.34 and 5.10 ppm indicating a benzyl group and the peak at 3.64 ppm was ascribed to an OH terminal group. A second distribution of peaks is associated with ethoxy (–OCH2CH3) and hydroxyl (–OH) end groups, which could also be observed in the 1H NMR spectra (Fig. S12, ESI†). This result indicated that an ethoxide niobium complex was also present and involved in the ROP process via a coordination–insertion mechanism.26,27 The successive series showed that there was a m/z difference of 114 between neighbouring peaks, which corresponds to the molecular weight of the monomer. The end group of PCL isolated using 1 in the absence of BnOH was also investigated by MALDI-TOF, and the spectrum (Fig. S13, ESI†) confirmed that PCL with an ethoxy and hydroxyl (–OH) end groups was formed.
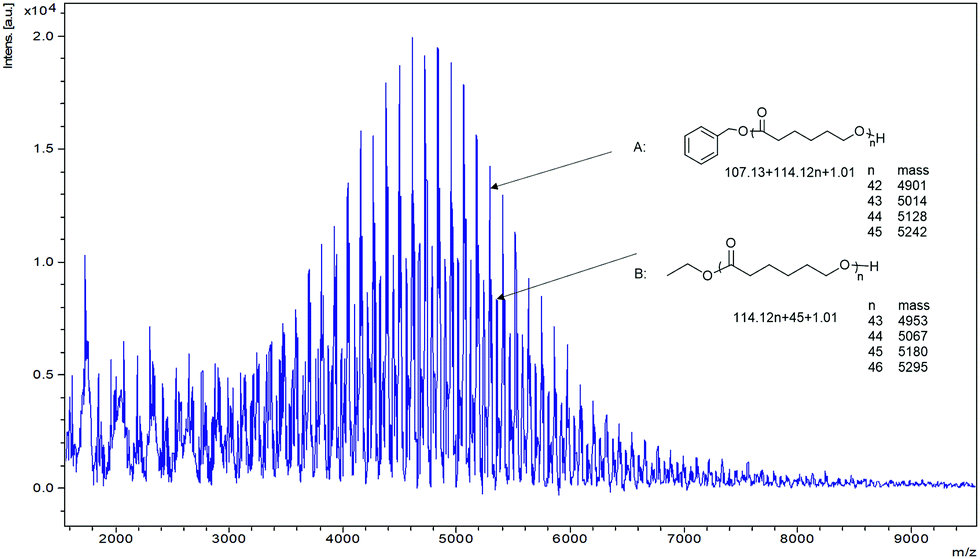 | ||
| Fig. 6 MALDI-TOF spectrum of the PCL catalysed by complexes 1 (Table 1, entry 1); n is the degree of polymerization. | ||
Catalytic behaviour toward ROP of rac-lactide (r-LA)
Complexes 1–4 have also been screened for the ROP of r-LA using the [LA]0:[Nb/Ta]0:[BnOH] ratio 250:1:1 over 24 h. The results are depicted in Table 2. The reaction temperature was first investigated, and it was observed that when the temperature was 130 °C, only the L1H2 derived complexes 1 and 2·0.5 MeCN were active towards the ROP of r-LA, and the monomer conversion reached 40% after 24 h for 1. It was evident that complexes derived from L2H3 were inactive at 130 °C even after 24 h (Table 2, entries 1–4). When the temperature was increased to 160 °C, the activities substantially increased in all cases (Table 2, entries 9–12). For example, a 92% conversion was achieved using 4·2.25MeCN affording a polymer with Mn = 9000 with good control (PDI = 1.0) after 24 h (Table 2, entry 12). We also tested the catalytic behaviour of complexes 1–4 in the absence of BnOH at 130 °C and 160 °C. Interestingly, the results suggested that addition of BnOH has a negative effect on the polymerization rate and molecular weight (Table 2, entries 5–8 and 13–16). For a plot of conversion and time (min) for the polymerization of r-LA using 1–4, see Fig S14, ESI.† The kinetic study of the ROP of r-LA using 1–4 is shown in Fig. 7. The plot exhibited first order dependence on the r-LA concentration. The rate order observed is 4·2.25MeCN > 1 > 3·2MeCN > 2·0.5MeCN. The L2H3 derived Ta complex 4 showed the highest rate [Kobs = 17 × 10−4 min−1] followed by the L1H2 derived Nb complex 1 [Kobs = 11 × 10−4 min−1], L2H3 derived Nb complex 3·2MeCN [Kobs = 8.2 × 10−4 min−1] and the L1H2 derived Ta complex 2 [Kobs = 5.6 × 10−4 min−1]. Interestingly, for the ROP of r-LA, these results show that the Ta-based complex performs best when bound by the L2H3 derived ligand set, whereas the Nb system is superior in the case of the L1H2 derived ligand.
| Run | Cat. | [LA]0:[cat.]0:[BnOH]a |
T (°C) | Conv.b (%) | M n (calc.) | M n (obs.) | PDId | Pr |
|---|---|---|---|---|---|---|---|---|
| a [r-LA] = 10.00 mmol, [catalyst 1 or 2] = 0.01 mmol, [3 or 4] = 0.02 mmol, [BnOH] = 0.04 mmol (0.01 M in toluene), 24 h. b Determined by 1H NMR spectroscopy. c M n(calc.) = 144 × [LA]0/[Nb/Ta]0 × conv.% + Mendgroup, assuming one propagation chain per metal atom. d M n(obs) and PDI obtained by GPC in THF relative to polystyrene standards corrected by the Mark-Houwink correction factor Mnobs = MnGPC raw data × 0.58. e Pr is the probability of r dyad as determined by 2D homo J-resolved NMR spectroscopic analysis. | ||||||||
| 1 | 1 | 250:1:1 |
130 | 40 | 3708 | 1200 | 1.4 | — |
| 2 | 2 | 250:1:1 |
130 | 25 | 2358 | 900 | 1.6 | — |
| 3 | 3 | 250:1:1 |
130 | 3 | — | — | — | — |
| 4 | 4 | 250:1:1 |
130 | 11 | — | — | — | — |
| 5 | 1 | 250:1:0 |
130 | 72 | 6526 | 2700 | 1.3 | — |
| 6 | 2 | 250:1:0 |
130 | 39 | 3556 | 2000 | 1.2 | — |
| 7 | 3 | 250:1:0 |
130 | 4 | — | — | — | — |
| 8 | 4 | 250:1:0 |
130 | 46 | 8326 | 2300 | 1.3 | — |
| 9 | 1 | 250:1:1 |
160 | 77 | 7038 | 4500 | 1.1 | 0.50 |
| 10 | 2 | 250:1:1 |
160 | 55 | 5058 | 2000 | 1.2 | 0.46 |
| 11 | 3 | 250:1:1 |
160 | 65 | 11808 |
9000 | 1.2 | 0.61 |
| 12 | 4 | 250:1:1 |
160 | 92 | 16668 |
9000 | 1.0 | 0.36 |
| 13 | 1 | 250:1:0 |
160 | 93 | 8416 | 7200 | 1.3 | 0.51 |
| 14 | 2 | 250:1:0 |
160 | 87 | 7876 | 4000 | 1.9 | 0.50 |
| 15 | 3 | 250:1:0 |
160 | 89 | 16066 |
8000 | 1.2 | 0.59 |
| 16 | 4 | 250:1:0 |
160 | 99 | 17866 |
10000 |
1.4 | 0.35 |
 | ||
| Fig. 7 Plots of ln([LA]0/[LA]t) versus time catalyzed by complexes 1–4; Reaction conditions: [LA]0:[Nb/Ta]0:[BnOH] = 250:1:1, [LA] = 10 mmol, [catalyst 1 or 2] = 0.01 or 0.02 mmol (3 or 4), [BnOH] = 0.04 mmol (0.01 M in toluene), reaction temperature: 160 °C. | ||
To determine the stereo-chemical microstructure of the PLA polymers, 2D homo J-resolved was employed and peaks were assigned by reference to the literature.28,29 For example, complex 4 gives isotactic PLA (Table 2, run 12 and 16, Pr = 0.35–0.36), while 3 shows a selectivity for heterotactic PLA (Fig. S15–S22, ESI†).
Polymer end groups were analysed by 1H NMR spectroscopy and MALDI-TOF mass spectra. For example, for the ROP of r-LA using 2·0.5 in the absence of BnOH in a 250:1:0 ratio, a series of peaks were observed with the main peaks identified as CH3CH2O– and OH end groups. The set of peaks, namely 144.13 n + 45 + 1.01 + 22.99, is attributed to (LA)n + –OCH2CH3 + H + Na+ (Fig. 8). From the 1H NMR spectrum, a quartet peak (e) at δ 4.15–4.20 ppm and a triple peak (f) at 1.27 ppm are consistent with the presence of the end group OCH2CH3, whilst the quartet peak (a) at δ 4.32–4.37 is the methine group of the other end group (Fig. 9).
 | ||
| Fig. 8 MALDI-TOF spectrum of the PLA obtained using 2·0.5MeCN (Table 2, entry 14). | ||
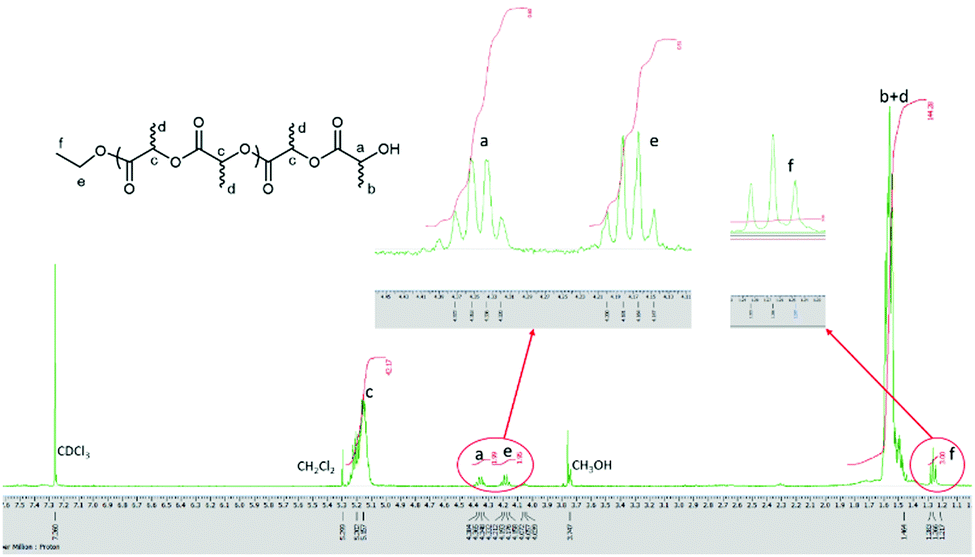 | ||
| Fig. 9 1H NMR spectrum (400 MHz, CDCl3) of the PLA obtained using 2·0.5MeCN (Table 2, entry 14). | ||
Copolymerization of ε-CL and r-LA
Based on the results of the homopolymerizations of r-LA and ε-CL (Table 1, run 9 and Table 2, run 16), 4·2.25MeCN was selected to further explore the ability for the controlled synthesis of diblock PLA-b-PCL or PCL-b-PLA and random copolymers. The copolymerization of ε-CL and r-LA was studied using 4·2.25MeCN with different feeding sequences at 160 °C and with a ratio of [CL]:[LA]:[cat] = [125]:[125]:[1] (i.e. CL:LA = 50:50). Results are summarized in Table 3.
| Run | First stage polymerization | Second stage polymerization | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| First monomer | Time (h) | Conversion first monomer | Second monomer | Time (h) | Conv. CLa (%) | Conv. LAa (%) | M n(calc.) | M n(obs.) | PDIb | |
| [LA] = 3.46 mmol, [CL] = 3.46 mmol, [complex 4] = 0.028 mmol; solvent: toluene = 2 mL; temperature: 160 °C; reaction time: 48 h.a calculated from 1H NMR spectroscopy.b Mn(obs) obtained by GPC in THF relative to polystyrene standards corrected by the Mark–Houwink correction factor.c Mn(calc.) = MLA × [LA]0/[Ta] × %conv + MCL × [CL]0/[Ta] × %conv + Mendgroup. | ||||||||||
| 1 | CL | 12 | 52 | LA | 36 | 68 | 44 | 8851 | 6000 | 1.1 |
| 2 | CL | 24 | 88 | LA | 24 | 95 | 59 | 12079 |
8000 | 1.3 |
| 3 | LA | 6 | 74 | CL | 42 | 99 | 99 | 16000 |
11000 |
1.5 |
| 4 | LA | 12 | 86 | CL | 36 | 37 | 95 | 11232 |
8000 | 1.5 |
| 5 | LA | 24 | 94 | CL | 24 | 59 | 99 | 13160 |
9000 | 1.2 |
| 6 | LA + CL | 48 | — | — | — | 94 | 95 | 15294 |
13000 |
1.4 |
To find an optimum reaction time for the copolymerization, the relationship between the conversion of the monomers and the reaction time following the addition of the first monomer was studied. It was found that when ε-CL was the first monomer added (12 h polymerization time), then following addition of r-LA and an additional 36 h, the conversions of ε-CL and r-LA were 68% and 44% respectively. On increasing the first monomer (ε-CL) reaction time to 24 h, the final conversion of ε-CL and r-LA reached 95% and 59%, respectively. It was evident that there was always an amount of unreacted r-LA when ε-CL was the first monomer added no matter how prolonged was the reaction time. It was evident here that the ROP of ε-CL was somewhat more difficult than that of r-LA. This is typified by the homopolymerization results whereby the activity of complex 4·2.25MeCN for ε-CL polymerization is rather low, and required 24 h to convert 250 equiv. of ε-CL to 88% monomer conversion at 160 °C, whilst 18 h was needed to convert 99% of the same amount of r-LA. The 13C NMR spectrum of the copolymer when ε-CL was the first monomer added and reacted for 24 h exhibits two carbonyl signals at 173.59 and 169.37 ppm, corresponding to the PCL and PDLLA block, respectively (Fig. 10(b)). The absence of any other peaks between these two carbonyl groups suggests no transesterification occurring in the polymerization.30
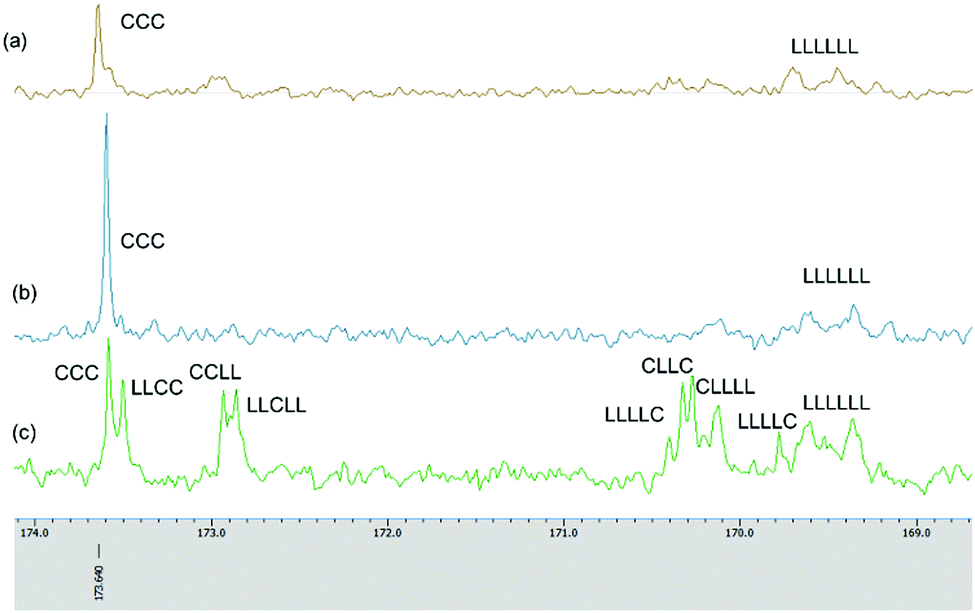 | ||
| Fig. 10 Expanded 13C NMR spectra of copolymers prepared in sequential copolymerization of r-LA and ε-CL catalysed by 4·2.25MeCN. (a) Table 3, entry 3; (b) Table 3, entry 2; (c) Table 3, entry 6. | ||
In contrast, when r-LA was added first and then after 6 h ε-CL was introduced, 4·2.25MeCN was found to be an efficient initiator and could produce a block copolymer after 48 h with conversions for ε-CL and r-LA of 99% and 99%, respectively (Table 3, entry 3). Increasing the r-LA polymerization time to 12 h and 24 h reduced the conversion of ε-CL and led to transesterification. In the 1H NMR of PLA-b-CL (Fig. S23, ESI†), the expected signals for the copolymer were observed; the end groups of CH3CH2O– and OH were present. The 13C NMR spectrum of the copolymer exhibits two carbonyl signals at 173.63 and 169.45 ppm, corresponding to the PCL and PDLLA block, respectively (Fig. 10(a)).
For entry 6, we attempted the one-pot copolymerization of r-LA and ε-CL. After 48 h, the two monomers achieved high conversions of 94% for ε-CL and 95% for r-LA. The percentage of CL–LA heterodiads could be calculated from the 1H NMR spectrum by comparing the relative intensity of the methylene protons for CL–LA and CL–CL (Fig. S24, ESI†), and the percentage of CL–LA heterodiads was 94% suggesting the copolymers had random sequences.31 The chain microstructure of the copolymer was studied by 13C NMR spectrum (Fig. 10(c)); the carbonyl sequences were assigned according to the literature.30–34 The average lengths of lactidyl unit LLA and caproyl unit LCL can be calculated according to the equations as described by Kasperczyk.35 For PCL-co-PDLLA (Table 3, entry 6), LLA = 2.5 and LCL = 1.9, while LLA is slightly longer. The signal at 171 ppm, relating to the C–L–C sequence was not detected indicating the absence of second mode of transesterification.35 The molecular weights of the copolymers were lower than their theoretical values, with polydispersity in the range 1.1–1.5.
Experimental
All manipulations were carried out under an atmosphere of dry nitrogen using Schlenk and cannula techniques or in a conventional nitrogen-filled glove box. ε-Caprolactone was dried over molecular sieves (3 Å). Toluene were dried over sodium/benzophenone, and acetonitrile from calcium hydride. All solvents were distilled and degassed prior to use. All chemicals were purchased from Sigma Aldrich or TCI UK and used as received. IR spectra (nujol mulls, KBr windows) were recorded on a Nicolet Avatar 360 FT IR spectrometer. Elemental analyses were performed by the elemental analysis service at the London Metropolitan University, or the Department of Chemistry at the University of Hull. MALDI-TOF mass spectra were acquired by averaging at least 100 laser shots. Molecular weights were calculated from the experimental traces using the OmniSEC software. NMR spectra were recorded at 400 MHz on a JEOL ECZ 400S spectrometer.Crystallography‡
Full sets of X-ray diffraction intensity data were collected using modern X-ray diffractometers at the National Crystallography Service in Southampton, UK. Routine processing of raw intensity data and multi-scan absorptions corrections were applied. The structures were solved using dual-space methods within SHELXT and full-matrix least squares refinement was carried out using SHELXL-201836via program Olex2.37 All non-hydrogen positions were located in the direct and difference Fourier maps and refined using anisotropic displacement parameters.Kinetic method
Kinetic experiments were carried out using a Youngs tap NMR tube at the required temperature. For example, under nitrogen, ε-CL (1.74 mmol, 0.19 mL), toluene-d8 (0.5 mL) and BnOH in toluene (0.01 M, 0.69 mL) were added to the NMR tube containing 0.12 mL of a stock solution of complex in toluene (0.033 mL mg−1). The sample was then analyzed at 15 minute intervals.Conclusions
In this work, we have isolated and structurally characterized two different families of complexes on reacting the acids Ph2C(X)CO2H (L1H2, X = OH; L2H3 X = NH2) with the group V metal alkoxides Nb(OEt)5 (M = Nb, Ta). In the case of benzilic acid (L1H2), the products were tetranuclear complexes of the form [M4(OEt)8(L1)4(μ-O)2], whereas for 2,2′-diphenylglycine (L2H3), dinuclear complexes of the type [M2(OEt)4(L2H2)4(μ-O)] were produced. The benzilic acid derived complexes 1 and 2·0.5MeCN exhibited good activities in the ROP of ε-CL in the presence of BnOH (conversions were somewhat lower in the absence of BnOH), with the Nb system affording superior conversion. Complexes 1–4 were all active for the ROP of r-LA in the absence of BnOH at 160 °C. The Ta-based complex performs best when bound by the L2H3 derived ligand set, whereas the Nb system is superior in the case of the L1H2 derived ligand. The analysis of the stereoselective indicated complex 4 affords an isotactic PLA (Pr = 0.35). End group analysis confirmed the alkoxide moiety of the complex initiated the ROP of ε-CL or r-LA by a coordination–insertion mechanism. Copolymerizations of ε-CL or r-LA, in the presence of 4·2.25MeCN, were conducted using different feed sequences. Block copolymers of PLA-b-CL and PCL-b-LA and random copolymers PLA-co-CL were successfully synthesized by adjusting the feed sequence.The two new families of niobium and tantalum complexes reported herein, add to the sparse number of examples reported to-date that utilize these metals in catalysts for the ring opening polymerization of cyclic esters.
Author contributions
Xin Zhang conducted the synthetic work, Timothy J. Prior conducted the crystallography and Carl Redshaw supervised the synthetic work and acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by UKRI Creative Circular Plastic Grant (EP/S025537/1). We thank the University of Hull for sponsorship (of X. Z.). We also thank the EPSRC National Crystallographic Service at Southampton for data.Notes and references
- K. Stridsberg, M. Ryner and A. Albertsson, Adv. Polym. Sci., 2002, 157, 41–65 CrossRef CAS.
- D. J. Mooney, G. Organ, J. P. Vacanti and R. Langer, Cell Transplant., 1994, 3, 203–210 CrossRef CAS PubMed.
- A. Gupta and V. Kumar, Eur. Polym. J., 2007, 43, 4053–4074 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- J. C. Bogaert and P. Coszach, Macromol. Symp., 2000, 153, 287–303 Search PubMed.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- A. L. Sisson, D. Ekinci and A. Lendlein, Polymer, 2013, 54, 4333–4350 CrossRef CAS.
- S. Sriputtirat, W. Boonkong, S. Pengprecha, A. Petsom and N. Thongchul, Adv. Chem. Eng. Sci., 2012, 02(No. 01), 14 Search PubMed.
- F. Ebrahimi and H. Ramezani Dana, Int. J. Polym. Mater. Polym. Biomater., 2021, 1–14 CAS.
- O. Santoro, X. Zhang and C. Redshaw, Catalysts, 2020, 10, 800 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC.
- A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- R. Xu and C. Chen, Prog. Chem., 2012, 24, 1519–1525 CAS.
- R. Mehta, V. Kumar, H. Bhunia and S. N. Upadhyay, J. Macromol. Sci., Polym. Rev., 2005, 45, 325–349 CrossRef.
- Y. Al-Khafaji, X. Sun, T. J. Prior, M. R. Elsegood and C. Redshaw, Dalton Trans., 2015, 44, 12349–12356 RSC.
- Y. F. Al-Khafaji, M. R. J. Elsegood, J. W. A. Frese and C. Redshaw, RSC Adv., 2017, 7, 4510–4517 RSC.
- Y. F. Al-Khafaji, T. J. Prior, L. Horsburgh, M. R. J. Elsegood and C. Redshaw, ChemistrySelect, 2017, 2, 759–768 CrossRef.
- J. Collins, O. Santoro, T. J. Prior, K. Chen and C. Redshaw, J. Mol. Struct., 2021, 1224 Search PubMed.
- J. L. Mata-Mata, J. A. Gutiérrez, M. A. Paz-Sandoval, A. R. Madrigal and A. Martínez-Richa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6926–6942 CrossRef CAS.
- Q. Zhang, W. Zhang, N. M. Rajendran, T. Liang and W. H. Sun, Dalton Trans., 2017, 46, 7833–7843 RSC.
- L. E. Breyfogle, C. K. Williams, V. G. Young, Jr., M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2006, 928–936 RSC.
- M. Hayatifar, F. Marchetti, G. Pampaloni and S. Zacchini, Inorg. Chem., 2013, 52, 4017–4025 CrossRef CAS PubMed.
- M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2014, 6, 1892–1898 CrossRef CAS.
- C. Ludwig and M. Viant, Phytochem. Anal., 2010, 21, 22–32 CrossRef CAS PubMed.
- D. Dakshinamoorthy and F. Peruch, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2161–2171 CrossRef CAS.
- D. Pappalardo, L. Annunziata and C. Pellecchia, Macromolecules, 2009, 42, 6056–6062 CrossRef CAS.
- D. J. Darensbourg and O. Karroonnirun, Macromolecules, 2010, 43, 8880–8886 CrossRef CAS.
- M. Naddeo, A. Sorrentino and D. Pappalardo, Polymers, 2021, 13 Search PubMed.
- D. J. Gilmour, R. L. Webster, M. R. Perry and L. L. Schafer, Dalton Trans., 2015, 44, 12411–12419 RSC.
- J. Kasperczyk and M. Bero, Makromol. Chem., 1993, 194, 913–925 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Alternative views of 1–4; IR and mass spectra; conversion v time plot; 1H NMR spectra of the polymers including 2D J resolved spectra of the PLA. CCDC 2150211–2150214. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2nj02527b |
| ‡ Crystallographic data for 1–4. Crystal data for compound 1: C72H80Nb4O22, M = 1669, Monoclinic, space group C2/c, a = 22.2498(4), b = 14.4513(2), c = 25.8064(5) Å, V = 7575.3(3) Å3, Z = 4, Dc = 1.463 g cm−3, F(000) = 3408, T = 100(2) K, μ = 0.661 mm−1, λ(Mo-Kα) = 0.71075 Å, θmax = 57.396°, R1 ([I > 2σ(I)] = 0.0317, wR2 (all data) = 0.0856. For 2·0.5MeCN: C73H81.5N0.5Ta4O22, M = 2041.68, Monoclinic, space group C2/c, a = 22.2617(4), b = 14.3187(2), c = 25.8451(8) Å, V = 7491.0(3) Å3, Z = 4, Dc = 1.810 g cm−3, F(000) = 3964, T = 100(2) K, μ = 5.896 mm−1, λ(Mo-Kα) = 0.71075 Å, θmax = 57.400°, R1 ([I > 2σ(I)] = 0.0194, wR2 (all data) = 0.0475. For 3·2MeCN: C68H74N6Nb2O13, M = 1369.15, Monoclinic, space group C2/c, a = 54.0723(4), b = 10.74623(10), c = 22.47203(18) Å, V = 13041.45(19) Å3, Z = 8, Dc = 1.395 g cm−3, F(000) = 5680, T = 100(2) K, μ = 0.413 mm−1, λ(Mo-Kα) = 0.71075 Å, θmax = 57.400°, R1 ([I > 2σ(I)] = 0.0340, wR2 (all data) = 0.0881. For 4·2.25MeCN: C68.5H74.75N6.25Ta2O13, M = 1555.49, Monoclinic, space group C2/c, a = 54.1856(8), b = 10.7080(2), c = 22.4972(4) Å, V = 13038.2(4) Å3, Z = 8, Dc = 1.585 g cm−3, F(000) = 6236, T = 100(2) K, μ = 3.416 mm−1, λ(Mo-Kα) = 0.71075 Å, θmax = 58.702°, R1 ([I > 2σ(I)] = 0.0561, wR2 (all data) = 0.1510. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |