
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RAFT solution copolymerization of styrene and 1,3-butadiene and its application as a tool for block copolymer preparation†
Abdullah
Gunaydin
ab,
Patrick
Grysan
a,
Daniel
F. Schmidt
 a,
Reiner
Dieden
a,
Marc
Weydert
c and
Alexander
S. Shaplov
*a
a,
Reiner
Dieden
a,
Marc
Weydert
c and
Alexander
S. Shaplov
*a
aLuxembourg Institute of Science and Technology (LIST), 5 Avenue des Hauts-Fourneaux, L-4362 Esch-sur-Alzette, Luxembourg. E-mail: alexander.shaplov@list.lu
bUniversity of Luxembourg, 2 Avenue de l’Université, L-4365 Esch-sur-Alzette, Luxembourg
cGoodyear Innovation Center Luxembourg, Avenue Gordon Smith, L-7750 Colmar-Berg, Luxembourg
First published on 25th July 2022
Abstract
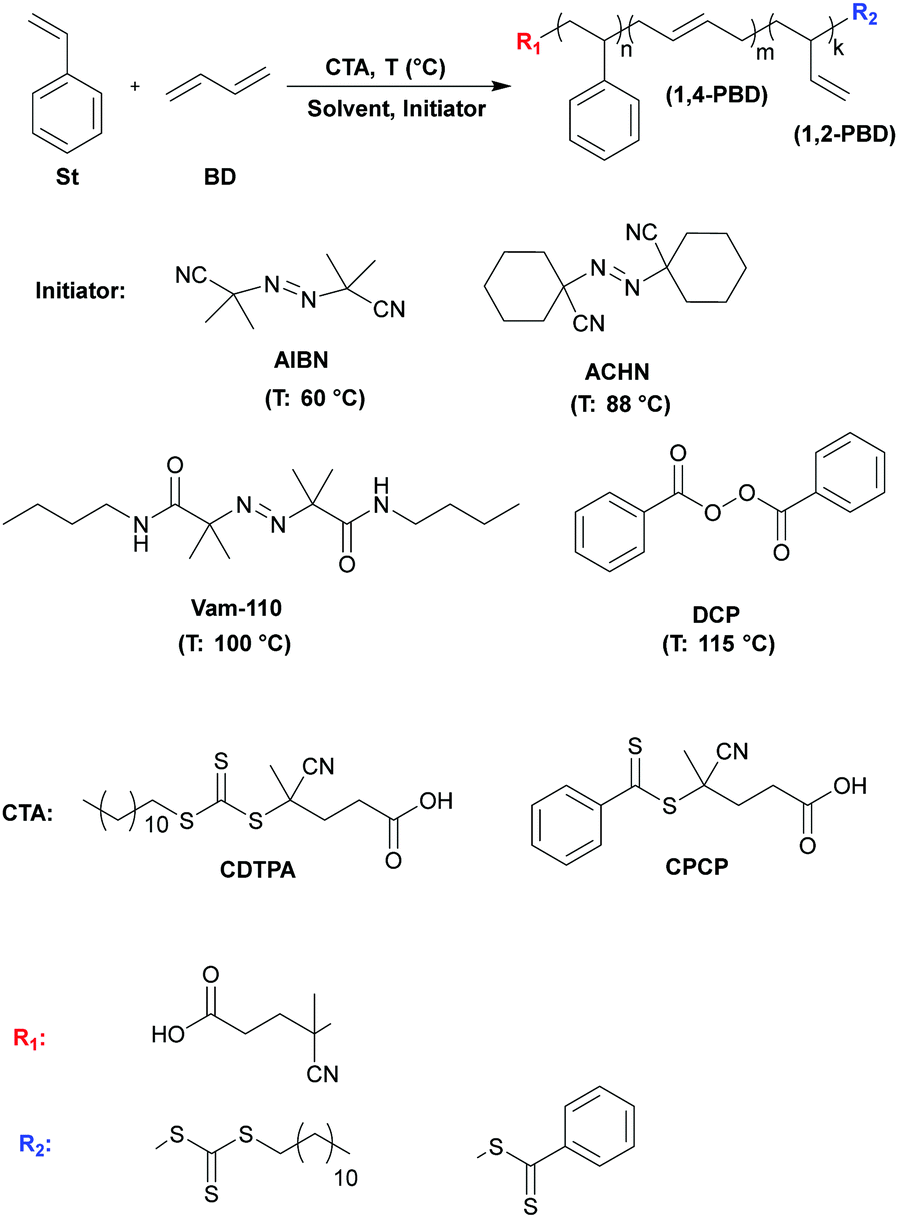
For the first time, random copolymers of styrene (St) and 1,3-butadiene (Bd) (poly(Stn-r-Bdm), styrene butadiene rubber, SBR) were successfully prepared via solution reversible addition-fragmentation-transfer (RAFT) polymerization by employing dithio- and trithiocarbonate chain transfer agents (CTAs). The influence of various reaction parameters such as temperature and duration of polymerization, type of CTA, solvent and initiator, on molecular weight, molecular weight distribution (Mw/Mn) and the yield of the copolymers was investigated in detail. Determination of optimal reaction conditions allowed for the successful preparation of linear poly(Stn-r-Bdm) having Mn of up to 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and Mw/Mn ≤ 1.6, with an isolated yield of up to 39 wt%. According to NMR the obtained copolymers were random and did not contain any styrene blocks (more than 5 units in sequence). The composition of poly(St-r-Bd) was found to be nearly independent of reaction conditions and consisted of 19.6–24.0, 15.0–15.5 and 60.5–64.5 wt% of styrene, (1,2)-Bd and (1,4)-Bd units, respectively. The glass transition temperature (Tg) of the copolymers (measured via DSC) varied between −55 and −62 °C, while Tonset (measured via TGA) ranged between 385 and 390 °C. The optimized synthetic method for production of poly(Stn-r-Bdm) copolymers was then extended to produce various poly[Xn-b-(Stm-r-Bdk)] block copolymers, where X represents different methacrylic or styrenic monomeric units. The molecular weight of the poly[Xn-b-(Stm-r-Bdk)] block copolymers was mainly dependent on the molar mass of the starting poly(Xn) macro-CTA and reached as high as 72000 g mol−1, with the SBR segment varying between 11800 and 39600 g mol−1. These materials, believed to be the first of their kind reported in the literature, show clear evidence of nanostructure formation via AFM and promise unique and attractive combinations of stiffness, toughness, thermomechanical performance and chemical reactivity. This work opens up new avenues for the synthesis of novel copolymers with exceptional levels of structural control, thus providing additional tools to the polymer research community as far as the design and creation of materials with new and useful properties is concerned.
000 g mol−1 and Mw/Mn ≤ 1.6, with an isolated yield of up to 39 wt%. According to NMR the obtained copolymers were random and did not contain any styrene blocks (more than 5 units in sequence). The composition of poly(St-r-Bd) was found to be nearly independent of reaction conditions and consisted of 19.6–24.0, 15.0–15.5 and 60.5–64.5 wt% of styrene, (1,2)-Bd and (1,4)-Bd units, respectively. The glass transition temperature (Tg) of the copolymers (measured via DSC) varied between −55 and −62 °C, while Tonset (measured via TGA) ranged between 385 and 390 °C. The optimized synthetic method for production of poly(Stn-r-Bdm) copolymers was then extended to produce various poly[Xn-b-(Stm-r-Bdk)] block copolymers, where X represents different methacrylic or styrenic monomeric units. The molecular weight of the poly[Xn-b-(Stm-r-Bdk)] block copolymers was mainly dependent on the molar mass of the starting poly(Xn) macro-CTA and reached as high as 72000 g mol−1, with the SBR segment varying between 11800 and 39600 g mol−1. These materials, believed to be the first of their kind reported in the literature, show clear evidence of nanostructure formation via AFM and promise unique and attractive combinations of stiffness, toughness, thermomechanical performance and chemical reactivity. This work opens up new avenues for the synthesis of novel copolymers with exceptional levels of structural control, thus providing additional tools to the polymer research community as far as the design and creation of materials with new and useful properties is concerned.
Introduction
Styrene butadiene rubber (SBR) is a synthetic rubber copolymer that finds widespread use in the tire industry. In addition to tires, many other industrial products (hoses, belts, flooring, shoe soles, mats, etc.) have benefited from SBR's introduction.1 Generally, the application area of a given SBR depends on its composition, thermal and tensile properties.2 SBR is industrially produced via the emulsion radical or solution anionic copolymerization of styrene (St) and 1,3-butadiene (Bd).3 Each of these methods possesses certain advantages and limitations.Despite the commercial realization of the methods mentioned above, many studies have explored other approaches for the preparation of SBR-based block copolymers.4–8 Among these, reversible addition-fragmentation-transfer (RAFT) polymerization is promising given its ability to provide a high level of control over molecular weight and unprecedented tolerance of functional groups.9,10 Moreover, in comparison with anionic polymerization, RAFT is less sensitive to moisture,11 while compared to emulsion radical polymerization, RAFT offers reduced branching and narrower molecular weight distributions.12–14 Although attempts to produce Bd-based copolymers using several chain transfer agents (CTA) and macro-CTA agents have been carried out via emulsion RAFT polymerization15–17 (for a recent review, see18), or via photo RAFT polymerization in the continuous-flow reactor,19 the studies dedicated to Bd RAFT (co)polymerization in solution are few and far between. As of this writing, the authors are aware of only seven published reports describing the solution RAFT homopolymerization of Bd20–22 and its copolymerization with acrylonitrile.23–25
Boutevin and coworkers20 conducted RAFT polymerization of Bd in acetonitrile and isopropanol with various fluorinated dithioester CTAs. Reactions were carried out using tert-butyl peroxy-3,5,5-trimethylhexanoate (Trigonox 42 S) and di-tert-butyl peroxide initiators at 105 and 150 °C, respectively.20 In spite of high reaction temperatures, the polymerization of Bd resulted in polymers with low molecular weights (Mn = 1300–1400 g mol−1), albeit with narrow molecular weight distributions (Mw/Mn = 1.2–1.5). The conversion was limited as well (12–16 wt%).20 Transitioning to a trithiocarbonate-based CTA (2-(dodecylthiocarbonothioylthio)propanoic acid, DoPAT) allowed for an increase in the molecular weight of resultant poly(Bd) to 3200 g mol−1 (Mw/Mn ≈ 1.5), although in this case polymerization was performed at a lower temperature (7022vs. 105–150 °C20) with 2,2′-azobis(2-methylpropionitrile) (AIBN) as initiator and toluene as solvent.22 The kinetics of the polymerization were found to be extremely slow, explained partly by the low kp value and partly by the low initiator concentration in the system (the Bd:CTA:AIBN molar ratio was 1077:1:0.2).22 By switching to another trithiocarbonate-based CTA, namely 2-(dodecyl thiocarbonothioylthio)-2-methylpropanoic acid (DDMAT), increasing the reaction temperature to 95 °C and using dicumyl peroxide (DCP) as the initiator, Abdollahi et al.21 were able to increase the molecular weight of the resultant poly(Bd) up to ∼11000 g mol−1 (Mw/Mn ≈ 1.4–1.9). At all studied Bd:DDMAT:DCP ratios Mn was found to be linearly dependent on conversion, but after 35 h the conversion remained limited to 20%. In contrast to DoPAT,20 however, DDMAT21 did not retard or inhibit polymerization and provided good control over chain growth. Thus, the type of the CTA and the temperature of the reaction were found to be the most crucial parameters influencing Bd solution RAFT polymerization.20–22
Solution RAFT copolymerization of Bd and acrylonitrile (AN) was studied by the group of Barner-Kowollik et al.23–25 The monomers were employed in their azeotropic ratio of AN:Bd = 38:62 and the overall monomer concentration was fixed at 9.4 M. In their first publication, copolymerization was carried out in N,N-dimethylacetamide (DMAc) at temperatures as high as 100 °C using 1,10-azobis(cyclohexanecarbonitrile) (ACHN, Vazo 88) as initiator.24 Both trithiocarbonate-(DoPAT and dibenzyltrithiocarbonate (DiBenT)) and dithioacetate-based (cumyl phenyl dithioacetate (CPDA)) CTAs were tested. The influence of the initiator to RAFT-agent ratio as well as the influence of the CTA and ACHN concentrations on the evolution of conversion with time was explored, and near-linear trends were observed under the investigated reaction conditions.24 Linear poly(Ann-r-Bdm) copolymers were obtained with Mn up to 59700 g mol−1 and Mw/Mn ratios of 1.2–2.0 depending on the extent of conversion.24 CTA type did not significantly affect the overal conversion (42–51%), but did influence the molecular weight of the obtained poly(Ann-r-Bdm) copolymer: DoPAT (Mn = 58500 g mol−1) > CPDA (42000) > DiBenT (31600).24 For a 9 h reaction time, the presence of acrylonitrile increased conversion (55%) as compared to the case of Bd homopolymerization (42–51%).20 Finally, the theoretically predicted values of molecular weight were found to be in agreement with those determined experimentally. In their next study, Barner-Kowollik et al.24 investigated the influence of various solvents and initiators on the rate of Bd:AN copolymerization and the molecular weight of the obtained poly(Ann-r-Bdm) copolymers. Copolymerization was studied in DMAc, chlorobenzene (PhCl), 1,4-dioxane, tert-butanol, isobutyronitrile, toluene, trimethylacetonitrile, dimethyl carbonate, acetonitrile, methyl acetate, acetone and methyl tert-butyl ether (MTBE) at 100 °C with 2,2′-azobis(N-butyl-2-methylpropionamide) (VAm-110). Conversions after 22 h ranged from 15 wt% for methyl acetate to 35 wt% for DMAc.24 Such differences were explained by changes in the decomposition behaviour of the employed VAm-110 azo initiator as studied by ultraviolet-visible (UV-vis) spectroscopy. While copolymerization in DMAc with VAm-110 led to a conversion-dependent Mn values very close to theoretical expectations, the utilization of ACHN or 1-[(1-cyano-1-methylethyl)azo]formamide (V30) resulted in a strong deviation from linearity. The highest conversion (34%) and molecular weight (Mn = 100000 g mol−1) for poly(Ann-r-Bdm) were achieved when polymerization was conducted in DMAc with DoPAT and VAm-110 as CTA and initiator, respectively.24 Finally, attempts were made to increase NBR molecular weight through the combination of the RAFT technique with either copper-mediated alkyne–azide cycloaddition25 or Diels–Alder reactions.26 The optimal parameters for poly(Ann-r-Bdm) synthesis as determined in24 were used to produce NBR segments with Mn in the range of 1000–42000 g mol−1 and having either propargyl or cyclopentene end groups. These NBR building blocks were further reacted with 1,4-bis(azidomethyl)benzene or polymer segments with pyridine end groups to produce NBRs with molecular weights of up to 97000 g mol−1.25,26
In the present study, we report for the first time the controlled random copolymerization of Bd with styrene (St) via the solution RAFT technique (Scheme 1). Optimization of the reaction parameters (type of CTA and initiator, CTA:initiator ratio, reaction temperature and time) enabled the preparation of linear poly(Stn-r-Bdm) copolymers with molecular weights as high as Mn(SEC) = 29500 g mol−1 and Mw/Mn values in the range of 1.3–1.6. Furthermore, this approach was successfully applied to the preparation of various high molecular weight (Mn(SEC) = 52000–72400 g mol−1) poly[Xn-b-(Stm-r-Bdk)] block copolymers, where X represents different methacrylic or styrenic monomeric units. The latter highlights the versatility of this method and demonstrates how it may be used to prepare novel random and block copolymers with complex, highly engineered sequence distributions that promise interesting performance profiles. While practical application of this approach would require significant improvements in reaction kinetics and yield, this work nonetheless demonstrates a simpler, more accessible route to elastomeric block copolymers than traditional anionic polymerization.
 | ||
| Scheme 1 Synthesis of poly(Stn-r-Bdm) via solution RAFT polymerization. | ||
Experimental section
Materials
Chlorobenzene (PhCl, 99+%, Acros), anhydrous N,N-dimethylacetamide (DMAc, 99.8+%, Acros), anhydrous N,N dimethylformamide (DMF, 99.8%, Acros), scandium(III) trifluoromethanesulfonate (Sc(OTf)3, 99%, Aldrich), 4-cyano-4-[(dodecylsulfanyl thiocarbonyl)sulfanyl]pentanoic acid (CDTPA, 97%, Aldrich), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPCP), (97%, Aldrich), 1,1′-azobis(cyclohexanecarbonitrile) (ACHN, 98%, Aldrich), dicumyl peroxide (DCP, 98%, Aldrich), 2,2′-azobis(N-butyl-2-methylpropionamide) (VAm-110, >95%, Wako) and 4-methoxyphenol (99%, Aldrich) were used without further purification. Anhydrous tetrahydrofuran (THF) and toluene (>99%, Acros) were produced using an SPS solvent purification system (MBraun, Germany, aluminum oxide and 4 Å sieves columns). 1,1,2-Trichloroethane (TCA, 97%, Aldrich) and 1-methylnaphthalene (95%, Aldrich) were purified by distillation over CaH2. Trihexyltetradecyl phosphonium chloride (≥95.0%, Aldrich) was dried at 55 °C/1 mbar for 24 hours. 2,2′-Azobis(2-methylpropionitrile) (AIBN, 98%, Aldrich) and 4,4′-azobis(4-cyanovaleric acid) (ACVA, ≥98.0%, Aldrich) were crystallized from methanol. 1,3-Butadiene (Bd, ≥99.3%, Linde Gas Benelux B. V.) was distilled through a column filled with 5 Å molecular sieves and activated aluminum oxide spherical balls (BASF F-200). Isobornyl methacrylate (IBOMA, VISIOMER® Terra IBOMA, 99%, EVONIK Operations GmbH), methyl methacrylate (MMA, ≥99%, Aldrich) and styrene (St, 99.9%, Aldrich) were distilled over CaH2 prior to use.Methods
NMR spectra were recorded on an AMX-600 spectrometer (Bruker, Germany) at 25 °C in the indicated deuterated solvents and listed in ppm. The signal corresponding to the residual protons of the deuterated solvent was used as an internal standard for 1H and 13C NMR. Signal assignment was performed using 2D NMR techniques: heteronuclear single quantum coherence (HSQC), heteronuclear multiple bond correlation (HMBC), H–H correlation spectroscopy (H–H COSY). IR spectra were acquired on a Bruker Tensor 27 Fourier IR-spectrometer (Bruker, USA) using ATR technology (128 scans, 2 cm−1 resolution).A 1260 Infinity II gel permeation chromatograph (GPC, Agilent Technologies, USA) was used to determine Mn, Mw and Mw/Mn of the polymers. The chromatograph was equipped with an integrated IR detector, a PLgel 5 mm MIXED-C, PLgel 5 mm MIXED-D columns and a PLgel guard column (Agilent Technologies, USA). CHCl3 or THF was used as an eluent with a flow rate of 1.0 mL min−1 at 40 °C. Polystyrene standards (Agilent Technologies, Mp = 162–1500 × 103 g mol−1) were used to perform calibration for poly(Stn-r-Bdm) and poly[Stn-b-(Stm-r-Bdk)]. Polymethylmethacrylate (Agilent Technologies, Mp = 500–1500 × 103 g mol−1) were used to perform calibration for the poly(IBOMA), poly(MMA), poly[IBOMAn-b-(Stm-r-Bdk)] and poly[MMAn-b-(Stm-r-Bdk)].
Thermogravimetric analysis (TGA) was carried out in air on a TGA2 STARe System (Mettler Toledo, Switzerland), applying a heating rate of 5 °C min−1. The onset weight loss temperature (Tonset) was determined as the point in the TGA curve at which a significant deviation from the horizontal was observed. The resulting temperature was then rounded to the nearest 1 °C. DSC experiments were performed on a DSC3+ STARe System differential calorimeter (Mettler Toledo, Switzerland) with a heating rate of 10 °C min−1 in the range of −80 to 150 °C for SBR. Thermal mechanical analysis (TMA) of poly[Xn-b-(Stm-r-Bdk)] block copolymer samples was performed under inert atmosphere (He) using a DIL 402 select Expedis dilatometer (NETZSCH, Germany) with a constant load of 0.3 N at a heating rate of 3 °C min−1 in the range of −80 to 60 °C for the low Tg block (SBR block) and a heating rate of 10 °C min−1 in the range of −80 to 200 °C for the high Tg block (PIBOMA, PMMA, and PS).
Atomic force microscopy (AFM) images were recorded with an MFP-3D Infinity microscope (Asylum Instruments/Oxford Instruments, United Kingdom) in tapping mode (−20 °C, in air). AC160TS-R3 (Olympus, Japan) cantilevers were applied with a stiffness of 26 N m−1 and resonance frequency of 300 KHz. The domain periodicity was evaluated from three different 1 × 1 μm2 images. On each image, two profiles were taken, and for each, the distance over ten consecutive periods was recorded. The images were recorded in the so-called ‘soft tapping mode’, to avoid deformation and indentation of the polymer surface by the tip. All the images were collected with the maximum available number of pixels (512) in each direction. The general procedure for the preparation of the samples for AFM was as follows: 150 mg of block copolymer were dissolved in 1.5 ml of chloroform and cast onto a glass slide at 22 °C. An inverted glass funnel with the neck filled with cotton was then place over top of the glass slide in order to ensure gradual evaporation (over the course of hours), thus enabling reorganization of the films to achieve (near-)equilibrium morphologies. Finally, the films were dried at 55 °C/1 mbar for 12 h. This approach was chosen in preference to standard thermal of the films (as commonly practiced in the block copolymer literature) given the tendency of the SBR blocks to undergo thermally induced crosslinking.
Solution RAFT copolymerization of styrene and 1.3-butadiene
Poly(Stn-r-Bdm) copolymers were prepared varying reaction conditions via the RAFT technique. Depending on the initiator type, the temperature was set to 60, 88, 100, and 115 °C for AIBN, ACHN, VAm-110, and DCP, respectively. A typical procedure for the synthesis of SBR with CDTPA RAFT agent and VAm-110 initiator is given below as an example of copoly10 (Table 1, entry 10):| Entry | Poly(Stn-r-Bdm) | CTA | Solvent | Initiator | Temperature (°C) | M n(target) | M n(SEC) (g mol−1) | M w/Mnb | Yieldc (%) |
|---|---|---|---|---|---|---|---|---|---|
|
a Reaction time 72 h, [Bd]:[St] = 85:15 by weight, [Bd + St] = 50 wt%, [CTA]:[initiator] = 5:1 by mol.
b By GPC in CHCl3 at 40 °C (calibration with PS standards).
c Isolated yield.
d
M
n of soluble fraction.
e Reaction time 192 h (8 days).
f Addition of 3 portions of VAm-110.
g [CTA]:[initiator] = 5:5 by mol.
h Ionic liquid (IL): trihexyltetradecylphosphonium chloride, [DMAc]:[IL] = 50:50 by volume.
i [Sc(CF3SO3)3]:[styrene + butadiene] = 1:73 by mol.
|
|||||||||
| 1 | Copoly 1 | CPCP | Chlorobenzene | AIBN | 60 | 100000 |
6 800 | 1.3 | 3 |
| 2 | Copoly 2 | CPCP | Chlorobenzene | ACHN | 88 | 100000 |
14160 |
2.3 | 6 |
| 3 | Copoly 3 | CPCP | Chlorobenzene | VAm-110 | 100 | 100000 |
16000 |
4.1 | 8 |
| 4 | Copoly 4 | CPCP | Chlorobenzene | DCP | 115 | 100000 |
23000d |
3.3 | 20 |
| 5 | Copoly 5 | CPCP | DMF | VAm-110 | 100 | 100000 |
23300 |
3.1 | 6 |
| 6 | Copoly 6 | CPCP | DMAc | VAm-110 | 100 | 100000 |
19800 |
2.5 | 12 |
| 7 | Copoly 7 | CPCP | TCA | VAm-110 | 100 | 100000 |
18000 |
2.0 | 5 |
| 8 | Copoly 8 | CDTPA | THF | VAm-110 | 100 | 100000 |
11000 |
2.1 | 9 |
| 9 | Copoly 9 | CDTPA | Chlorobenzene | VAm-110 | 100 | 100000 |
12300 |
1.3 | 9 |
| 10 | Copoly 10 | CDTPA | DMAc | VAm-110 | 100 | 100000 |
22300 |
1.8 | 16 |
| 11 | Copoly 11 | CDTPA | TCA | VAm-110 | 100 | 100000 |
15500 |
1.3 | 8 |
| 12 | Copoly 12 | CDTPA | DMAc | DCP | 115 | 100000 |
28300d |
2.5 | 39 |
| 13 | Copoly 13 | CDTPA | TCA | DCP | 115 | 100000 |
26000d |
1.6 | 27 |
| 14 | Copoly 14 | CDTPAe | DMAc | VAm-110 | 100 | 100000 |
29500 |
1.6 | 22 |
| 15 | Copoly 15 | CDTPA | DMAc | VAm-110 | 100 | 50000 |
12400 |
1.4 | 14 |
| 16 | Copoly 16 | CDTPA | DMAc | VAm-110 | 100 | 150000 |
27600 |
1.5 | 22 |
| 17 | Copoly 17 | CDTPA | DMAc | VAm-110 | 115 | 100000 |
18300 |
1.3 | 10 |
| 18 | Copoly 18 | CDTPAf | DMAc | VAm-110 | 100 | 100000 |
19500 |
1.5 | 13 |
| 19 | Copoly 19 | CDTPAg | DMAc | VAm-110 | 100 | 100000 |
20600 |
1.7 | 21 |
| 20 | Copoly 20 | CDTPA | DMAc/ILh | VAm-110 | 100 | 100000 |
16000 |
2.0 | 8 |
| 21 | Copoly 21 | CDTPA | DMAc/Sc(CF3SO3)3i | VAm-110 | 100 | 100000 |
17000 |
1.7 | 19 |
Bd (20.00 g, 370 mmol) was distilled into a pressure stable glass reactor (Büchiglasuster, Switzerland) pre-cooled at −20 °C under vacuum. The autoclave was equipped with a manometer, a gas inlet valve, a sampling valve closed with septum, a security disk and a magnetic stirrer. After the distillation of Bd, the reactor was filled with an inert atmosphere (Ar) up to 0.1 bar overpressure.
A solution of styrene (3.53 g, 34 mmol), CDTPA (0.0950 g, 0.235 mmol) and VAm-110 (0.0735 g, 0.235 mmol, CDTPA:Vam −110 = 1:1 by mol) in 31 ml of anhydrous DMAc was placed into a separate Schlenk flask and degassed via three freeze-pump-thaw cycles. This solution was further injected via syringe into the reactor containing Bd under overpressure of the inert atmosphere (Ar) at −20 °C. The reactor was heated to 100 °C under vigorous stirring (300–350 rpm), and the reaction was continued for 72 h (Caution: high pressure (5.5 bars) is reached very quickly). The course of the reaction was monitored by the pressure drop caused by 1,3-butadiene consumption. After the completion of the reaction, the glass autoclave was cooled down to 50 °C, the unreacted Bd was released from the reactor, and 0.06 g (0.48 mmol) of 4-methoxyphenol (inhibitor) in 4 ml of dichloromethane was added to quench polymerization. The resultant polymer was purified by double precipitation into the methanol excess, collected by decantation and dried at 55 °C/1 mbar for 12 hours. Yield: 4.96 g (21%); Mn(SEC) = 22300 g mol−1; Mw/Mn = 1.8; 1H NMR (600.2 MHz, CDCl3): δ = 7.45–6.80 (br. m, 5H, H6), 5.48 (br. m, H, H13), 5.40–5.00 (br. m, 2H, H9), 5.00–4.77 (br. m, 2H, H14), 3.90 (d, 2H, J = 7.3Hz, H7′), 3.27 (t, 2H, J = 7.5Hz, Alk-5), 2.48 (br. m, 2H, H8), 2.19 (br. m, 2H, H7), 2.10–1.68 (br. m, 6H, H10, H12), 1.63 (br. m, 2H, Alk-4), 1.35 (br. m, 2H, Alk-3), 1.28–1.03 (br. m, 9H, H1′, H1′, H2, H15), 0.80 (t, 3H, J = 14.0 Hz, H1); 13C NMR (150.9 MHz, CDCl3): δ = 145.4 (br. m, C6i), 142.7 (br. m, C13), 132.5–125.3 (br. m, C6, C9), 114.2 (br. m, C14), 45.7 (br. m, C8), 43.5 (br. m, C12), 42.9–42.0 (br. m, C15a), 40.1 (br. m, C7), 38.1 (br. s, C11), 36.9 (s, Alk-5), 35.6 (br. m, C16), 32.7 (br. m, C10b), 31.9 (s, Alk-1′′), 29.6–28.8 (multiple s, Alk-2, Alk-3), 28.0 (s, Alk-4), 27.4 (br. m, C10a), 25.5–24.7 (br. m, C15), 22.7 (s, Alk-1′), 14.1 (s, Alk-1) (see ESI† file for full assignment); IR (ATR-mode): 3063 (w, aromatic CH), 3025 (w, aromatic CH), 3003 (w, cis CH), 2915 (s, CH), 2843 (m, CH), 1712 (w, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (CTA)), 1639 (w, cis –CHCH–), 1602 (w, CC aromatic), 1493 (m, CC aromatic), 1449 (s), 1436 (s), 1349 (w), 1308 (w), 1261 (w), 1074 (m), 1029 (w), 993 (m, vinyl CH2), 965 (vs, trans –CHCH–), 910 (vs, vinyl CH2), 805 (m), 759 (m, CH aromatic), 699 (vs, CH aromatic) cm−1; Tg = −58 °C (DSC); Tonset = 387 °C (TGA).
O (CTA)), 1639 (w, cis –CHCH–), 1602 (w, CC aromatic), 1493 (m, CC aromatic), 1449 (s), 1436 (s), 1349 (w), 1308 (w), 1261 (w), 1074 (m), 1029 (w), 993 (m, vinyl CH2), 965 (vs, trans –CHCH–), 910 (vs, vinyl CH2), 805 (m), 759 (m, CH aromatic), 699 (vs, CH aromatic) cm−1; Tg = −58 °C (DSC); Tonset = 387 °C (TGA).
Synthesis of macro-chain transfer agents (macro-CTA)
A set of macro-CTAs, namely, poly(IBOMA)12K, poly(IBOMA)26K, poly(IBOMA)36K, poly(IBOMA)46K, poly(MMA)40K and poly(St)43K was prepared by solution RAFT polymerization. A typical procedure for the synthesis of a macro-CTA is described below by an example of poly(MMA)40K (Table 3, entry 6) preparation:MMA (10.00 g, 100 mmol), CDTPA (0.0504 g, 0.125 mmol) and AIBN (0.0051 g, 0.031 mmol, CDTPA:AIBN = 4:1 by mol) were dissolved in 7 ml of anhydrous TCA in a Schlenk flask. The solution was deoxygenated by three freeze-pump-thaw cycles and sealed under an inert atmosphere (Ar). The reaction was conducted at 60 °C for 24 h. Polymerization was quenched by the injection of 0.06 g (0.48 mmol) of 4-methoxyphenol (inhibitor) solution in 4 ml of dichloromethane, and then the reaction mixture was diluted with dichloromethane. The resultant polymer was isolated by double precipitation into the excess of methanol and dried at 55 °C/1 mbar for 12 h. Yield: 9.5 g (95%); Mn(SEC) = 40300 g mol−1; Mw/Mn = 1.1; 1H NMR (600.2 MHz, CDCl3): δ = 3.54 (s, 3H, H8), 2.04–1.93 (br. m, 2H, H10), 1.93–1.69 (br. m, 2H, H6), 1.20 (br. s, 2H, H11), 1.15 (br. t, 3H, J = 6 Hz, H7mm), 0.96 (s, 3H, H7mr), 0.79 (s, 3H, H7rr); 13C NMR (150.9 MHz, CDCl3): δ = 178.3–176.1 (br. m, C13), 55.0–52.1 (br. m, C6), 51.8 (s, C8), 45.5 (s, C14mm), 44.8 (s, C14mr), 44.5 (s, C14rr), 31.7 (s, C11), 29.6–28.7, (m, Alk-2, Alk-3, Alk-4), 22.6 (s, Alk-1′), 21.0, (s, C7mm), 18.7 (s, C7mr), 16.4 (s, C7rr), 14.0 (s, Alk-1) (see ESI† file for full assignment); IR (ATR-mode): 2993 (w, CH), 2950 (m, CH), 2844 (w, CH), 1722 (vs, CO), 1479 (m, αCH2), 1435 (s), 1387 (w,–OCH3), 1266 (m), 1239 (s, asC–O–C), 1189 (s), 1143 (vs, sC–O–C), 1063 (m), 986 (m), 965 (m), 917 (w), 842 (m), 749 (m, αCH2) cm−1; Tg = 130 °C (TMA); Tonset = 267 °C (TGA).
Synthesis of poly[Xn-b-(Stm-r-Bdk)] block copolymers
A set of poly[Xn-b-(Stm-r-Bdk)] block copolymers was synthesized from the respective macro-CTAs by conducting solution RAFT random copolymerization of styrene and 1,3-butadiene. A typical procedure is provided by the example of the poly[MMA40K-b-(St-r-Bd)23K] synthesis (Table 3, entry 6):Bd (20.00 g, 370 mmol) was distilled into a pressure stable glass reactor (Büchiglasuster, Switzerland) pre-cooled at −20 °C under vacuum. Afterwards, the reactor was filled with an inert atmosphere (Ar) up to 0.1 bar overpressure. A solution of styrene (3.53 g, 34 mmol), poly(MMA)40K (9.41 g, 0.235 mmol), VAm-110 (0.0735 g, 0.235 mmol), poly(MMA)40K:VAm-110 = 1:1 (molar ratio), in 136 ml of anhydrous TCA was placed into a separate Schlenk flask and degassed via three freeze-pump-thaw cycles. The degassed solution was transferred via syringe into the reactor containing Bd at −20 °C. The mixture was stirred until the formation of a clear solution and the reactor was heated to 100 °C (Caution: Pressure reached 5.5 bars in 20 minutes). Polymerization was continued under stirring at 100 °C for 72 h. Then the reactor was cooled down to 50 °C, the unreacted Bd was released and a solution of 0.06 g (0.48 mmol) of 4-methoxyphenol (inhibitor) in 4 ml of dichloromethane was added to quench polymerization. The resultant viscous solution was diluted with dichloromethane and the copolymer was purified by double precipitation into the methanol excess. Product, representing yellow powder, was dried at 55 °C/1 mbar for 12 hours. Yield: 11.2 g (34%); Mn(SEC) = 63800 g mol−1; Mw/Mn = 1.4; 1H NMR (600.2 MHz, CDCl3): δ = 7.30–6.98 (br. m, 5H, H6), 5.54 (br. m, 1H, H13), 5.48–5.15 (br. m, 2H, H9), 5.03–4.84 (br. m, 2H, H14), 3.95 (d, 2H, J = 6Hz, H17), 3.58 (s, 3H, H19), 2.53 (s, 2H, H8), 2.24 (s, 2H, H7), 2.05–1.80 (br. m, H10, H12, H21), 1.24 (br. s, H22), 1.19 (br. t, 3H, J = 13 Hz, H16mm), 1.00 (br. t, 3H, H16mr), 0.83 (br. t, 3H, H16rr); 13C NMR (150.9 MHz, CDCl3): δ = 178.4–176.2 (br. m, C18), 145.3 (s, C6i), 142.6 (br. m, C13), 131.6–125.8 (br. m, C6, C9), 114.8–113.8 (br. m, C14), 54.7–52.2 (m, C15), 51.7 (s, C19), 45.7 (br. m, C8), 45.5 (s, C17mm), 44.9 (s, C17mr), 44.52 (s, C17rr), 43.5 (br. m, C12), 40.0 (br. m, C7), 38.1 (br. m, C11), 35.7 (br. m, C21), 32.7 (br. m, C10b, C22), 32.0–31.8 (br. m, C1′′), 30.5–29.9 (br. m, C2, C3, C4), 27.3 (s, C10a), 22.6 (s, C1′), 21.1 (br. m, C16mm), 18.7 (br. m, C16rm), 16.5 (s, C16rr) (see ESI† for full assignment); IR (ATR-mode): 2999 (w, CH), 2946 (m, CH), 2916 (m, CH), 2843 (m, CH), 1725 (vs, CO), 1640 (w, cis –CHCH–), 1483 (m, αCH2), 1435 (s), 1387 (m, –OCH3), 1364 (w), 1269 (m), 1241 (s, asC–O–C), 1190 (s), 1145 (vs, sC–O–C), 1064 (m), 993 (w, vinyl CH2), 965 (s, trans –CHCH–), 911 (m, vinyl CH2), 842 (w), 810 (w), 781 (w), 749 (w, αCH2), 730 (w), 700 (m, CH aromatic) cm−1. Tg1 = −8.5 °C and Tg2 = 132.1 °C (TMA).
Results and discussion part
Choosing a proper RAFT or chain transfer (CTA) agent is essential to achieve an effective control over molecular weight, realize a narrow molecular weight distribution, and construct macromolecules with well-defined architectures, including random, block or gradient copolymers.27 The goal of the study was to use RAFT polymerization as a versatile technique for the preparation of SBR-based block copolymers. Thus, the selection of the RAFT agent was limited by the condition that it should be able to polymerize monomers having different vinyl groups, namely, styrene, 1,3-butadiene and various methacrylates. As discussed in the introduction, for (co)polymerization of Bd the various RAFT agents based on dithioesters, trithiocarbonates and dithiobenzoates have been successfully applied.20–25 Similar CTAs were found to be quite effective in the controlled polymerization of styrene.28,29 However, not all of the above mentioned RAFT agents are capable of effectively polymerizing (meth)acrylates. Only those trithiocarbonate and dithiobenzoate CTAs (R–S–CS–Z), that possess an R leaving group with tertiary carbon having three different substituents including one cyano (CN) group, were reported to polymerize (meth)arcylates in a controlled manner.30–32 Such leaving groups with a tertiary radical show higher transfer constants and produce more stable radical species in comparison to groups containing primary or secondary carbons. Moreover, the electron-withdrawing effect of CN substituents increases the capability of the R group to reinitiate monomers as an expelled radical, leading to better control over the polymerization.33,34 Thus, to fulfill the requirement for effective and controlled polymerization of the various monomers mentioned above, the 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoic acid (CDTPA, trithiocarbonate-type) and 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPCP, dithiobenzoate-type) having a tertiary carbon with a cyano group were selected (Scheme 1). Afterwards, the study continued with the investigation of the influence of various reaction parameters (the type of RAFT agent, solvent and initiator, the temperature and the duration of polymerization, etc.) on solution RAFT copolymerization of styrene and butadiene, enabling the determination of the optimum conditions for the synthesis of poly(Stn-r-Bdm) with highest molecular weight and in highest yield (Table 1). Because of this goal, the results of the various polymerizations will be compared in terms of molecular weight, Mw/Mn ratios and yield. Finally, in order to ensure that the poly(Stn-r-Bdm) would possess the desired elastomeric character and low Tg, it was decided to fix the Bd:St ratio at 85:15 (w/w).
 | ||
| Scheme 2 Synthetic approach for the preparation of poly[Xn-b-(Stm-r-Bdk)] block copolymers. | ||
Effect of the RAFT agent
The influence of the RAFT agent on the synthesis of poly(Stn-r-Bdm) was examined in two different solvents, namely in N,N-dimethylacetamide (DMAc) and chlorobenzene (PhCl), at 100 °C with the VAm-110 (Table 1, entries 3, 6, 9 and 10) initiator. In terms of SBR molecular weight, the results obtained were ambiguous. The utilization of CDTPA in DMAc led to the synthesis of poly(Stn-r-Bdm) with higher molecular weight (Mn(SEC) = 22300 g mol−1) in comparison with SBR prepared in the same solvent with CPCP (19800 g mol−1) (Table 1, entries 6 and 10). In contrast, in chlorobenzene poly(Stn-r-Bdm) synthesized with CPCP showed higher molecular weight than with CDTPA: 16000 and 12300 g mol−1, respectively (Table 1, entries 3 and 9). It was observed that both RAFT agents influenced the Mw/Mn ratios of the resultant poly(Stn-r-Bdm). While the Mw/Mn ratios obtained with CDTPA were quite satisfactory (∼1.3–1.8), the dispersity of SBR synthesized with CPCP indicated a loss of control (Mw/Mn = 2.5 and 4.1 in DMAc and PhCl, respectively). In both solvents, the application of CDTPA resulted in slightly higher isolated yields of poly(Stn-r-Bdm) (16 and 9%) in comparison with utilization of CPCP (12 and 8%, correspondingly) (Table 1, entries 3,6 and 9,10). The discussed trends can be summarized as follows:| CPCP (Mn(SEC) = 19800 g mol−1) < CDTPA (22300) in DMAc |
| CDTPA (Mn(SEC) = 12300 g mol−1) < CPCP (16000) in PhCl |
| CDTPA (Mw/Mn = 1.8) < CPCP (2.5) in DMAc |
| CDTPA (Mw/Mn = 1.3) < CPCP (4.1) in PhCl |
It can be concluded that for the RAFT synthesis of poly(Stn-r-Bdm), the CDTPA agent was more preferable due to the achievement of a copolymer with lower molecular weight distribution and in higher yield. These results were found to be in agreement with those reported previously for the styrene RAFT homopolymerization.35
Effect of [CTA]/[initiator] ratio
The [CTA]/[initiator] ratio is a critical parameter that can affect the control over polymerization.36 The increase in the CTA:initiator ratio from 5:1 to 5:5 resulted in the improvement of the SBR's yield from 16 to 21%, but at the same time led to the decrease in Mn from 22300 to 20600 g mol−1 (Table 1, entries 10 and 19). Thus, for all future experiments the CTA:initiator ratio was fixed as 5:1 (Table 1). A reduction in initiator concentration might be expected to increase the livingness of the polymerization and the molecular weight of the product. However, this would also tend to reduce the yield still further, and was therefore not considered in the scope of the current effort.
Effect of initiator
The influence of initiator type on the copolymerization of styrene and Bd was studied through use of a set of azo-compounds, namely AIBN, ACHN and VAm-110, as well as DCP, as a representative of peroxide type initiators. The reaction temperatures were selected based on the initiator's 10 h half-life temperatures and ranged from 60 °C to 110 °C (Scheme 1). Utilization of azo-initiators in PhCl in combination with CPCP RAFT agent resulted in the synthesis of poly(Stn-r-Bdm) with the yields ranging from 3 to 8% (Table 1, entries 1–3). The overall evolution of molecular weight, molecular weight distribution and yields of SBRs in accordance with the nature of initiator used can be summarized as follows:| AIBN (SBR Mn(SEC) = 6800 g mol−1) < ACHN (14160) < Vam −110 (16000) |
| VAm-110 (Mw/Mn= 4.1) > ACHN (2.3) > AIBN (1.3) |
| AIBN (SBR yield: 3%) < ACHN (6) < VAm-110 (8) |
It was found that among azo-initiators, the VAm-110 provided the SBR with highest molecular weight (16000 g mol−1) and in highest yield (8%), although simultaneously with the broadest molecular weight distribution (Mw/Mn= 4.1) (Table 1, entries 1–3).
With the aim to study the influence of other initiators, the peroxide type DCP, having a working temperature closed to that of VAm-110, was selected. The utilization of DCP allowed for a significant increase in the molecular weight (up to 23000 g mol−1) and yield (up to 20%) of poly(Stn-r-Bdm) (Table 1, entries 1–4). One explanation for this is that DCP was promoting partial cross-linking as was supported by the presence of insoluble polymer parts. To confirm this conclusion, the copolymerization was conducted in the presence of DCP in two other solvents (Table 1, entries 12 and 13). In both TCA and DMAc, the utilization of DCP significantly increased the copolymer's yield and molecular weight, but again resulted in partial cross-linking. Analyzing all obtained results (Table 1), it is possible to conclude that higher polymerization temperatures (associated with different initiators) resulted in higher molecular weights and yields of SBR copolymers.
Keeping in mind that the duration of the reaction (72 h) was far longer than the half-life (10 h) of the VAm-110 at the chosen temperature (100 °C), the low copolymers yields were at first explained by the insufficient amount of initiating radicals/species. To test this assumption, additional amounts of VAm-110 solution in DMAc were injected at equal intervals, i.e. after 24 and 48 h of reaction (Table 1, entry 18). This led only to little change in copolymers yield (16 to 13%) and Mn (22300 to 19500 g mol−1) (Table 1, entry entries 10 and 18). The first possible explanation for low Mn and yield can be the low reactivity of 1.3-butadiene in radical polymerizations.22 For example, the kinetic study of 1,3-butadiene RAFT homopolymerization conducted by P. Xu and coworkers in toluene with AIBN initiator and DoPAT RAFT agent at 70 °C revealed that even for a target Mn of 3000 the conversion reached only 6% within 45 h of reaction, indicating the rate of homopolymerization was notably slow.22 In another study, the RAFT solution homopolymerization of 1.3-butadiene in acetonitrile with tert-butyl peroxy-3.5.5-trimethylhexonoate and 3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl-2-phenyl-2-((phenylcarbonothioyl)thio)ace-tate at higher temperature (110 °C) over 50 h resulted in an Mn of only 1400 g mol−1 and 16% conversion.20 The second possible reason for low molecular weight of SBR as detected by GPC may be branching of the obtained copolymer thanks to the presence of double bonds in the Bd repeats, though proving this would require an in-depth study beyond the scope of the current investigations.
Effect of solvent
The effect of solvent was investigated by first conducting the RAFT copolymerization in PhCl, DMF, DMAc and TCA with VAm-110 initiator and CPCP RAFT agent (Table 1, entries 3, 5, 6, and 7). The copolymerization at 100 °C for 72 h resulted in the preparation of SBRs with molecular weights ranging from 16000 to 23300 g mol−1. An acceptably narrow molecular weight distribution (Mw/Mn = 2.0) was obtained only in TCA, while the use of PhCl, DMF, and DMAC provided broader molecular weight distributions (Mw/Mn = 4.1, 3.1, and 2.5, respectively). Although by conducting the reaction in DMAc it was possible to increase the yield of the copolymer up 12%, the range of the obtained yields (5–12%) was generally low (Table 1, entries 3, 5, 6, and 7). The impact of solvent type on molecular weight, molecular weight distribution and yield of obtained poly(Stn-r-Bdm) copolymers can be summarized as follows:| PhCl (Mn = 16000 g mol−1) < TCA (18000) < DMAc (19800) < DMF (23300) |
| PhCl (Mw/Mn = 4.1) > DMF (3.1) > DMAc (2.5) > TCA (2.0) |
| TCA (Yield of SBR: 5%) < DMF (6) < PhCl (8) <DMAc (12) |
As previously noted, CDTPA was found to be the most effective RAFT agent for the copolymerization of Bd with styrene. Thus, the investigation of solvent effects was continued in the same set of solvents, but in the presence of the CDTPA RAFT agent (Table 1, entries 8–11). As a common observation, the transfer to CDTPA led to the slight increase in the molecular weight and yields of the obtained copolymers. At the same time, much lower Mw/Mn values in the range of 1.3–2.1 were observed.
Consistent with this observation, the GPC traces of poly(Stn-r-Bdm) copolymers produced in this fashion became remarkably narrow (Fig. 1). The obtained results can be arranged in the following order:
| THF (Mn = 11000 g mol−1) < PhCl (12300) < TCA (15500) < DMAc (22300) |
| THF (Mw/Mn = 2.1) > DMAc (1.8) > PhCl (1.3) ∼ TCA (1.3) |
| TCA (Yield of SBR: 8%) < THF (9) ∼ PhCl (9) < DMAc (16). |
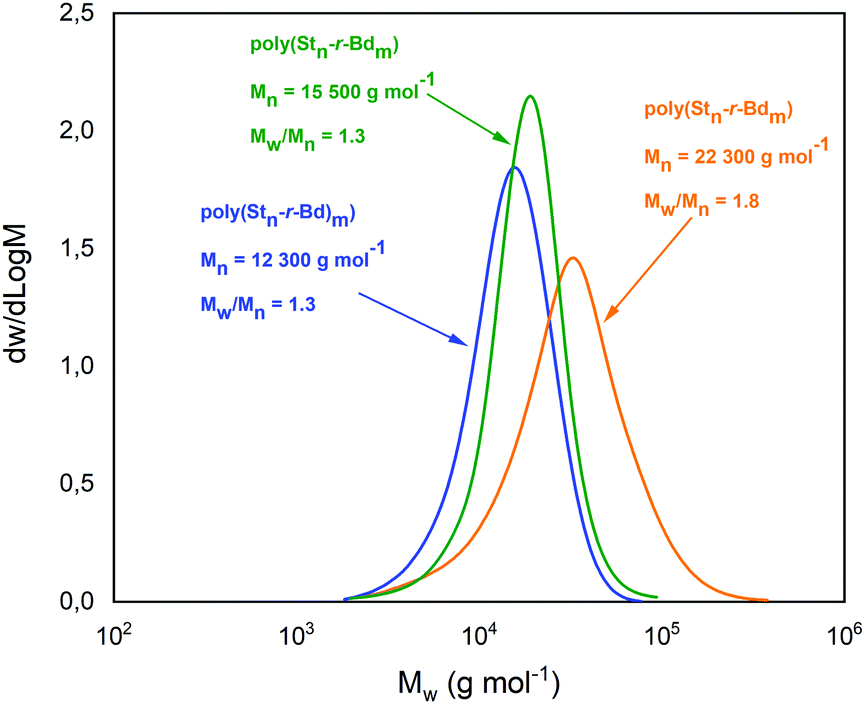 | ||
| Fig. 1 GPC traces of poly(Stn-r-Bdm) copolymers (CHCl3, PS standards) obtained by solution RAFT process (Table 1, entries 9–11). | ||
Providing a combination of high molecular weight and high yield at the cost of a small increase in Mw/Mn ratio, DMAc was identified as the optimal solvent for RAFT copolymerization of styrene and 1,3-butadiene at a given conditions (Table 1, entry 10).
Effect of temperature
The influence of temperature on the synthesis of poly(Stn-r-Bdm) copolymers was briefly mentioned in the section on the initiator effects. It was established that the increase in reaction temperature from 60 to 100 °C increased copolymer molecular weight and yield (Table 1, entries 1–3). While the recommended working temperature (the temperature of the 10 h half-life) for VAm-110 initiator is 110 °C, one additional experiment was performed in DMAc with CDTPA and VAm-110 at 115 °C (Table 1, entry 17). The increase of reaction temperature from 100 °C to 115 °C led to a reduction in the molecular weight (18300 g mol−1), molecular weight distribution (Mw/Mn: 1.3), and the yield (10%) in comparison to the polymerization performed at 100 °C (Table 1, entries 10 and 17). Thus, 100 °C was found to be optimal for copolymerization.
Effect of reaction duration
Although, 72 h of polymerization can be considered as excessive, especially taking into account the half-life of VAm-110 initiator equal to approximately 25 h at 100 °C, another experiment was carried out for 192 h (Table 1, entry 14). The prolongation of the reaction duration up to 8 days gave an increase in the molecular weight from 22300 to 29500 g mol−1, and the yield from 16 to 22% (Table 1, entries 10 and 14). This is consistent with the fact that, even after more than 8 days (approximately 8 half-lives), we would still expect to have (½)8 = 0.4% of unreacted VAm-110 remaining – meaning that VAm-110 decomposition and initiation is continuously occurring during the entire period. Taking into account the low reactivity of Bd vs. radical polymerization reactions, this provides additional time for molecular weight increase. Furthermore, it should also be noted that the presence of the RAFT agent ensures the formation of more stable, longer-lived radicals than would otherwise be generated through VAm-110 decomposition alone.
Effect of additives
Several attempts to increase the molecular weight and the yield of poly(Stn-r-Bdm) copolymer by application of various catalytic additives were also made.It was established previously that the conducting of polymerization in ionic liquids (ILs) as reaction medium37–41 or adding small amounts of ILs during bulk polymerization42 led to pronounced effects: (1) significant increase in the molecular weight of the resultant polymer; (2) improved yields of polymers (close to quantitative); (3) accelerated reaction rate. Thus, the first catalytic additive to test was the trihexyltetradecylphosphonium chloride IL. As styrene was not soluble in the neat IL, a [DMAc]:[IL] = 50:50 v/v ratio mixture was used for RAFT copolymerization (Table 1, entry 20). Unfortunately, however, the obtained poly(Stn-r-Bdm) copolymer precipitated during the reaction, decreasing both the Mn and yield of the SBR in comparison with work performed in neat DMAc (Table 1, entries 10 and 20).
Moad et al.43 investigated the modulation of the activity of an acid/base switchable dithiocarbamate RAFT agent, cyanomethyl (4-fluorophenyl)(pyridin-4-yl)carbamodithioate, with the Lewis acid scandium trifluoromethanesulfonate. It was revealed that Sc(OTf)3 was able to deliver improved control over RAFT copolymerization of methyl acrylate and vinyl acetate, Mn close to the target one and decreased Mw/Mn ratios of copolymers.43 Therefore, the effect of Sc(OTf)3 on styrene/BD RAFT copolymerization was studied as well (Table 1, entry 21). The introduction of Sc(OTf)3 to the RAFT system reduced the molecular weight from 22300 to 17000 g mol−1 and produced a bimodal molecular weight distribution as observed in the GPC curve (Table 1, entries 21 and 10), but slightly increased copolymer yield (from 16 to 19%).
Optimal conditions
Based on a detailed analysis of all the experiments in Table 1, it can be concluded that the optimal reaction conditions for the synthesis of poly(Stn-r-Bdm) copolymer with highest possible molecular weight and in highest yield are as follows: DMAc (solvent), CDTPA (RAFT agent), VAm-110 (initiator), [BD + St] = 50 wt%, [CTA]:[initiator] = 5:1 by mol, 100 °C and 192 h (Table 1, entry 14).
At this point it is important to briefly address the question of absolute yields in more detail, given that the maximum yield reported in Table 1 is 39%. While this may appear low in comparison to other investigations, the origins of the limited yields observed here stem from the initial choice of the Bd:St ratio as 85:15 (w/w). This is because, in addition to already slow reaction kinetics of St RAFT polymerization (Fig. S1, ESI†), it is well-known that Bd polymerizes even more slowly via radical processes. For these reasons, the overall propagation kinetics are very slow, the yields are low, and St units are overrepresented in the final copolymer composition as compared to the composition of the monomer feed (Table 2). To address the issue of low yields, three strategies are envisioned. First, we note that prior work involving the RAFT solution copolymerization of acrylonitrile and Bd at a 64:36 w/w ratio23–25 resulted in conversions of up to 62–64%. This shows that the presence of larger amounts of a more reactive monomer (such as St or acrylonitrile) favors greater conversion and a higher yield of the resulting copolymer. Second, while the additives studied here did not produce the desired increases in yield (Table 1, entries 20 and 21), this approach nonetheless deserves further attention, given the potential for novel additives to provide better performance. Third, in the case where some level of crosslinking may be tolerate in the final block copolymer, the use of peroxide initiators during the growth of the second block can provide significant increases in yield as well (Table 1, entry 4).
| Poly(Stn-r-Bdm) | M n(SEC) (g mol−1) | Stb (wt%) | (1,2)-Bdb (wt%) | (1,4)-Bdbc (wt%) | T g (°C) | T onset (°C) |
|---|---|---|---|---|---|---|
| a By GPC in CHCl3 at 40 °C (calibration with PS standards). b Microstructure determined by 1NMR using procedure published in.44 c cis and trans content were not separated. d T g determined by DSC. e T onset determined by TGA under inert atmosphere. | ||||||
| Copoly 9 | 12300 |
20.0 | 15.5 | 64.5 | −60 | 315 |
| Copoly 10 | 22300 |
19.6 | 15.4 | 65.0 | −62 | 310 |
| Copoly 11 | 15500 |
24.0 | 15.5 | 60.5 | −55 | 310 |
| Copoly 19 | 20600 |
21.0 | 15.0 | 64.0 | −58 | 315 |
Living character of (co)polymerization
Ideally, the living character of a RAFT (co)polymerization is revealed through a detailed study of the reaction kinetics. In this case, however, the low reactivity of Bd requires its use in high concentrations. This, in turn, means all (co)polymerizations must take place at high pressures (5.5–6 bar), which makes the collection of samples for the purpose of a traditional kinetic analysis impossible. Nonetheless, given the critical importance of establishing whether the (co)polymerization is living or not, we have studied the effect of the monomer/initiator ratio on molecular weight (Mn(SEC)) obtained from GPC. On the one hand, the expected linear relationship between the monomer/initiator ratio and the measured molecular weight is indeed observed, confirming the living character of the polymerization. However, the absolute values of the measured molecular weights are approximately four times lower than would be expected based on theoretical concerns. This relationship may be seen graphically in Fig. S9 (see ESI†).This data is correlated with the experiments presented in Table 1 (entries 10, 15 and 16). The discrepancy in molecular weights can be explained by several reasons: (1) the slope of the theoretical line is given at 100% of conversion, while the real conversions varied between 14 and 25%; (2) the obtained poly(Stn-r-Bdm) copolymer may be branched due to the presence of residual double bonds (though proving this is challenging and beyond the scope of the current work); (3) slow polymerization kinetics of both Bd and St in the given conditions. Indeed, the study of styrene RAFT polymerization kinetics performed in the optimal conditions (Fig. S1, ESI†) revealed that after 72 h of reaction the conversion reached only 62% and the actual molecular weight was still lower than the calculated one (63000 and 100000 g mol−1, respectively).
Properties of poly(Stn-r-Bdm) copolymers
Several poly(Stn-r-Bdm) copolymers, namely, copoly9-copoly11 and copoly19, have been selected from Table 1 for the investigation of their physical chemical properties (Table 2). These polymers were chosen as samples prepared in different solvents and with different [CTA]:[Initiator] ratios. At first, their structure and purity were proved by 1H and 13C NMR (Fig. 2 and Fig. S2, S3, ESI†). 1H NMR showed the presence of St (7.60–7.20, 7.09–6.89, 2.48, 2.19 ppm) and Bd units (5.55–5.12, 4.95–4.79, 2.06–1.80 ppm) as well as the peaks attributed to the remains of CDTPA agent (Fig. S2, ESI†). Further on, the composition or the microstructure of the copolymers was identified by 1H NMR in accordance with the previously published procedure.44 The only deviation from above mentioned technique44 was the choice of the deuterated solvent: instead of CS2:1,1,2,2-tetrachloroethane-d2 (2:1 v:v ratio) mixture the 1H NMR analysis in this work was performed in deuterated chloroform (Fig. S2, ESI†). All selected poly(Stn-r-Bdm) copolymers were found to be random. This statement is done based on the absence of any peak related to styrene blockiness (more than 5 units of styrene in a row) at 6.75–6.30 ppm (Fig. 2 and Fig. S2, ESI†). The styrene content slightly varied in the range of 19.6–24.0 wt%, while the vinyl content ((1,2)-Bd units) was practically fixed in between 15.0 and 15.5 wt% (Table 2). Accordingly, the (1,4)-Bd units fraction narrowly ranged from 60.5 to 65.0 wt%. Therefore, it can be concluded that the microstructure of poly(Stn-r-Bdm) copolymers within the same loading of monomers was nearly independent on the reaction conditions. The structure of the obtained random copolymers was additionally confirmed by IR spectroscopy (Fig. S4, ESI†). The following characteristic absorption bands were observed: 699 and 759 cm−1 (C–H deformation vibrations from aromatic ring), 910 and 993 cm−1 (vinyl CH2 vibrations), 964 cm−1 (trans –CHCH– vibrations), 1493 and 1602 cm−1 (CC vibrations from aromatic ring), 1639 cm−1 (cis –CHCH– vibrations), 1712 cm−1 (CO vibrations from RAFT agent), 2915 and 2843 cm−1 (aliphatic CH vibrations), 3003 (cis CH vibrations), 3025 and 3063 cm−1 (aromatic CH vibrations).
All tested copolymers were found to have similar solubility in organic solvents: they were soluble in chlorinated solvents (CHCl3, TCA, CH2Cl2), hydrocarbons (cyclohexane, toluene, hexane (at 50 °C)), some polar solvents (THF, diethyl ether, DMAc (at 60 °C)). They were found to be insoluble in acetonitrile, acetone, some aprotic polar solvents (DMF, DMSO, NMP) and alcohols (MeOH, EtOH). The observed solubility of poly(Stn-r-Bdm) copolymers was found to be almost identical to that of high molecular weight linear poly(Stn-r-Bdm) prepared via traditional solution anionic polymerization.
 | ||
| Fig. 2 1H NMR of poly(Stn-r-Bdm) copolymer (Table 1, entry 11) produced by solution RAFT copolymerization (more detailed assignment is presented in Fig. S3a and S3b, see ESI†). | ||
Thermal properties of copolymers were studied by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) (Table 2 and Fig. S10, S11, ESI†). According to DSC, poly(Stn-r-Bdm) copolymers showed one single glass transition temperature (Tg) ranging from −55 to −62 °C (Fig. S10, ESI†). As the vinyl content in the copolymers was nearly unaltered (15.0–15.5 wt%), the obtained Tg values were well correlated with the styrene content.45 Thus, the lower was the styrene fraction, the lower was the observed Tg (Table 2).
The thermal degradation behaviour of copolymers was assessed by TGA under inert atmosphere (Table 2). The weight loss profile of all copolymers revealed one-step degradation mechanism (Fig. S11, ESI†). It was found that the weight loss onset temperature Tonset varied in the range of 310–315 °C and the thermal stability of poly(Stn-r-Bdm) copolymers was nearly independent on the molecular weight.
Synthesis of poly[Xn-b-(Stm-r-Bdk)] block copolymers
To demonstrate the applicability of the suggested approach a set of different poly[Xn-b-(Stm-r-Bdk)] block copolymers was prepared via solution RAFT method (Table 3 and Fig. S4, S7, S8, ESI†). To narrow the dispersity and to obtain the block copolymers with as little branching as possible it was decided to start the process with the synthesis of well-defined macro-CTAs and then to perform chain extension with random copolymerization of St and Bd (Scheme 2). Both methacrylic (poly(IBOMA), poly(MMA)) and styrenic (poly(St)) types of macro-CTAs were used to show the versatility of method. MMA and St were selected as “classical” representatives, while IBOMA was chosen because the isobornyl bicyclic structure gives rise to methacrylate polymers with enhanced thermal stability and outstanding heat-resistance with Tg > 191 °C.46,47| Entry | A-block | B-block | poly[Xn-b-(Stm-r-Bdk)] (A-b-B copolymer) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Macro-CTA | M n(SEC) (g mol−1) | M w/Mnb | M n (g mol−1) | Block copolymers | M n(SEC) (g mol−1) | M w/Mnb | Yieldc (%) | T g1 (°C) | T g2 (°C) | Type of morphologye | Characteristic size A (nm) | Characteristic size B (nm) | |
|
a Reaction conditions: VAm-110, 1,1,2-trichloroethane (TCA), [Bd]:[St] = 85:15 wt%, [Bd + St] = 13 wt%, [CTA]:[VAm-110] = 5:1 by mol, 100 °C, 72 h, Mn(target) for SBR block = 100000 g mol−1.
b By GPC in THF at 40 °C (calibration with PS standards).
c Isolated yield.
d Determined by TMA.
e Determined by AFM: L-lamellar, D-disordered, ML-modulated lamellar (for detailed size characterisation see ESI).
|
|||||||||||||
| 1 | Poly(MMA)40K | 40300 |
1.1 | 23500 |
poly[MMA40K-b-(St-r-Bd)23K] | 63800 |
1.4 | 34 | −9 | 132 | L | 72 ± 11 | — |
| 2 | Poly(IBOMA)12K | 12400 |
1.1 | 39600 |
poly[IBOMA12K-b-(St-r-Bd)39K] | 52000 |
1.6 | 28 | −68 | 152 | D | — | — |
| 3 | Poly(IBOMA)26K | 26000 |
1.2 | 28700 |
poly[IBOMA26K-b-(St-r-Bd)28K] | 54700 |
1.6 | 28 | −6 | 184 | ML | 185 ± 96 | 89 ± 21 |
| 4 | Poly(IBOMA)36K | 36700 |
1.2 | 26300 |
poly[IBOMA36K-b-(St-r-Bd)26K] | 63000 |
1.5 | 33 | −6 | 187 | ML | 290 ± 72 | 74 ± 11 |
| 5 | Poly(IBOMA)46K | 46500 |
1.2 | 25900 |
poly[IBOMA46K-b-(St-r-Bd)25K] | 72400 |
1.5 | 40 | −5 | 185 | ML | 201 ± 95 | 51 ± 14 |
| 6 | Poly(St)43K | 43500 |
1.4 | 15000 |
poly[St43K-b-(St-r-Bd)15K] | 58500 |
1.6 | 36 | 4 | 102 | ML | 176 ± 44 | 64 ± 13 |
The various macro-CTAs were synthesized with CDTPA agent employing the optimal conditions determined previously.47 For comparison three macro-CTAs were prepared with the same Mn of around 40000 g mol−1 (Table 3, entries 1, 5, 6, ESI†), while for poly(IBOMA) the molecular weight was varied from 12400 to 46500 g mol−1 (Table 3, entries 1, 2–5). For methacrylic type macro-CTAs the Mw/Mn ratio did not exceed 1.2, implying excellent control over the polymerization. For poly(St)43K it was found to be slightly higher at Mw/Mn = 1.4 (Table 3, entry 6). The isolated macro-CTAs were further chain extended via the random copolymerization of Bd and St at an 85:15 monomer ratio (Scheme 2 and Table 3). The optimal conditions determined above were applied for this synthesis with the exception that DMAc was replaced with TCA due to the insolubility of some of macro-CTAs in DMAc. For all block copolymers SEC analysis showed monomodal peaks that were continuously shifted towards shorter elution times indicating the successful growth of the poly(St-r-Bd) chain (Fig. 3).
 | ||
| Fig. 3 GPC traces of macro-CTAs and respective block copolymers obtained via solution RAFT polymerization. | ||
The Mw/Mn ratio, ranging between 1.4 and 1.6, was considered satisfactory for RAFT polymerization. The molecular weight of the obtained block copolymers was in the range of 52000–72400 g mol−1 (Table 3). It was monotonously increasing from 25900 to 39600 g mol−1 with decreases in poly(IBOMA) macro-CTA Mn from 46500 to 12400 g mol−1, respectively (Table 3, entries 2–5). The molar mass of the block copolymer was found to be dependent of the type of methacrylic macro-CTA: in both experiments with poly(IBOMA)46K and poly(MMA)40K the grown Mn of SBR block reached 23500–39600 g mol−1 (Table 3, entries 1 and 5). Compared to RAFT copolymerization of poly(Stn-r-Bdm) alone, these molecular weights are higher, as expected, due to the higher initial viscosity of the reaction medium, which helps to suppress termination. At the same time, use of the styrenic macro-CTA allowed growth of only a 15000 g mol−1 poly(St-r-Bd) block (Table 3, entry 6).
The Tgs of poly[Xn-b-(Stm-r-Bdk)] block copolymers were evaluated using thermomechanical analysis (TMA). TMA method (Fig. S12, ESI†) was applied due to the uncertainty in high Tg determination via DSC as was reported previously for poly(IBOMA-b-Bd-b-IBOMA) triblock copolymers.48 The resultant copolymers displayed two distinct Tg temperatures on the thermomechanical curve, consistent with the formation of block copolymers (Table 3 and Fig. S12, ESI†). The transition at low temperatures (from −68 to 4 °C) was ascribed to the poly(St-r-Bd) block, whereas the Tg of poly(IBOMA), poly(MMA) and poly(St) blocks were observed in the high-temperature region at 184–187, 132 and 102 °C, respectively. The pronounced difference in the Tg of the poly(St-r-Bd) block can be explained by the variation in its size (Mn).
While the observation of a single GPC peak in combination with two separate Tg values, as detected here via TMA, is indeed consistent with the formation of a block copolymer, it is also true that a physical blend of homopolymers with similar molecular weights could also produce such a result. In order to fully exclude this possibility, one approach is to examine the phase behaviour of such systems. In the case of a blend of homopolymers, macroscopic phase-separation is expected on length-scales of tens to hundreds of microns or larger. In the case of block copolymers, on the other hand, phase-separation is inherently limited by molecular architecture resulting in the formation of nanoscale domains instead. Traditionally, such nanostructures are most often characterized either by microscopy (SEM, TEM, AFM, etc.) or scattering methods (SAXS, SANS, etc.), which are sensitive to the presence of such domains. Thus, to provide further evidence of successful poly[Xn-b-(Stm-r-Bdk)] block copolymer formation, thin films were solvent cast onto glass substrates with slow solvent evaporation to encourage the formation of (near-)equilibrium morphologies, then studied by atomic force microscopy (AFM). AFM images of phase shift (Fig. 4 and Fig. S14, ESI†) revealed that phase separation mostly occurred at the nano-scale and the self-assembly can be attributed to three categories.49 A clear lamellar morphology was observed for the poly[MMA40K-b-(St-r-Bd)23K] copolymer with a domain size of ∼72 nm (Fig. 4(a) and (b)). For poly[IBOMA-b-(St-r-Bd)] copolymers the type of morphology was dependent on the ratio between the lengths of both blocks (Fig. 4(c)–(h)). When the poly(IBOMA) content was small in poly[IBOMA12K-b-(St-r-Bd)39K] the phase separation was found to be disordered. However, the increase in poly(IBOMA) content led to segregation in a modulated lamellar (ML) morphology (Fig. 4(c)–(h)). Here, the diameter of the ribbon-like structures decreased from 89 to 51 nm on transition from poly[IBOMA26K-b-(St-r-Bd)28K] to poly[IBOMA46K-b-(St-r-Bd)25K]. A similar ML morphology was demonstrated by the poly[St43K-b-(St-r-Bd)15K] block copolymer.
 | ||
| Fig. 4 AFM phase images of block copolymers: poly[MMA40K-b-(St-r-Bd)23K] (a) and (b), poly[IBOMA26K-b-(St-r-Bd)28K] (c) and (d), poly[IBOMA36K-b-(St-r-Bd)26K] (e) and (f), poly[IBOMA46K-b-(St-r-Bd)25K] (g), (h) and poly[St43K-b-(St-r-Bd)15K] (i) and (j). The dark areas show the soft part of the sample, while the bright domains represent the hard part. | ||
Conclusions
For the first time, the suitability in principle of a solution RAFT process (in contrast to the emulsion RAFT method) for the random copolymerization of styrene (St) and 1,3-butadiene (Bd) (poly(Stn-r-Bdm)) was shown. It was demonstrated that the use of trithiocarbonate (4-Cyano-4 [(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (CDTPA)) chain transfer agent (CTA) leads to the desired copolymers in higher yield and with higher molecular weight in comparison with dithiocarbonate CTA. The optimization of reaction parameters such as polymerization temperature and time, type of CTA, solvent and initiator allowed for the successful preparation of soluble poly(Stn-r-Bdm) copolymers in 39% isolated yield and with Mn values of up to 29500 g mol−1 and Mw/Mn ≤ 1.6. All obtained copolymers were random and did not contain any styrene blocks. Their composition was nearly independent of reaction conditions and consisted of 19.6–24.0, 15.0–15.5 and 60.5–64.5 wt% of styrene, (1,2)-Bd and (1,4)-Bd units, respectively.
To demonstrate the applicability and versatility of this approach, a range of poly[Xn-b-(Stm-r-Bdk)] block copolymers was prepared via the solution RAFT method starting from well-defined macro-CTAs synthesized from various methacrylic (poly(IBOMA), poly(MMA)) and styrenic (poly(St)) monomers. Successful block copolymer synthesis was confirmed by different methods including SEC, TMA and AFM analyses. The molecular weight of poly[Xn-b-(Stm-r-Bdk)] block copolymers was mainly dependent on the molar mass of the starting poly(Xn) macro-CTA and reached as high as 72000 g mol−1, with the attached SBR segment extension varying between 11800 and 39600 g mol−1.
In sum, this approach provides a novel, readily accessible means of preparing copolymers with complex architectures based on a range of monomers, including those with functional groups, in order to generate high performance materials with tailored properties. The advantages of this method include (1) the relative simplicity of the reaction (no need for extreme purification of the monomers, less sensitivity towards moisture), (2) the control over the copolymer molecular weight and molecular weight distribution, and (3) the tolerance towards functional groups in monomers. This unique combination of useful characteristics highlights the promise of this approach (in spite of the long reaction times) as a new tool for the synthesis of next-generation copolymers with specifically designed and highly attractive performance profiles, where one of the blocks will possess rubbery properties and can be additionally cross-linked.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Luxembourg National Research Fund (FNR) through the IPBG project TireMat-Tech (Grant No. 16/11514551). Authors would like to thank Prof. Barner-Kowollik (Queensland University of Technology (QUT), Brisbane, Australia) for providing valuable advices and discussions. EVONIK Operations GmbH (Germany, Essen, http://www.visiomer.com) is greatly acknowledged for the supplying of isobornyl methacrylate (VISIOMER® Terra IBOMA, IBOMA). Authors would like to warmly thank Benoit Marcolini (LIST) and Régis Vaudemont (LIST) for their help and advices related to materials characterization.Notes and references
- J. A. Brydson, in Developments in Rubber Technology-2: Synthetic Rubbers, ed. A. Whelan and K. S. Lee, Springer, Netherlands, Dordrecht, 1981, pp.21–49, DOI: DOI:10.1007/978-94-009-8108-9_2.
- B. Rodgers and A. Halasa, in Encyclopedia of Polymer Blends, ed. A. I. Isayev, 2011, ch. 4, pp.163–206, DOI: DOI:10.1002/9783527805242.ch4.
- M.-C. Iovu, S. Mapolie and A. G. Britchi, Macromol. Symp., 2001, 165, 55–62 CrossRef CAS.
- C. A. da Silva, H. Budde, M. Menzel, U. Wendler, M. Bartke, M. Weydert and M. Beiner, RSC Adv., 2016, 6, 50460–50470 RSC.
- E. Kim, E. Lee, I. Park and T. Chang, Polym. J., 2002, 34, 674–681 CrossRef CAS.
- J. E. Kennedy, D. M. Devine, J. G. Lyons, L. M. Geever and C. L. Higginbotham, J. Mater. Sci., 2009, 44, 889–896 CrossRef CAS.
- L. Lei, L. Han, H. Ma, R. Zhang, X. Li, S. Zhang, C. Li, H. Bai and Y. Li, Macromolecules, 2021, 54, 2691–2702 CrossRef CAS.
- E. Passaglia and F. Donati, Polymer, 2007, 48, 35–42 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, 2008, pp.235–284 Search PubMed.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- S. Pragliola, T. Acierno and P. Longo, Polym. J., 2013, 45, 904–908 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- K. Nieswandt, P. Georgopanos, M. Held, E. Sperling and V. Abetz, Polymers, 2021, 14, 62 CrossRef.
- R. Wei, Y. Luo, W. Zeng, F. Wang and S. Xu, Ind. Eng. Chem. Res., 2012, 51, 15530–15535 CrossRef CAS.
- G. Bar-Nes, R. Hall, V. Sharma, M. Gaborieau, D. Lucas, P. Castignolles and R. G. Gilbert, Eur. Polym. J., 2009, 45, 3149–3163 CrossRef CAS.
- G. Moad, Polym. Int., 2017, 66, 26–41 CrossRef CAS.
- F. Lauterbach, M. Rubens, V. Abetz and T. Junkers, Angew. Chem., Int. Ed., 2018, 57, 14260–14264 CrossRef CAS PubMed.
- P. Lebreton, B. Ameduri, B. Boutevin and J.-M. Corpart, Macromol. Chem. Phys., 2002, 203, 522–537 CrossRef CAS.
- P. Ganjeh-Anzabi, V. Haddadi-Asl, M. Salami-Kalajahi and M. Abdollahi, J. Polym. Res., 2013, 20, 248 CrossRef.
- R. Wei, Y. Luo and P. Xu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2980–2989 CrossRef CAS.
- A. Kaiser, S. Brandau, M. Klimpel and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1616–1621 CrossRef CAS PubMed.
- C. J. Dürr, S. G. J. Emmerling, A. Kaiser, S. Brandau, A. K. T. Habicht, M. Klimpel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 174–180 CrossRef.
- C. J. Dürr, S. G. J. Emmerling, P. Lederhose, A. Kaiser, S. Brandau, M. Klimpel and C. Barner-Kowollik, Polym. Chem., 2012, 3, 1048–1060 RSC.
- C. J. Dürr, L. Hlalele, A. Kaiser, S. Brandau and C. Barner-Kowollik, Macromolecules, 2013, 46, 49–62 CrossRef.
- E. Rizzardo, G. Moad and S. H. Thang, Handbook of RAFT Polymerization, 2008, pp.189–234, DOI: DOI:10.1002/9783527622757.ch6.
- A. Goto, K. Sato, Y. Tsujii, T. Fukuda, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2001, 34, 402–408 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- M. W. M. Fijten, R. M. Paulus and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3831–3839 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- X. Zhang, O. Giani, S. Monge and J.-J. Robin, Polymer, 2010, 51, 2947–2953 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- A. Favier and M.-T. Charreyre, Macromol. Rapid Commun., 2006, 27, 653–692 CrossRef CAS.
- S. Harrisson, S. R. Mackenzie and D. M. Haddleton, Macromolecules, 2003, 36, 5072–5075 CrossRef CAS.
- Y. S. Vygodskii, O. A. Mel’nik, E. I. Lozinskaya, A. S. Shaplov, I. A. Malyshkina, N. D. Gavrilova, K. A. Lyssenko, M. Y. Antipin, D. G. Golovanov, A. A. Korlyukov, N. Ignat’ev and U. Welz-Biermann, Polym. Adv. Technol., 2007, 18, 50–63 CrossRef CAS.
- I. Woecht, G. Schmidt-Naake, S. Beuermann, M. Buback and N. García, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1460–1469 CrossRef CAS.
- P. Kubisa, Prog. Polym. Sci., 2009, 34, 1333–1347 CrossRef CAS.
- E. Andrzejewska, M. Podgorska-Golubska, I. Stepniak and M. Andrzejewski, Polymer, 2009, 50, 2040–2047 CrossRef CAS.
- S. A. Chesnokov, M. Y. Zakharina, A. S. Shaplov, E. I. Lozinskaya, I. A. Malyshkina, G. A. Abakumov, F. Vidal and Y. S. Vygodskii, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2388–2409 CrossRef CAS.
- A. Tselepy, T. L. Schiller, S. Harrisson, C. Guerrero-Sanchez, G. Moad and D. J. Keddie, Macromolecules, 2018, 51, 410–418 CrossRef CAS.
- A. F. Halasa, C. Jasiunas, B. Hsu, S. Henning and K. S. Seo, J. Appl. Polym. Sci., 2013, 127, 2116–2120 CrossRef CAS.
- A. F. Halasa, J. Prentis, B. Hsu and C. Jasiunas, Polymer, 2005, 46, 4166–4174 CrossRef CAS.
- A. Matsumoto, K. Mizuta and T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2531–2539 CrossRef CAS.
- A. Gunaydin, C. Mugemana, P. Grysan, C. Eloy Federico, R. Dieden, D. F. Schmidt, S. Westermann, M. Weydert and A. S. Shaplov, Polymers, 2021, 13, 1626 CrossRef CAS PubMed.
- J. M. Yu, P. Dubois and R. Jérôme, Macromolecules, 1996, 29, 7316–7322 CrossRef CAS.
- F. Bates, M. Schulz, A. Khandpur, S. Förster and J. Rosedale, Faraday Discuss., 1994, 98, 7 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Kinetics of styrene RAFT polymerization, assigned 1H, 13C NMR and IR spectra of poly(MMA)n macro-CTA, poly(Stn-r-Bdm) and poly[MMAn-b-(Stm-r-Bdk)] copolymers, TGA plot of poly(Stn-r-Bdm). See DOI: https://doi.org/10.1039/d2nj02286a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |